Discovery of Four Novel Viruses Associated with Flower Yellowing Disease of Green Sichuan Pepper (Zanthoxylum armatum) by Virome Analysis

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Preparation and HTS
2.3. HTS Data Analysis and Virus Identification
2.4. Determination of Virus Genomes
2.5. HTS Data Reanalysis
2.6. Sequence and Phylogenetic Analysis
2.7. RT-PCR Protocols and Field Surveys
2.8. Graft Transmission
3. Results
3.1. Virus Discovery by Homology Analysis
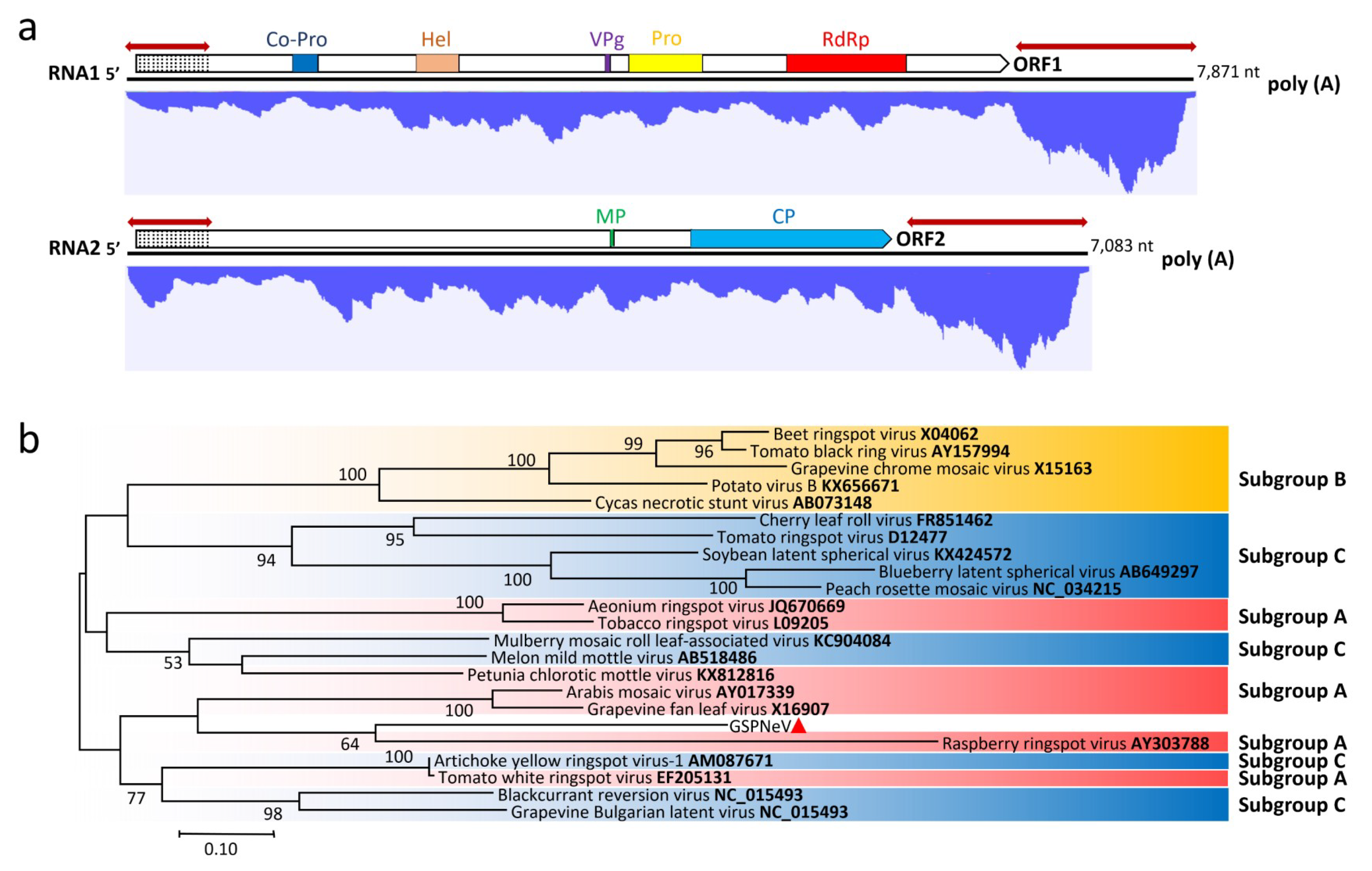
3.2. Genome Characterization and Classification of GSPNeV
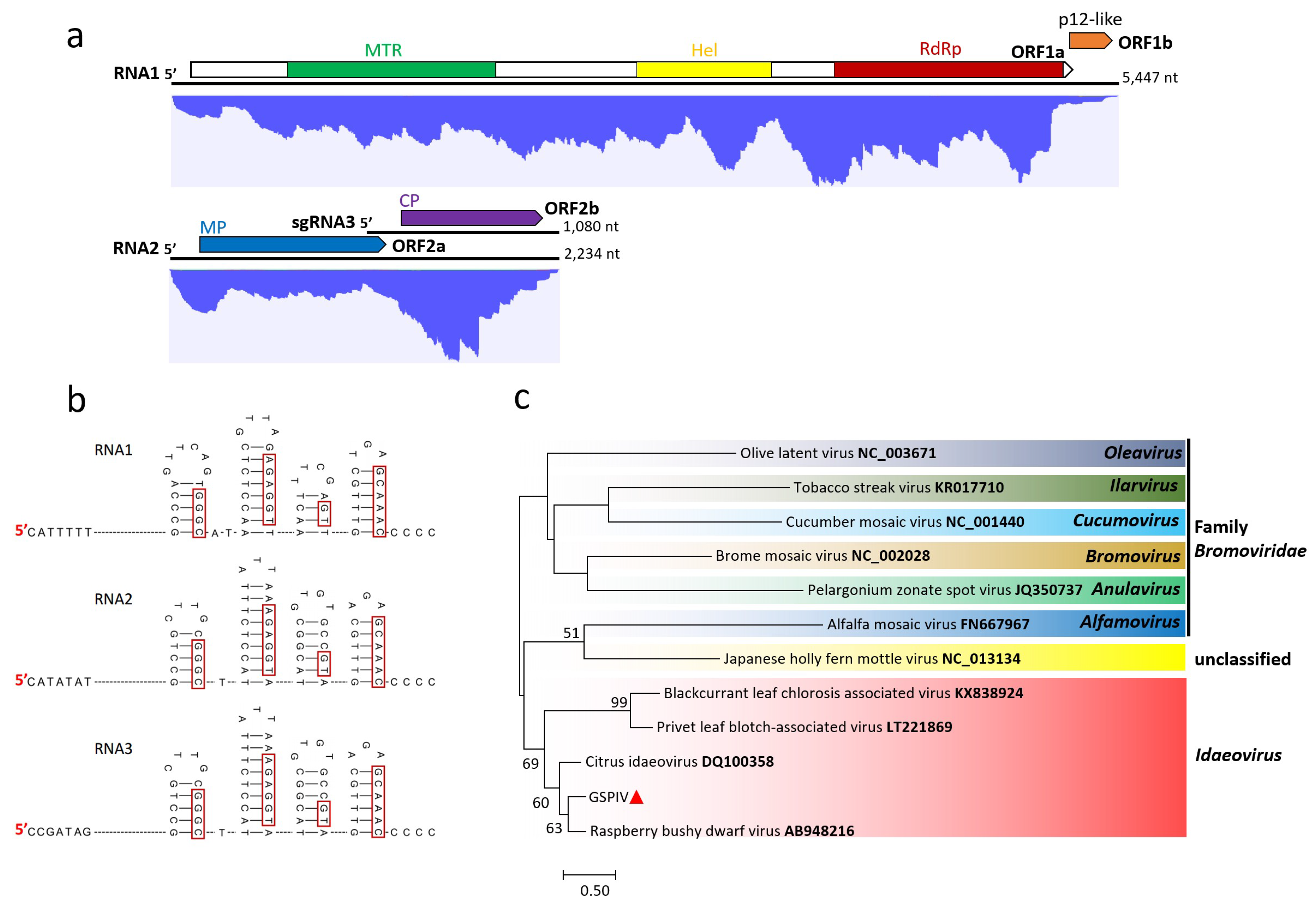
3.3. Genome Characterization and Classification of GSPIV
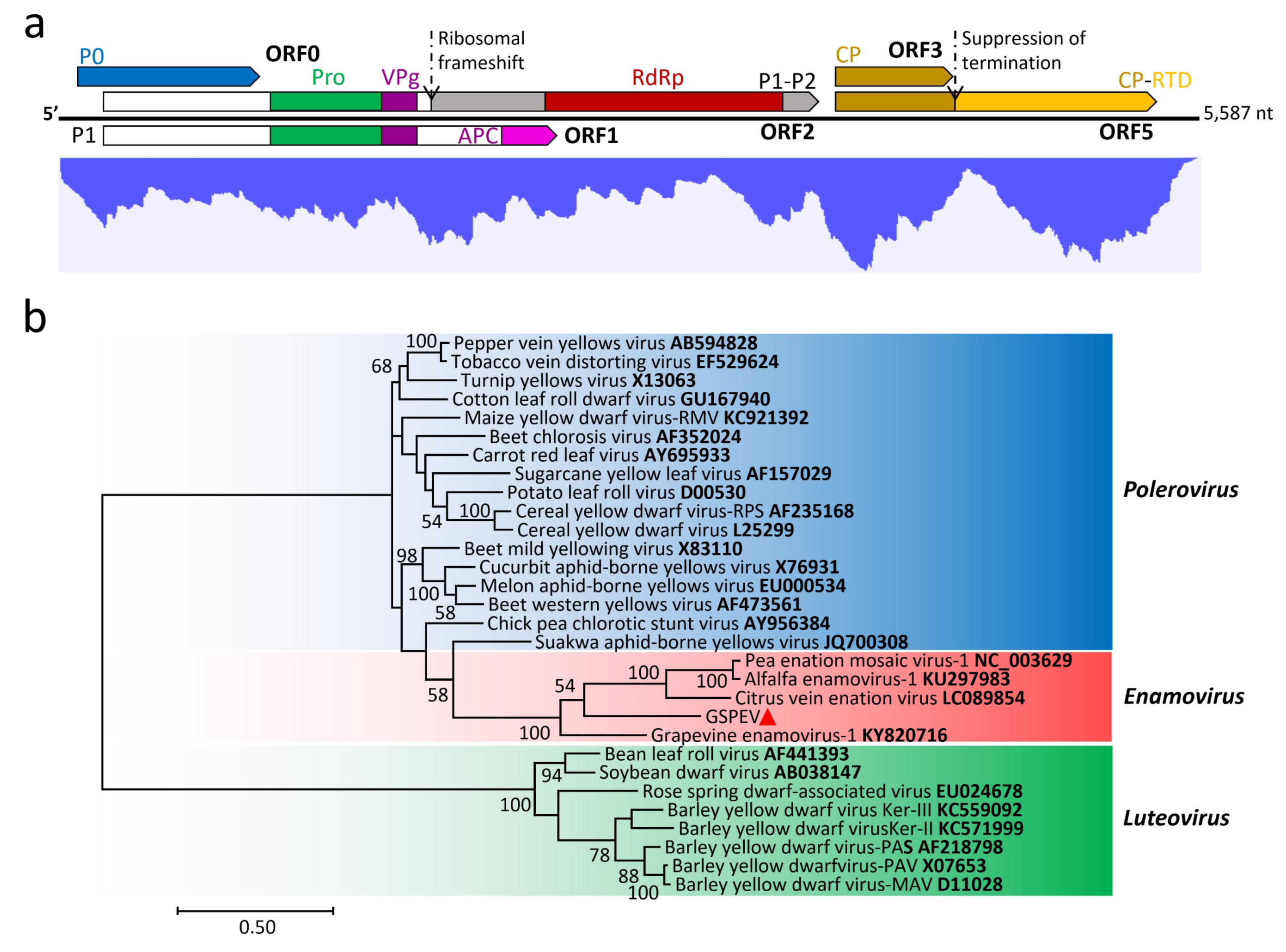
3.4. Genome Characterization and Classification of GSPEV
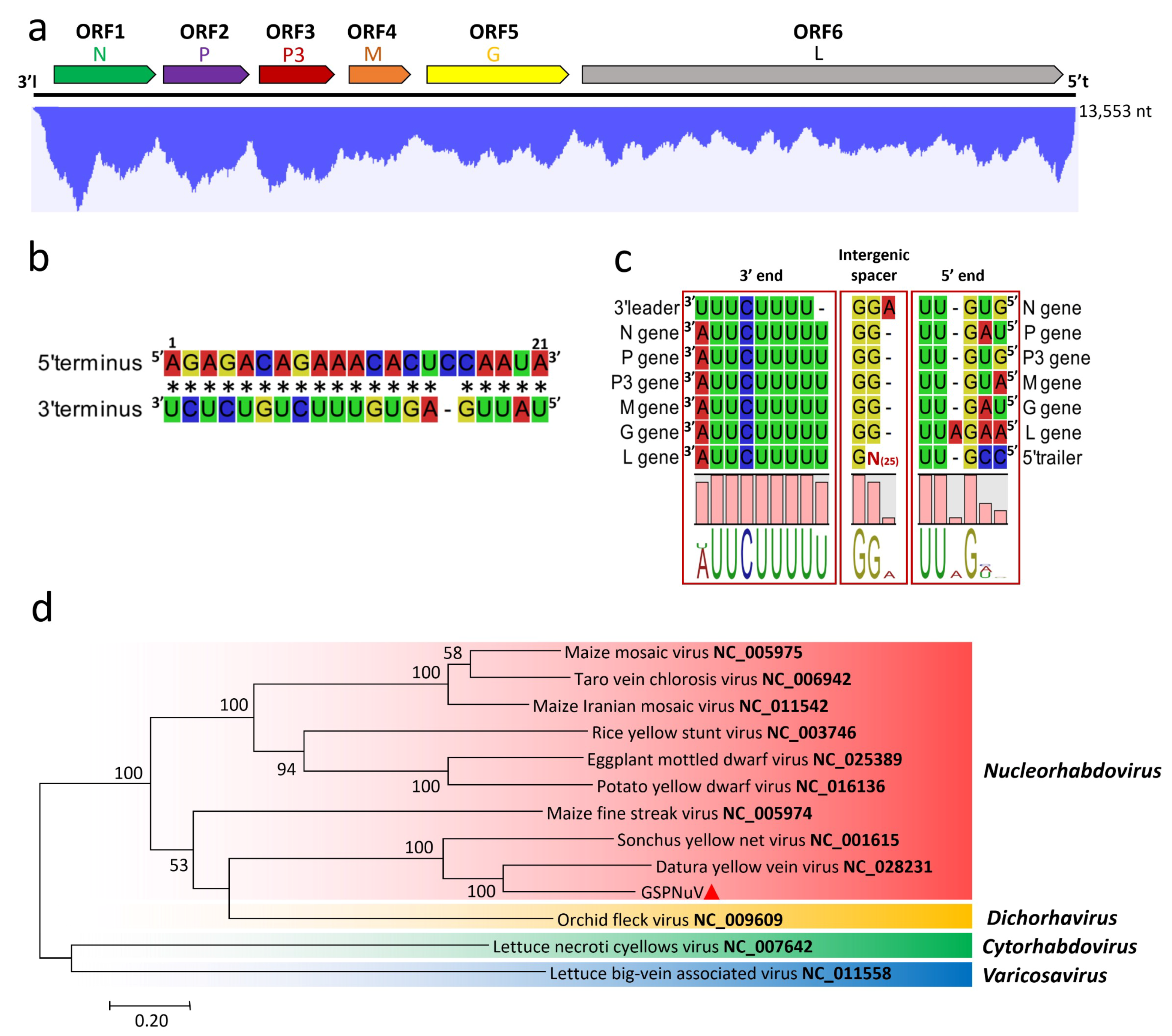
3.5. Genome Characterization and Classification of GSPNuV
3.6. Transcriptomic Analysis
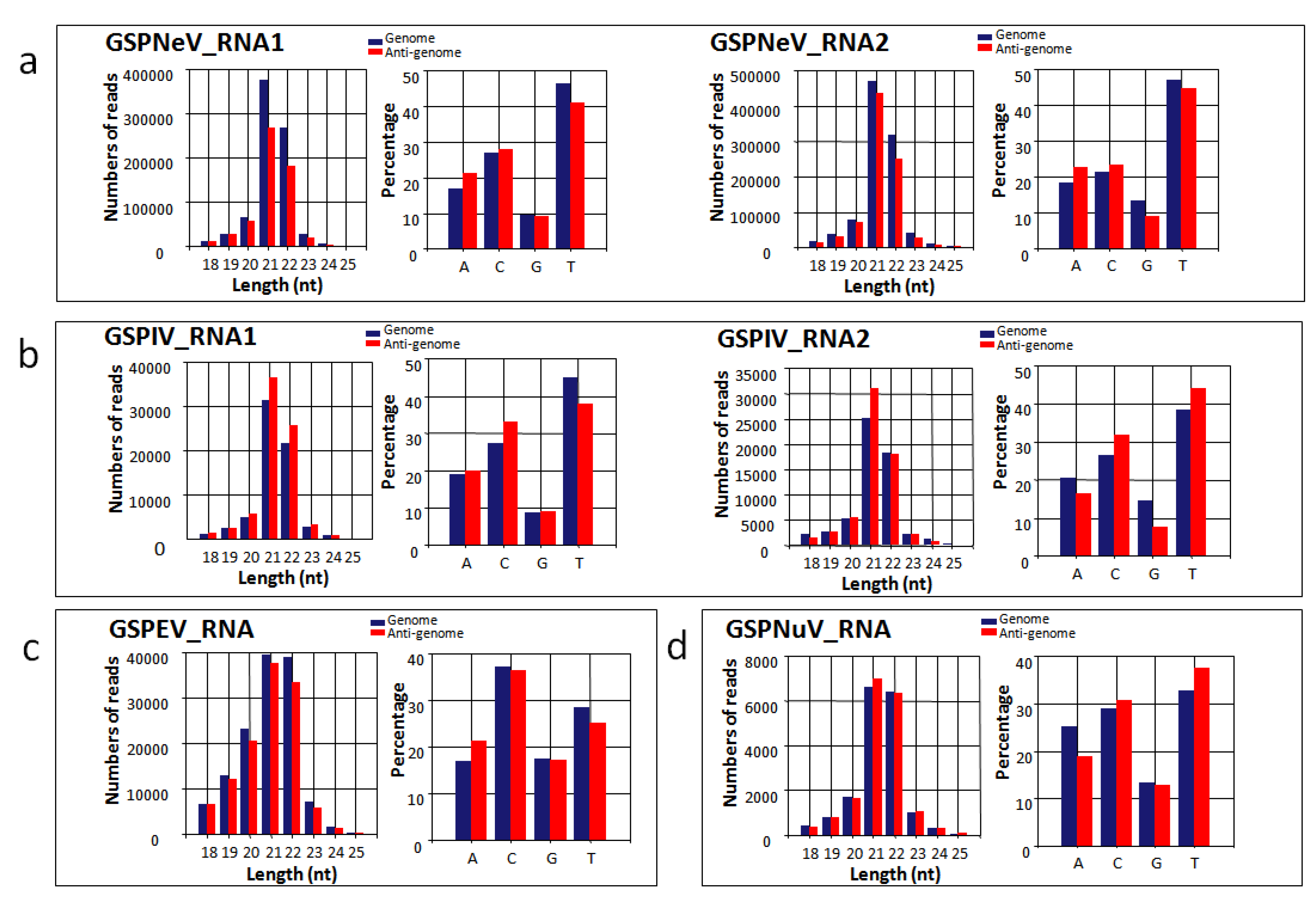
3.7. Small RNA Analysis
3.8. Virus Detection in Fields
3.9. Graft and Natural Transmissible
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Appelhans, M.S.; Reichelt, N.; Groppo, M.; Paetzold, C.; Wen, J. Phylogeny and biogeography of the pantropical genus Zanthoxylum and its closest relatives in the proto-Rutaceae group (Rutaceae). Mol. Phylgen. Evol. 2018, 126, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Negi, J.S.; Bisht, V.K.; Bhandari, A.K.; Singh, P.; Sundriyal, R.C. Chemical constituents and biological activities of the genus zanthoxylum: A review. Afr. J. Pure Appl. Chem. 2011, 5, 412–416. [Google Scholar]
- Zhu, H.; Huang, Y.J.; Ji, X.P.; Su, T.; Zhou, Z.K. Continuous existence of Zanthoxylum (Rutaceae) in Southwest China since the Miocene. Quatern. Int. 2016, 392, 224–232. [Google Scholar] [CrossRef]
- Phuyal, N.; Jha, P.K.; Raturi, P.P.; Rajbhandary, S. Zanthoxylum armatum DC.: Current knowledge, gaps and opportunities in Nepal. J. Ethnopharmacol. 2019, 229, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Tian, C.; Liang, Y.; Wang, P. An investigation of pricklyash diseases in shaanxi and gansu provinces. J. Northwest. For. Univ. 1994, 9, 139–143. [Google Scholar]
- Tang, W.; Xie, Q.; Guan, J.; Jin, S.; Zhao, Y. Phytochemical profiles and biological activity evaluation of Zanthoxylum bungeanum, maxim seed against asthma in murine models. J. Ethnopharmacol. 2014, 152, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, K.H.; Shin, H.D. First report of Septoria pachyspora causing leaf blotch of Zanthoxylum schinifolium. Plant Pathol. 2008, 57, 383. [Google Scholar] [CrossRef]
- Han, S.S.; Lee, K.J.; Kamala-Kannan, S. Detection of aster yellows phytoplasma (16SrI) associated with prickly ash (Zanthoxylum schinifolium S. et Z.) witches′ broom disease in Korea. J. Phytopathol. 2013, 161, 582–585. [Google Scholar] [CrossRef]
- Yang, W.X.; Liu, F.; Zhang, N.; Ren, X.D.; Liu, D.Q. First report of Althernaria alternata causing blight on Zanthoxylum piperitum in China. Plant Dis. 2013, 97, 840. [Google Scholar]
- Ning, P.; Liang, P.; Wang, X.; Huang, S.; Huang, Y.; Shi, A.; Chen, C.; Li, Q.; Hsiang, T. First report of leaf spot caused by Alternaria alternata on Zanthoxylum dissitum in China. Plant Dis. 2016, 100, 1233. [Google Scholar] [CrossRef]
- Zhou, X.; O’Donnell, K.; Aoki, T.; Smith, J.; Kasson, M.T.; Cao, Z.M. Two novel fusarium species that cause canker disease of prickly ash (Zanthoxylum bungeanum) in northern China form a novel clade with Fusarium torreyae. Mycologia 2016, 108, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, P.; Tyson, G.W. Microbiology: Metagenomics. Nature 2008, 455, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- Allander, T.; Tammi, M.T.; Eriksson, M.; Bjerkner, A.; Tiveljunglindell, A.; Andersson, B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc. Natl. Acad. Sci. USA 2005, 102, 12891–12896. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic analysis of the viromes of three North American bat species: Viral diversity among different bat species that share a common habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef]
- Roossinck, M.J. Plant virus metagenomics: Biodiversity and ecology. Annu. Rev. Genet. 2012, 46, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, B.; Freeborough, M.J.; Maree, H.J.; Celton, J.M.; Rees, D.J.; Burger, J.T. Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 2010, 400, 157–163. [Google Scholar] [CrossRef]
- Jo, Y.; Choi, H.; Kim, S.M.; Kim, S.L.; Lee, B.C.; Cho, W.K. The pepper virome: Natural co-infection of diverse viruses and their quasispecies. BMC Genomics 2017, 18, 453. [Google Scholar] [CrossRef]
- Wu, Q.; Ding, S.W.; Zhang, Y.; Zhu, S. Identification of viruses and viroids by next-generation sequencing and homology-dependent and homology-independent algorithms. Annu. Rev. Phytopathol. 2015, 53, 425–444. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using clustalw and clustalx. Curr. Protoc. Bioinform. 2002. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Sanfacon, H.; Iwanami, T.; Karasev, A.; Van der Vlugt, R.; Wellink, J.; Wetzel, T.; Yoshikawa, N. Secoviridae. In Virus Taxonomy: Ninth Report of the International Committee on the Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Press: Oxford, UK, 2011; pp. 881–899. [Google Scholar]
- Mayo, M.A.; Fritsch, C. A possible consensus sequence for VPg of viruses in the family Comoviridae. Febs Lett. 1994, 354, 129–130. [Google Scholar] [CrossRef]
- Rott, M.E.; Tremaine, J.H.; Rochon, D.M. Nucleotide sequence of tomato ringspot virus RNA1. J. Gen. Virol. 1995, 76, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Mushegian, A.R.; Ryabov, E.V.; Dolja, V.V. Diverse groups of plant RNA and DNA viruses share related movement proteins that may possess chaperone-like activity. J. Gen. Virol. 1991, 72, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Morris, T.J. Evolution and taxonomy of positive-strand RNA viruses: Implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef]
- Navarro, B.; Loconsole, G.; Giampetruzzi, A.; Aboughanem-Sabanadzovic, N.; Ragozzino, A.; Ragozzino, E.; Di Serio, F. Identification and characterization of privet leaf blotch-associated virus, a novel idaeovirus. Mol. Plant. Pathol. 2017, 18, 925–936. [Google Scholar] [CrossRef]
- James, D.; Phelan, J. Complete genome sequence and analysis of blackcurrant leaf chlorosis associated virus, a new member of the genus Idaeovirus. Arch. Virol. 2017, 162, 1705–1709. [Google Scholar] [CrossRef]
- Ziegler, A.; Mayo, M.A.; Murant, A.F. Proposed classification of the bipartite-genomed raspberry bushy dwarf idaeovirus, with tripartite-genomed viruses in the family Bromoviridae. Arch. Virol. 1993, 131, 483–488. [Google Scholar] [CrossRef]
- Chen, A.Y.S.; Watanabe, S.; Yokomi, R.; Ng, J.C.K. Nucleotide heterogeneity at the terminal ends of the genomes of two California Citrus tristeza virus strains and their complete genome sequence analysis. Virol. J. 2018, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Valverde, R.A.; Sabanadzovic, S.A. novel plant virus with unique properties infecting Japanese holly fern. J. Gen. Virol. 2009, 90, 2542–2549. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, A.F.; Correa, R.L.; Nakasugi, K.; Jackson, C.; Kawchuk, L.; Vaslin, M.F.S.; Waterhouse, P.M. The Enamovirus P0 protein is a silencing suppressor which inhibits local and systemic RNA silencing through AGO1 degradation. Virology 2012, 426, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Preitner, N.; Quan, J.; Dan, W.N.; Hancock, M.L.; Shi, J.; Tcherkezian, J.; Youngpearse, T.L.; Flanagan, J.G. APC is an RNA-binding protein and its interactome provides a link to neural development and microtubule assembly. Cell 2014, 158, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Vives, M.C.; Velazquez, K.; Pina, J.A.; Moreno, P.; Guerri, J.; Navarro, L. Identification of a new enamovirus associated with citrus vein enation disease by deep sequencing of small RNAs. Phytopathol. 2013, 103, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Giedroc, D.P.; Cornish, P.V. Frameshifting RNA pseudoknots: Structure and mechanism. Virus Res. 2009, 139, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sivakumar, S.; Wang, Z.; Bonning, B.C.; Miller, W.A. The readthrough domain of pea enation mosaic virus coat protein is not essential for virus stability in the hemolymph of the pea aphid. Arch. Virol. 2009, 154, 469–479. [Google Scholar] [CrossRef]
- Brown, C.M.; Dineshkumar, S.P.; Miller, W.A. Local and distant sequences are required for efficient readthrough of the barley yellow dwarf virus PAV coat protein gene stop codon. J. Virol. 1996, 70, 5884–5892. [Google Scholar]
- Dietzgen, R.G.; Innes, D.J.; Bejerman, N. Complete genome sequence and intracellular protein localization of Datura yellow vein nucleorhabdovirus. Virus Res. 2015, 205, 7–11. [Google Scholar] [CrossRef]
- Heaton, L.A.; Hillman, B.I.; Hunter, B.G.; Zuidema, D.; Jackson, A.O. Physical map of the genome of sonchus yellow net virus, a plant rhabdovirus with six genes and conserved gene junction sequences. Proc. Natl. Acad. Sci. USA 1989, 86, 8665–8668. [Google Scholar] [CrossRef]
- Walker, P.J.; Dietzgen, R.G.; Joubert, D.A.; Blasdell, K.R. Rhabdovirus accessory genes. Virus. Res. 2011, 162, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Rahmeh, A.; Morelli, M.; Whelan, S.P.J. A conserved motif in region V of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Gen. Virol. 2008, 82, 775–784. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, Y.; Wu, J. Biogenesis, function, and applications of virus-derived small RNAs in plants. Front. Microbiol. 2015, 6, 1237. [Google Scholar] [CrossRef]
- Mi, S.; Cai, T.; Hu, Y.; Chen, Y.; Hodges, E.; Ni, F.; Wu, L.; Li, S.; Zhou, H.; Long, C.; et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef]
- Villamor, D.E.V.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I.E. High throughput sequencing for plant virus detection and discovery. Phytopathology 2019. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.W. RNA-based antiviral immunity. Nat. Rev. Immunol. 2010, 10, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Du, P.; Wang, X.; Yu, Y.Q.; Qiu, Y.H.; Li, W.; Gal-On, A.; Zhou, C.; Li, Y.; Ding, S.W. Virus infection triggers widespread silencing of host genes by a distinct class of endogenous siRNAs in arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 14613–14618. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Wang, Y.; Cao, M.; Pantaleo, V.; Burgyan, J.; Li, W.X.; Ding, S.W. Homology-independent discovery of replicating pathogenic circular rnas by deep sequencing and a new computational algorithm. Proc. Natl. Acad. Sci. USA 2012, 109, 3938–3943. [Google Scholar] [CrossRef]
- Pecman, A.; Kutnjak, D.; Gutierrez-Aguirre, I.; Adams, I.; Fox, A.; Boonham, N.; Ravnikar, M. Next generation sequencing for detection and discovery of plant viruses and viroids: Comparison of two approaches. Front. Microbiol. 2017, 8, 1998. [Google Scholar] [CrossRef]
- MacFarlane, S.A. IdaeovIrus. In Virus Taxonomy: Ninth Report of the International Committee on the Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Press: Oxford, UK, 2011; pp. 1073–1075. [Google Scholar]
- Domier, L.L. Luteoviridae. In Virus Taxonomy: Ninth Report of the International Committee on the Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Press: Oxford, UK, 2011; pp. 1045–1053. [Google Scholar]
- Dietzgen, R.G.; Calisher, C.H.; Kurath, G.; Kuzmin, I.V.; Rodriguez, L.L.; Stone, D.M.; Tesh, R.B.; Tordo, N.; Walker, P.J.; Wetzel, T.; et al. Rhabdoviridae. In Virus taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Press: Oxford, UK, 2012; pp. 686–714. [Google Scholar]
- Brodersen, P.; Voinnet, O. The diversity of RNA silencing pathways in plants. Trends Genet. 2006, 22, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Cao, M.; Liu, W.; Ren, Y.; Zhou, X.; Wang, X. Two negative-strand RNA viruses identified in watermelon represent a novel clade in the order Bunyavirales. Front. Microbiol. 2017, 8, 1514. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Tian, X.; Zhang, S.; Ren, F.; Li, P.; Yu, Y.Q.; Li, R.; Zhou, C.; Cao, M. Molecular characterization of a novel luteovirus infecting apple by next-generation sequencing. Arch. Virol. 2018, 163, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Shen, P.; Li, M.; Tian, X.; Zhou, C.; Cao, M. Discovery of a novel geminivirus associated with camellia chlorotic dwarf disease. Arch. Virol. 2018. [Google Scholar] [CrossRef]
- Quito-Avila, D.F.; Martin, R.R. Real-time RT-PCR for detection of raspberry bushy dwarf virus, raspberry leaf mottle virus and characterizing synergistic interactions in mixed infections. J. Virol. Methods. 2012, 179, 38–44. [Google Scholar] [CrossRef]
- Ranieri, R.; Lister, R.M.; Burnett, P.A. Relationships between barley yellow dwarf virus titer and symptom expression in barley. Crop Sci. 1993, 33, 968–973. [Google Scholar] [CrossRef]
- Pataky, J.K.; Murphy, J.F.; D’Arcy, C.J. Resistance to maize dwarf mosaic virus, severity of symptoms, titer of virus, and yield of sweet corn. Plant Dis. 1990, 74, 359–364. [Google Scholar] [CrossRef]
- Yarwood, C.E. Latent period and generation time for two plant viruses. Am. J. Bot. 1952, 39, 613–618. [Google Scholar] [CrossRef]
- Bulger, M.A.; Stace-Smith, R.; Martin, R.R. Transmission and field spread of raspberry bushy dwarf virus. Plant. Dis. 1990, 74, 514–517. [Google Scholar] [CrossRef]
- Demler, S.A.; Zoeten, G.A.D.; Adam, G.; Harris, K.F. Pea enation mosaic enamovirus: Properties and aphid transmission. In The Plant Viruses, Polyhedral Virions and Bipartite RNA Genomes; Harrison, B.D., Murant, A.F., Eds.; Springer Press: Boston, MA, USA, 1996; pp. 303–344. [Google Scholar]
- Liu, Y.; Du, Z.; Wang, H.; Zhang, S.; Cao, M.; Wang, X. Identification and characterization of wheat yellow striate virus, a novel leafhopper-transmitted nucleorhabdovirus infecting wheat. Front. Microbiol. 2018, 9, 468. [Google Scholar] [CrossRef]
- Mansoor, S.; Amin, I.; Hussain, M.; Zafar, Y.; Briddon, R.W. Engineering novel traits in plants through RNA interference. Trends Plant Sci. 2006, 11, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Collinge, D.B.; Jørgensen, H.J.; Lund, O.S.; Lyngkjaer, M.F. Engineering pathogen resistance in crop plants: Current trends and future prospects. Annu. Rev. Phytopathol. 2010, 48, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.; Laimer, M.; Noris, E.; Schubert, J.; Wassenegger, M.; Tepfer, M. Strategies for antiviral resistance in transgenic plants. Mol. Plant pathol. 2008, 9, 73–83. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viruses | Contigs Amount | Contigs Size (bp) | Viral Reads | % of Total Reads | Average Coverage |
|---|---|---|---|---|---|
| [RNA-seq (RNA1&RNA2)]|[sRNA-seq (RNA1&RNA2)] | |||||
| GSPNeV | 2|12 | 6,945&6,170|145–391 | 4,152,530|3,050,449 | 7.15|10.42 | 34,717&49,160|3,502&5,164 |
| GSPIV | 2|40 | 5,433&2,372|58–241 | 3,078,099|254,692 | 5.3|0.87 | 24,850&145,377|532&2,515 |
| GSPEV | 1|11 | 4,729|83–152 | 226,501|213,707 | 0.39|0.73 | 6,129|784 |
| GSPNuV | 1|16 | 13,548|67–139 | 29,038|32,202 | 0.05|0.11 | 405|52 |
| Locations | Samples | Idaeovirus | Nepovirus | Nucleorhabdovirus | Enamovirus | |
|---|---|---|---|---|---|---|
| Bishan county | Fulu Town | 4 | − | + | − | + |
| 1 | − | − | − | − | ||
| 1* | − | + | − | − | ||
| Jiangjin district | Xianfeng Town | 1 | − | + | + | + |
| 3 1 | − | + | − | + | ||
| 1 | − | − | + | + | ||
| Ciyun Town | 6 2 | − | + | − | + | |
| 3* | − | + | − | − | ||
| 2 | − | − | − | − | ||
| Jiangjin district | 3 | + | + | + | + | |
| 2 | − | + | + | + | ||
| 5 | + | + | − | + | ||
| 2 | − | + | − | + | ||
| 1*1 | − | + | − | − | ||
| Changshou district | Dandu Town | 3 | − | − | − | − |
| 3* | − | + | − | − | ||
| Total | 41 | 8 | 34 | 7 | 27 | |
| Detection rate | 19.5% | 82.9% | 17.1% | 65.9% | ||
| Single virus infection | 0 | 8 | 0 | 0 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, M.; Zhang, S.; Li, M.; Liu, Y.; Dong, P.; Li, S.; Kuang, M.; Li, R.; Zhou, Y. Discovery of Four Novel Viruses Associated with Flower Yellowing Disease of Green Sichuan Pepper (Zanthoxylum armatum) by Virome Analysis. Viruses 2019, 11, 696. https://doi.org/10.3390/v11080696
Cao M, Zhang S, Li M, Liu Y, Dong P, Li S, Kuang M, Li R, Zhou Y. Discovery of Four Novel Viruses Associated with Flower Yellowing Disease of Green Sichuan Pepper (Zanthoxylum armatum) by Virome Analysis. Viruses. 2019; 11(8):696. https://doi.org/10.3390/v11080696
Chicago/Turabian StyleCao, Mengji, Song Zhang, Min Li, Yingjie Liu, Peng Dong, Shanrong Li, Mi Kuang, Ruhui Li, and Yan Zhou. 2019. "Discovery of Four Novel Viruses Associated with Flower Yellowing Disease of Green Sichuan Pepper (Zanthoxylum armatum) by Virome Analysis" Viruses 11, no. 8: 696. https://doi.org/10.3390/v11080696
APA StyleCao, M., Zhang, S., Li, M., Liu, Y., Dong, P., Li, S., Kuang, M., Li, R., & Zhou, Y. (2019). Discovery of Four Novel Viruses Associated with Flower Yellowing Disease of Green Sichuan Pepper (Zanthoxylum armatum) by Virome Analysis. Viruses, 11(8), 696. https://doi.org/10.3390/v11080696