The Kaposi’s Sarcoma-Associated Herpesvirus Protein ORF42 Is Required for Efficient Virion Production and Expression of Viral Proteins


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Plasmids
2.3. Bacterial Artificial Chromosome (BAC) Mutagenesis and Generation of KSHV-Infected Lines
2.4. Lentiviral Transduction and Transient Transfection
2.5. Virion Measurements by Flow Cytometry and qPCR
2.6. Measurement of Cell-Associated Virions
2.7. Measurement of Viral DNA Replication
2.8. Virion Isolation
2.9. Protein Isolation and Western Blotting
2.10. RNA Isolation and RT-qPCR
2.11. Nuclear-Cytoplasmic Fractionation
2.12. Metabolic Labeling
2.13. Statistical Analysis
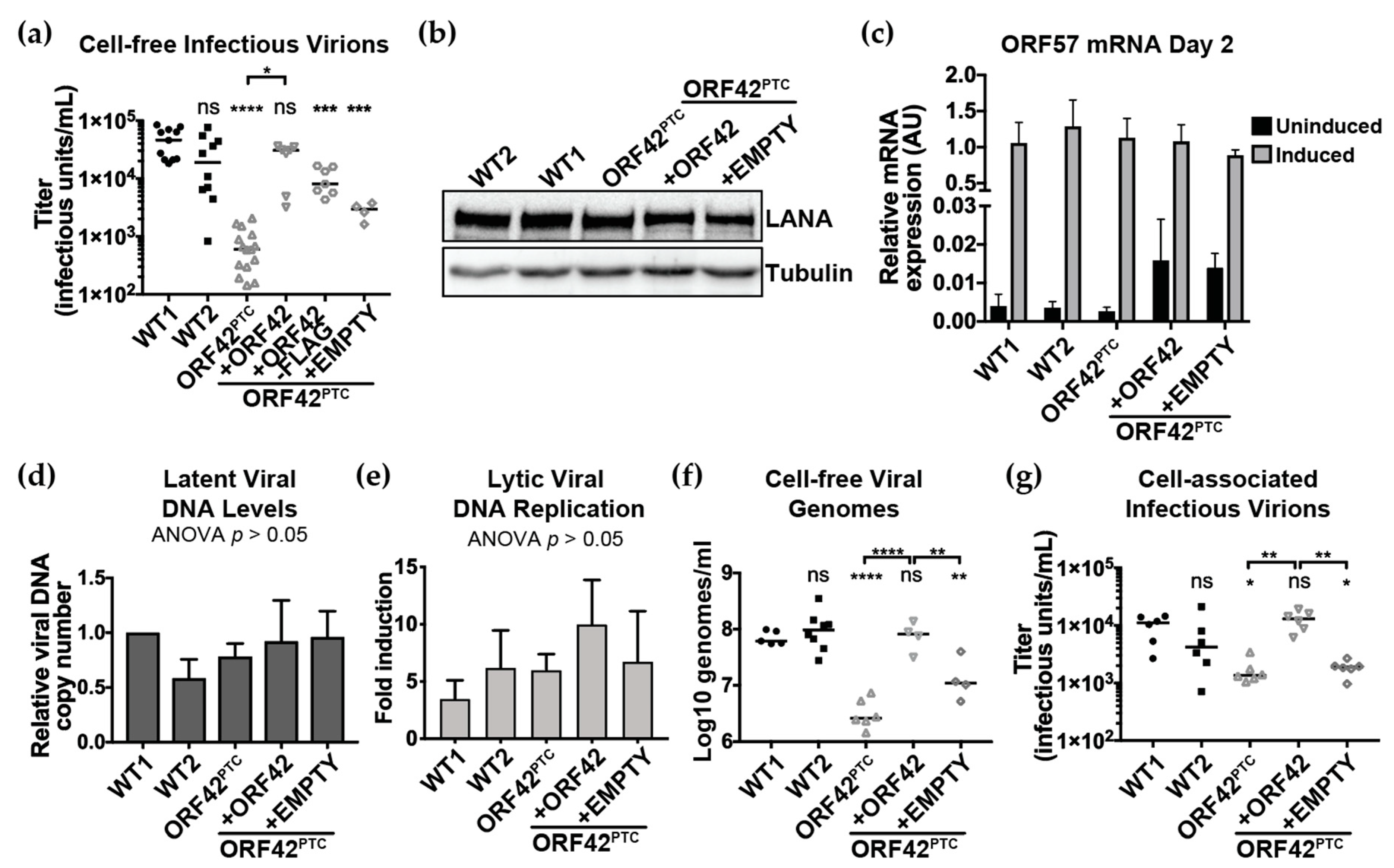
3. Results
3.1. Generation and Initial Characterization of KSHV ORF42PTC Infected Cells
3.2. KSHV ORF42 is Required for Efficient Production of Virions
3.3. ORF42 Is a Late Cytoplasmic Protein
3.4. ORF42 Is Required for Wild-Type Levels of Some Viral Proteins
3.5. ORF42 Activity May Potentiate Gene Expression at a Post-Transcriptional Level
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease (see comments). Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Uldrick, T.S.; Wang, V.; O’Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An Interleukin-6-Related Systemic Inflammatory Syndrome in Patients Co-Infected with Kaposi Sarcoma-Associated Herpesvirus and HIV but without Multicentric Castleman Disease. Clin. Infect. Dis. 2010, 51, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Kuppermann, B.D.; Wolitz, R.A.; Palestine, A.G.; Li, H.; Robinson, C.A. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N. Engl. J. Med. 1999, 340, 1063–1070. [Google Scholar] [CrossRef]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Loret, S.; Guay, G.; Lippé, R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 2008, 82, 8605–8618. [Google Scholar] [CrossRef]
- Nozawa, N.; Daikoku, T.; Yamauchi, Y.; Takakuwa, H.; Goshima, F.; Yoshikawa, T.; Nishiyama, Y. Identification and characterization of the UL7 gene product of herpes simplex virus type 2. Virus Genes 2002, 24, 257–266. [Google Scholar] [CrossRef]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G.; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, J.; Mocarski, E. Cytomegalovirus UL103 Controls Virion and Dense Body Egress. J. Virol. 2011, 85, 5125–5135. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, W.; Granzow, H.; Klopfleisch, R.; Klupp, B.G.; Rosenkranz, D.; Mettenleiter, T.C. The UL7 Gene of Pseudorabies Virus Encodes a Nonessential Structural Protein Which Is Involved in Virion Formation and Egress. J. Virol. 2005, 79, 11291–11299. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Ortiz, D.A.; Gurczynski, S.J.; Khan, F.; Pellett, P.E. Identification of human cytomegalovirus genes important for biogenesis of the cytoplasmic virion assembly complex. J. Virol. 2014, 88, 9086–9099. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sata, T.; Kawaguchi, Y. The product of the Herpes simplex virus 1 UL7 gene interacts with a mitochondrial protein, adenine nucleotide translocator 2. Virol. J. 2008, 5, 125. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Fan, S.; Zhou, J.; Zhang, Y.; Che, Y.; Cai, H.; Wang, L.; Guo, L.; Liu, L.; Li, Q. The mutated tegument protein UL7 attenuates the virulence of herpes simplex virus 1 by reducing the modulation of α-4 gene transcription. Virol. J. 2016, 13, 152. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Kedes, D.H. Mass Spectrometric Analyses of Purified Rhesus Monkey Rhadinovirus Reveal 33 Virion-Associated Proteins. J. Virol. 2006, 80, 1574–1583. [Google Scholar] [CrossRef]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef]
- Bortz, E.; Whitelegge, J.P.; Jia, Q.; Zhou, Z.H.; Stewart, J.P.; Wu, T.-T.; Sun, R. Identification of proteins associated with murine gammaherpesvirus 68 virions. J. Virol. 2003, 77, 13425–13432. [Google Scholar] [CrossRef]
- Bechtel, J.T.; Winant, R.C.; Ganem, D. Host and viral proteins in the virion of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 4952–4964. [Google Scholar] [CrossRef]
- Zhu, F.X.; Chong, J.M.; Wu, L.; Yuan, Y. Virion proteins of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Dai, X.; Xiao, Y.; Du, Y.; Chapa, T.J.; Johnson, J.R.; Li, X.; Krogan, N.J.; Deng, H.; Wu, T.-T.; et al. Virus-Like Vesicles of Kaposi’s Sarcoma-Associated Herpesvirus Activate Lytic Replication by Triggering Differentiation Signaling. J. Virol. 2017, 91, e00362-17. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.A.; Glassbrook, J.E.; Pellett, P.E. Protein-Protein Interactions Suggest Novel Activities of Human Cytomegalovirus Tegument Protein pUL103. J. Virol. 2016, 90, 7798–7810. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Malnati, M.S.; Scarlatti, G.; Gatto, F.; Salvatori, F.; Cassina, G.; Rutigliano, T.; Volpi, R.; Lusso, P. A universal real-time PCR assay for the quantification of group-M HIV-1 proviral load. Nat. Protoc. 2008, 3, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Speicher, D.J.; Johnson, N.W. Detection of human herpesvirus 8 by quantitative polymerase chain reaction: Development and standardisation of methods. BMC Infect. Dis. 2012, 12, 210. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, F.; Ye, F.; Gao, S.-J. Genetic Disruption of KSHV Major Latent Nuclear Antigen LANA Enhances Viral Lytic 2 Transcriptional Program. Virology 2008, 379, 234–244. [Google Scholar] [CrossRef]
- Abernathy, E.; Clyde, K.; Yeasmin, R.; Krug, L.T.; Burlingame, A.; Coscoy, L.; Glaunsinger, B. Gammaherpesviral Gene Expression and Virion Composition Are Broadly Controlled by Accelerated mRNA Degradation. PLoS Pathog. 2014, 10, e1003882. [Google Scholar] [CrossRef]
- Davis, Z.H.; Verschueren, E.; Jang, G.M.; Kleffman, K.; Johnson, J.R.; Park, J.; von Dollen, J.; Maher, M.C.; Johnson, T.; Newton, W.; et al. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 2015, 57, 349–360. [Google Scholar] [CrossRef]
- Brulois, K.F.; Chang, H.; Lee, A.S.-Y.; Ensser, A.; Wong, L.-Y.; Toth, Z.; Lee, S.H.; Lee, H.-R.; Myoung, J.; Ganem, D.; et al. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 2012, 86, 9708–9720. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.-C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.L.; Pulliam, T.H.; Dimaio, T.A.; Thalhofer, A.B.; Delgado, T.; Lagunoff, M. Glycolysis, Glutaminolysis, and Fatty Acid Synthesis Are Required for Distinct Stages of Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Davis, Z.H.; Hesser, C.R.; Park, J.; Glaunsinger, B.A. Interaction between ORF24 and ORF34 in the Kaposi’s Sarcoma-Associated Herpesvirus Late Gene Transcription Factor Complex Is Essential for Viral Late Gene Expression. J. Virol. 2016, 90, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.R.; Glaunsinger, B.A. Kaposi’s Sarcoma-Associated Herpesvirus ORF68 Is a DNA Binding Protein Required for Viral Genome Cleavage and Packaging. J. Virol. 2018, 92, 13. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-J.; Avey, D.; Li, W.; Gillen, J.; Fu, B.; Miley, W.; Whitby, D.; Zhu, F. ORF33 and ORF38 of Kaposi’s Sarcoma-Associated Herpesvirus Interact and Are Required for Optimal Production of Infectious Progeny Viruses. J. Virol. 2016, 90, 1741–1756. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, W.; Shao, Y.; Avey, D.; Fu, B.; Gillen, J.; Hand, T.; Ma, S.; Liu, X.; Miley, W.; et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe 2015, 18, 333–344. [Google Scholar]
- Toptan, T.; Fonseca, L.; Kwun, H.J.; Chang, Y.; Moore, P.S. Complex Alternative Cytoplasmic Protein Isoforms of the Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen 1 Generated through Noncanonical Translation Initiation. J. Virol. 2013, 87, 2744–2755. [Google Scholar] [CrossRef]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef]
- Virgin, H.W.; Latreille, P.; Wamsley, P.; Hallsworth, K.; Weck, K.E.; Canto, A.J.D.; Speck, S.H. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 1997, 71, 5894–5904. [Google Scholar] [PubMed]
- Tso, K.K.-Y.; Yip, K.Y.-L.; Mak, C.K.-Y.; Chung, G.T.-Y.; Lee, S.-D.; Cheung, S.-T.; To, K.-F.; Lo, K.-W. Complete genomic sequence of Epstein-Barr virus in nasopharyngeal carcinoma cell line C666-1. Infect. Agent Cancer 2013, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-K.; Cho, H.; Roh, S.W.; Kim, S.-J.; Myoung, J. Cell Type-Specific Interferon-γ-mediated Antagonism of KSHV Lytic Replication. Sci. Rep. 2019, 9, 2372. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Rämer, P.C.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV Infection Increases EBV-Associated Tumor Formation In Vivo via Enhanced EBV Lytic Gene Expression. Cell Host Microbe 2017, 22, 61–73.e7. [Google Scholar]
- Liang, Q.; Chang, B.; Brulois, K.F.; Castro, K.; Min, C.-K.; Rodgers, M.A.; Shi, M.; Ge, J.; Feng, P.; Oh, B.-H.; et al. Kaposi’s Sarcoma-Associated Herpesvirus K7 Modulates Rubicon-Mediated Inhibition of Autophagosome Maturation. J. Virol. 2013, 87, 12499–12503. [Google Scholar] [CrossRef] [PubMed]
- Bergson, S.; Itzhak, I.; Wasserman, T.; Gelgor, A.; Kalt, I.; Sarid, R. The Kaposi’s-sarcoma-associated herpesvirus orf35 gene product is required for efficient lytic virus reactivation. Virology 2016, 499, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Dünn-Kittenplon, D.; Kalt, I.; Lellouche, J.-P.; Sarid, R. The KSHV portal protein ORF43 is essential for the production of infectious viral particles. Virology 2019, 529, 205–215. [Google Scholar] [CrossRef]
- Li, W.; Avey, D.; Fu, B.; Wu, J.-J.; Ma, S.; Liu, X.; Zhu, F. Kaposi’s Sarcoma-Associated Herpesvirus Inhibitor of cGAS (KicGAS), Encoded by ORF52, Is an Abundant Tegument Protein and Is Required for Production of Infectious Progeny Viruses. J. Virol. 2016, 90, 5329–5342. [Google Scholar] [CrossRef]
- Lu, M.; Suen, J.; Frias, C.; Pfeiffer, R.; Tsai, M.-H.; Chuang, E.; Zeichner, S.L. Dissection of the Kaposi’s sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J. Virol. 2004, 78, 13637–13652. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Thompson, S.R.; Mathews, M.B.; Mohr, I. Translational Control in Virus-Infected Cells. Cold Spring Harb Perspect. Biol. 2019, 11, a033001. [Google Scholar] [CrossRef]
- Zhu, F.X.; Li, X.; Zhou, F.; Gao, S.-J.; Yuan, Y. Functional characterization of Kaposi’s sarcoma-associated herpesvirus ORF45 by bacterial artificial chromosome-based mutagenesis. J. Virol. 2006, 80, 12187–12196. [Google Scholar] [CrossRef] [PubMed]
- Stürzl, M.; Gaus, D.; Dirks, W.G.; Ganem, D.; Jochmann, R. Kaposi’s sarcoma-derived cell line SLK is not of endothelial origin, but is a contaminant from a known renal carcinoma cell line. Int. J. Cancer 2013, 132, 1954–1958. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. KSHV and the pathogenesis of Kaposi sarcoma: Listening to human biology and medicine. J. Clin. Invest. 2010, 120, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Oda, S.; Watanabe, M.; Oyama, M.; Kozuka-Hata, H.; Koyanagi, N.; Maruzuru, Y.; Arii, J.; Kawaguchi, Y. Roles of the Phosphorylation of Herpes Simplex Virus 1 UL51 at a Specific Site in Viral Replication and Pathogenicity. J. Virol. 2018, 92, e01035-18. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, A.H.; O’Hare, P. Regulation and cell-type-specific activity of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. J. Virol. 1990, 64, 3269–3279. [Google Scholar] [PubMed]
- O’Connor, C.M.; Shenk, T. Human cytomegalovirus pUL78 G protein-coupled receptor homologue is required for timely cell entry in epithelial cells but not fibroblasts. J. Virol. 2012, 86, 11425–11433. [Google Scholar] [CrossRef] [PubMed]
- Caviness, K.; Bughio, F.; Crawford, L.B.; Streblow, D.N.; Nelson, J.A.; Caposio, P.; Goodrum, F. Complex Interplay of the UL136 Isoforms Balances Cytomegalovirus Replication and Latency. mBio 2016, 7, e01986-15. [Google Scholar] [CrossRef] [PubMed]
- Kuang, E.; Tang, Q.; Maul, G.G.; Zhu, F. Activation of p90 Ribosomal S6 Kinase by ORF45 of Kaposi’s Sarcoma-Associated Herpesvirus and Its Role in Viral Lytic Replication. J. Virol. 2008, 82, 1838–1850. [Google Scholar] [CrossRef]
- Butnaru, M.; Gaglia, M.M. Transcriptional and post-transcriptional regulation of viral gene expression in the gamma-herpesvirus Kaposi’s sarcoma-associated herpesvirus. Curr. Clin. Micro. Rep. 2018, 5, 219–228. [Google Scholar] [CrossRef]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011, 9, 860–875. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct | Sense | Purpose | Sequence |
|---|---|---|---|
| pJP1-ORF42-Flag | F | Amplify ORF42-Flag and add AgeI site for restriction enzyme-based cloning | CGCCACCGGTATGTCCCTGGAAAGGGCCCTG |
| pJP1-ORF42-Flag | R | Amplify ORF42-Flag and add EcoRI site | GCGGGAATTCTTAAACGGGCCCCTTGTCGTCG |
| pJP1-ORF42 | R | Amplify ORF42 (untagged) and add EcoRI for restriction enzyme-based cloning | CGAGAATTCTTATTTTGAAAAAAGGGAAACAATGGGGGG |
| pJP1-ORF42stop | F | Introduce Ser25Stop mutation | CAGGCCTCTTCTCACTCGCGAGTCTTCG |
| pJP1-ORF42stop | R | Introduce Ser25Stop mutation | CGAAGACTCGCGAGTGAGAAGAGGCCTG |
| pCDNA4/TO-ORF42 (untagged) | F | Amplify ORF42 (untagged) and clone with Gibson into PmeI-digested pCDNA4/TO vector | CGATCCAGCCTCCGGACTCTAGC |
| pCDNA4/TO-ORF42 (untagged) | R | Amplify ORF42 (untagged) and clone with Gibson into PmeI-digested pCDNA4/TO vector | GAGGCTGATCAGCGGGTTTAAACTTATTTTGAAAAAAGGGAAACAATGG |
| ORF42-Flag BAC16 | F | Amplify Flag tag, add ORF42 sequence 5’ of tag and remove ORF42 stop codon | CCCCCCATTGTTTCCCTTTTTTCAAAATCAGGGCGGCCGCTCGAGGGA |
| ORF42-Flag BAC16 | R | Amplify Flag tag, add beginning of KanR gene 3’ to the Flag sequence | CTACTTATCGTCGTCATCCTTTAAACGGGCCCCTTGTC |
| ORF42-Flag BAC16 | F | Amplify KanR gene, add end of Flag seq 5’ to KanR | GACGACAAGGGGCCCGTTTAAAGGATGACGACGATAAGTAGGG |
| ORF42-Flag BAC16 | R | Amplify KanR gene, add beginning of Flag seq 3’ to the KanR gene (homologous to sequences at the 5’ end of KanR gene) | TTAAACGGGCCCCTTGTCGTCGTCGTCCTTGTAGTCGATGAACCAATTAACCAATTCTG |
| ORF42PTC BAC | F | Introduce Ser25Stop mutation | GGAGTGCCAATGAGTACTCATGCCCCGAAGACTCGCGAGTGAGAAGAGGCCTGTCCCGTATAGGATGACGACGATAAGTAGGG |
| ORF42PTC BAC | R | Introduce Ser25Stop mutation | GGGCACCACAGGGTGGGGGTATACGGGACAGGCCTCTTCTCACTCGCGAGTCTTCGGGGCAAACCAATTAACCAATTCTGATTAG |
| ORF42REV BAC | F | Revert the Ser25Stop mutation to the wild-type sequence | GGAGTGCCAATGAGTACTCATGCCCCGAAGACTCGCGAGTCAGAAGAGGCCTGTCCCGTATAGGATGACGACGATAAGTAGGG |
| ORF42REV BAC | R | Revert the Ser25Stop mutation to the wild-type sequence | GGGCACCACAGGGTGGGGGTATACGGGACAGGCCTCTTCTGACTCGCGAGTCTTCGGGGCAAACCAATTAACCAATTCTGATTAG |
| pCMV-ORF26 | F | Amplify ORF26 locus and add sequences homologous to vector for Gibson cloning | GGTTTAGTGAACCGTCAGATCCGCTAGCAGCTAACCCTTCTAGCGTTGG |
| pCMV-ORF26 | R | Amplify ORF26 locus and add sequences homologous to vector for Gibson cloning | TAACGCTTACAATTTACGCCTTAAGTTTTTAATCGTGGTGTAACCAGTG |
| LANA standard | F | PCR amplification | CCGGTGGAGGTAAAGGTGTTGCGGG |
| LANA standard | R | PCR amplification | GCAGTCCTGCCTGGGGCACCAATCAG |
| CCR5 standard | F | PCR amplification | GCACAGGGTGGAACAAGATGG |
| CCR5 standard | R | PCR amplification | CCCAAGAGTCTCTGTCACCTGCATAG |
| LANA qPCR [25] | F | Measurement of viral DNA levels | AGGATGGAGATCGCAGACAC |
| LANA qPCR [25] | R | Measurement of viral DNA levels | CCAGCAAACCCACTTTAACC |
| CCR5 qPCR [26] | F | Measurement of CCR5 DNA levels | ATGATTCCTGGGAGAGACGC |
| CCR5 qPCR [26] | R | Measurement of CCR5 DNA levels | AGCCAGGACGGTCACCTT |
| ORF26 qPCR [27] | F | Measurement of viral mRNA levels | AGCCGAAAGGATTCCACCATT |
| ORF26 qPCR [27] | R | Measurement of viral mRNA levels | TCCGTGTTGTCTACGTCCAGA |
| K8.1 qPCR [28] | F | Measurement of viral mRNA levels | AAAGCGTCCAGGCCACCACAGA |
| K8.1 qPCR [28] | R | Measurement of viral mRNA levels | GGCAGAAAATGGCACACGGTTAC |
| 18S qPCR [29] | F | Measurement of rRNA levels | GTAACCCGTTGAACCCCATT |
| 18s qPCR [29] | R | Measurement of rRNA levels | CCATCCAATCGGTAGTAGCG |
| ORF57 qPCR [30] | F | Measurement of viral mRNA levels | GGTGTGTCTGACGCCGTAAAG |
| ORF57 qPCR [30] | R | Measurement of viral mRNA levels | CCTGTCCGTAAACACCTCCG |
| Viral Gene: | WT1 | WT2 | ORF42PTC | ORF42PTC + ORF42 | ORF42PTC + Empty | |
|---|---|---|---|---|---|---|
| Latent | ||||||
| LANA | latent | 1.0 | 1.0 ± 0.2 | 0.9 ± 0.2 | 1.0 ± 0.2 | 0.9 ± 0.3 |
| Day 4 | ||||||
| ORF6 | early | 1.0 | 1.3 ± 0.5 | 0.8 ± 0.2 | 1.9 ± 1.3 | 1.2 ± 0.4 |
| ORF59 | early | 1.0 | 1.2 ± 0.3 | 0.7 ± 0.1 | 1.1 ± 0.2 | 0.8 ± 0.2 |
| ORF68 | early | 1.0 | 1.3 ± 0.5 | 1.1 ± 0.9 | 1.6 ± 1.6 | 0.9 ± 0.5 |
| ORF26 | late | 1.0 | 0.9 ± 0.5 | 0.6 ± 0.5 | 1.3 ± 1.0 | 0.7 ± 0.7 |
| K8.1 | late | 1.0 | 1.9 ± 1.0 | 0.5 ± 0.4 | 1.2 ± 0.7 | 0.8 ± 0.5 |
| ORF33 | late | 1.0 | 0.7 ± 0.4 | 0.8 ± 0.8 | 1.3 ± 0.8 | 0.8 ± 0.1 |
| ORF52 | late | 1.0 | 0.9 ± 0.3 | 1.1 ± 0.5 | 2.1 ± 0.6 | 1.5 ± 0.4 |
| Day 6 | ||||||
| ORF6 | early | 1.0 | 0.8 ± 0.6 | 0.6 ± 0.2 | 0.8 ± 0.5 | 0.7 ± 0.5 |
| ORF59 | early | 1.0 | 1.0 ± 0.4 | 0.7 ± 0.4 | 1.2 ± 0.7 | 0.8 ± 0.3 |
| ORF68 | early | 1.0 | 1.2 ± 0.4 | 0.5 ± 0.2 | 0.9 ± 0.4 | 0.6 ± 0.4 |
| ORF26 | late | 1.0 | 1.5 ± 1.0 | 0.3 ± 0.3 | 0.9 ±0.8 | 0.4 ± 0.5 |
| K8.1 | late | 1.0 | 1.8 ± 1.7 | 0.3 ± 0.4 | 1.3 ± 2.4 | 0.4 ± 0.6 |
| ORF33 | late | 1.0 | 0.9 ± 0.3 | 0.4 ± 0.1 | 1.2 ± 0.3 | 0.7 ± 0.3 |
| ORF52 | late | 1.0 | 2.2 ± 2.6 | 2.0 ± 2.3 | 3.3 ± 3.7 | 2.0 ± 2.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butnaru, M.; Gaglia, M.M. The Kaposi’s Sarcoma-Associated Herpesvirus Protein ORF42 Is Required for Efficient Virion Production and Expression of Viral Proteins. Viruses 2019, 11, 711. https://doi.org/10.3390/v11080711
Butnaru M, Gaglia MM. The Kaposi’s Sarcoma-Associated Herpesvirus Protein ORF42 Is Required for Efficient Virion Production and Expression of Viral Proteins. Viruses. 2019; 11(8):711. https://doi.org/10.3390/v11080711
Chicago/Turabian StyleButnaru, Matthew, and Marta Maria Gaglia. 2019. "The Kaposi’s Sarcoma-Associated Herpesvirus Protein ORF42 Is Required for Efficient Virion Production and Expression of Viral Proteins" Viruses 11, no. 8: 711. https://doi.org/10.3390/v11080711
APA StyleButnaru, M., & Gaglia, M. M. (2019). The Kaposi’s Sarcoma-Associated Herpesvirus Protein ORF42 Is Required for Efficient Virion Production and Expression of Viral Proteins. Viruses, 11(8), 711. https://doi.org/10.3390/v11080711