Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection


{kind=link}
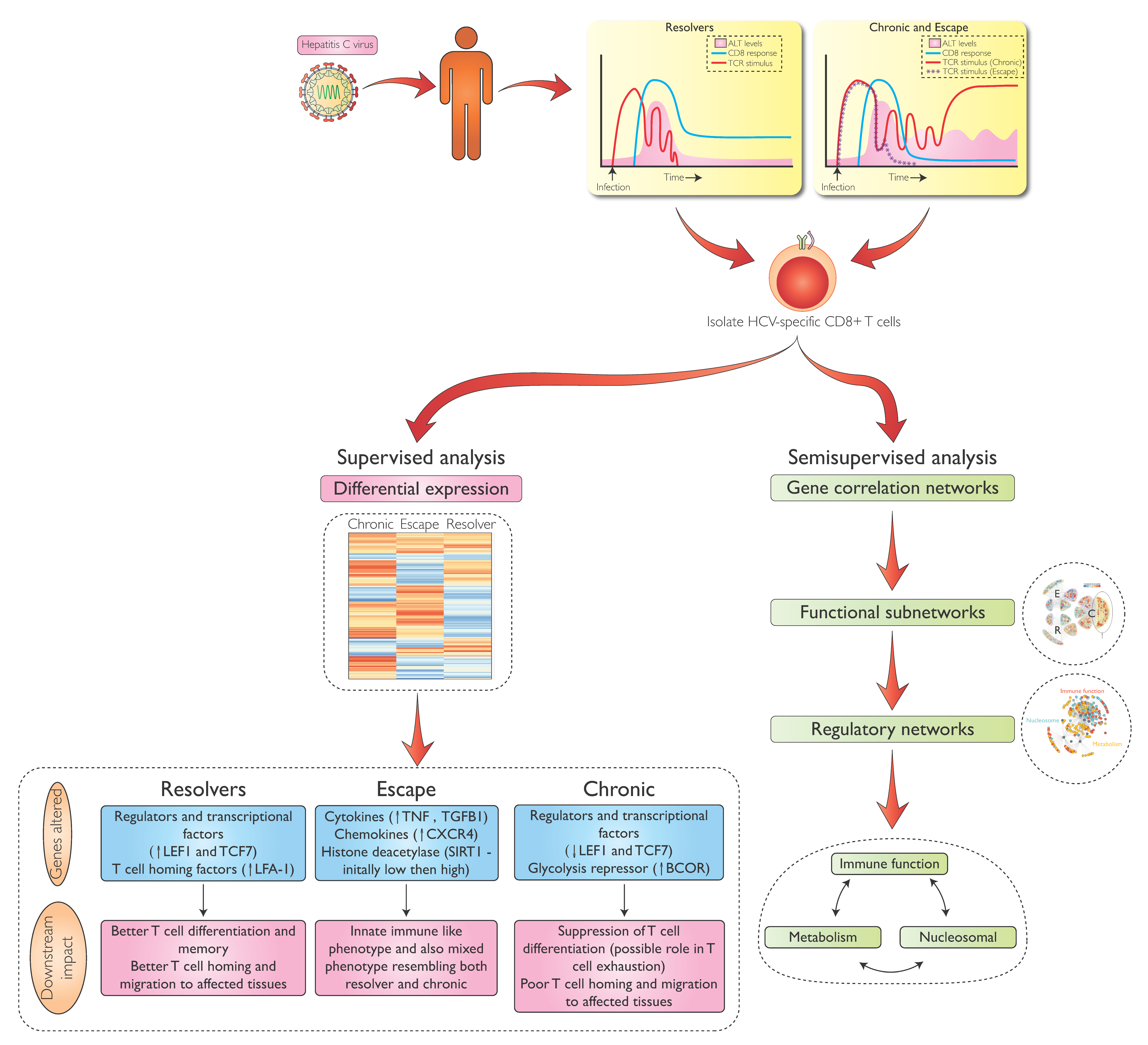
Abstract
1. Introduction
2. T Cells and the Outcome of HCV Infection
3. Hurdles to Deeper Analysis of HCV-Specific T-Cells
4. Transcriptional Analysis of HCV-Specific CD8 T-Cells During Early HCV Infection
5. Differential Gene Expression in HCV-Specific CD8 T-cells During Acute and Chronic Infection
6. Metabolic Regulation in HCV-Specific CD8 T-Cells
7. Transcriptional Regulation of HCV-Specific CD8 T-Cells Beyond Metabolism
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Lauer, G.M.; Walker, B.D. Hepatitis C Virus Infection. N. Engl. J. Med. 2001, 345, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Luxenburger, H.; Neumann-Haefelin, C.; Thimme, R. HCV-Specific T Cell Responses During and After Chronic HCV Infection. Viruses 2018, 10, 645. [Google Scholar] [CrossRef] [PubMed]
- Lauer, G.M.; Barnes, E.; Lucas, M.; Timm, J.; Ouchi, K.; Kim, A.Y.; Day, C.L.; Robbins, G.K.; Casson, D.R.; Reiser, M.; et al. High resolution analysis of cellular immune responses in resolved and persistent hepatitis C virus infection. Gastroenterology 2004, 127, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.K.H.; Dudley, D.D.; Afdhal, N.H.; Dienstag, J.; Rice, C.M.; Wang, L.; Houghton, M.; Walker, B.D.; Koziel, M.J. Liver-Derived CTL in Hepatitis C Virus Infection: Breadth and Specificity of Responses in a Cohort of Persons with Chronic Infection. J. Immunol. 1998, 160, 1479–1488. [Google Scholar] [PubMed]
- Grakoui, A.; Shoukry, N.H.; Woollard, D.J.; Han, J.-H.; Hanson, H.L.; Ghrayeb, J.; Murthy, K.K.; Rice, C.M.; Walker, C.M. HCV persistence and immune evasion in the absence of memory T cell help. Science 2003, 302, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Shoukry, N.H.; Grakoui, A.; Houghton, M.; Chien, D.Y.; Ghrayeb, J.; Reimann, K.A.; Walker, C.M. Memory CD8+ T Cells Are Required for Protection from Persistent Hepatitis C Virus Infection. J. Exp. Med. 2003, 197, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Walker, C.M.; Lemon, S.M. The Chimpanzee Model of Viral Hepatitis: Advances in Understanding the Immune Response and Treatment of Viral Hepatitis. ILAR J. 2017, 58, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Oldach, D.; Chang, K.-M.; Steiger, C.; Ray, S.C.; Chisari, F.V. Determinants of Viral Clearance and Persistence during Acute Hepatitis C Virus Infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef]
- Gerlach, J.T.; Diepolder, H.M.; Jung, M.C.; Gruener, N.H.; Schraut, W.W.; Zachoval, R.; Hoffmann, R.; Schirren, C.A.; Santantonio, T.; Pape, G.R. Recurrence of hepatitis C virus after loss of virus-specific CD4(+) T-cell response in acute hepatitis C. Gastroenterology 1999, 117, 933–941. [Google Scholar] [CrossRef]
- Schulze zur Wiesch, J.; Ciuffreda, D.; Lewis-Ximenez, L.; Kasprowicz, V.; Nolan, B.E.; Streeck, H.; Aneja, J.; Reyor, L.L.; Allen, T.M.; Lohse, A.W.; et al. Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J. Exp. Med. 2012, 209, 61–75. [Google Scholar] [CrossRef]
- Schulze zur Wiesch, J.; Lauer, G.M.; Day, C.L.; Kim, A.Y.; Ouchi, K.; Duncan, J.E.; Wurcel, A.G.; Timm, J.; Jones, A.M.; Mothe, B.; et al. Broad Repertoire of the CD4+ Th Cell Response in Spontaneously Controlled Hepatitis C Virus Infection Includes Dominant and Highly Promiscuous Epitopes. J. Immunol. 2005, 175, 3603–3613. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L.; Mosbruger, T.; Lauer, G.M.; Pardoll, D.; Thomas, D.L.; Ray, S.C. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 2005, 42, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Kasprowicz, V.; Schulze zur Wiesch, J.; Kuntzen, T.; Nolan, B.E.; Longworth, S.; Berical, A.; Blum, J.; McMahon, C.; Reyor, L.L.; Elias, N.; et al. High Level of PD-1 Expression on Hepatitis C Virus (HCV)-Specific CD8+ and CD4+ T Cells during Acute HCV Infection, Irrespective of Clinical Outcome. J. Virol. 2008, 82, 3154–3160. [Google Scholar] [CrossRef] [PubMed]
- Urbani, S.; Amadei, B.; Tola, D.; Massari, M.; Schivazappa, S.; Missale, G.; Ferrari, C. PD-1 Expression in Acute Hepatitis C Virus (HCV) Infection Is Associated with HCV-Specific CD8 Exhaustion. J. Virol. 2006, 80, 11398–11403. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Lauer, G.M.; Kavanagh, D.G.; Sheridan, I.; Kim, A.Y.; Lucas, M.; Pillay, T.; Ouchi, K.; Reyor, L.L.; Zur Wiesch, J.S.; et al. CD8 Epitope Escape and Reversion in Acute HCV Infection. J. Exp. Med. 2004, 200, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.L.; Mosbruger, T.; Mao, Q.; Liu, Z.; Wang, X.-H.; Yang, H.-C.; Sidney, J.; Sette, A.; Pardoll, D.; Thomas, D.L.; et al. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 2005, 201, 1741–1752. [Google Scholar] [CrossRef]
- Lauer, G.M.; Lucas, M.; Timm, J.; Ouchi, K.; Kim, A.Y.; Day, C.L.; zur Wiesch, J.S.; Paranhos-Baccala, G.; Sheridan, I.; Casson, D.R.; et al. Full-Breadth Analysis of CD8+ T-Cell Responses in Acute Hepatitis C Virus Infection and Early Therapy. J. Virol. 2005, 79, 12979–12988. [Google Scholar] [CrossRef]
- Van Leeuwen, E.M.M.; Koning, J.J.; Remmerswaal, E.B.M.; van Baarle, D.; van Lier, R.A.W.; ten Berge, I.J.M. Differential Usage of Cellular Niches by Cytomegalovirus versus EBV- and Influenza Virus-Specific CD8+ T Cells. J. Immunol. 2006, 177, 4998–5005. [Google Scholar] [CrossRef]
- Nitschke, K.; Flecken, T.; Schmidt, J.; Gostick, E.; Marget, M.; Neumann-Haefelin, C.; Blum, H.E.; Price, D.A.; Thimme, R.; Diamond, M.S. Tetramer Enrichment Reveals the Presence of Phenotypically Diverse Hepatitis C Virus-Specific CD8+ T Cells in Chronic Infection. J. Virol. 2015, 89, 25–34. [Google Scholar] [CrossRef]
- Wolski, D.; Foote, P.K.; Chen, D.Y.; Lewis-Ximenez, L.L.; Fauvelle, C.; Aneja, J.; Walker, A.; Tonnerre, P.; Torres-Cornejo, A.; Kvistad, D.; et al. Early Transcriptional Divergence Marks Virus-Specific Primary Human CD8+ T Cells in Chronic versus Acute Infection. Immunity 2017, 47, 648–663. [Google Scholar] [CrossRef]
- Kim, J.; Chang, D.-Y.; Lee, H.W.; Lee, H.; Kim, J.H.; Sung, P.S.; Kim, K.H.; Hong, S.-H.; Kang, W.; Lee, J.; et al. Innate-like Cytotoxic Function of Bystander-Activated CD8+ T Cells Is Associated with Liver Injury in Acute Hepatitis A. Immunity 2018, 48, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Barbarin, A.; Cayssials, E.; Jacomet, F.; Nunez, N.G.; Basbous, S.; Lefèvre, L.; Abdallah, M.; Piccirilli, N.; Morin, B.; Lavoue, V.; et al. Phenotype of NK-Like CD8(+) T Cells with Innate Features in Humans and Their Relevance in Cancer Diseases. Front. Immunol. 2017, 8, 1114. [Google Scholar] [CrossRef] [PubMed]
- Seyda, M.; Elkhal, A.; Quante, M.; Falk, C.S.; Tullius, S.G. T Cells Going Innate. Trends Immunol. 2016, 37, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Li, F.; Zeng, Z.; Zhao, Y.; Yu, S.; Shan, Q.; Li, Y.; Phillips, F.C.; Maina, P.K.; Qi, H.H.; et al. Tcf1 and Lef1 transcription factors establish CD8+ T cell identity through intrinsic HDAC activity. Nat. Immunol. 2016, 17, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Yamazaki, M.; Abe, M.; Sakimura, K.; Iwai, Y. Basic leucine zipper transcription factor, ATF-like (BATF) regulates epigenetically and energetically effector CD8 T-cell differentiation via Sirt1 expression. Proc. Natl. Acad. Sci. USA 2011, 108, 14885–14889. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494. [Google Scholar] [CrossRef]
- Gillum, M.P.; Erion, D.M.; Shulman, G.I. Sirtuin-1 regulation of mammalian metabolism. Trends Mol. Med. 2011, 17, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Finkelstein, D.; McCormick, L.L.; Chi, H.; Munger, J.; Green, D.R. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef]
- Boroughs, L.K. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- Ballesteros-Tato, A.; León, B.; Graf, B.A.; Moquin, A.; Adams, P.S.; Lund, F.E.; Randall, T.D. Interleukin-2 Inhibits Germinal Center Formation by Limiting T Follicular Helper Cell Differentiation. Immunity 2012, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, K.J.; Mohn, S.E.; Weinmann, A.S. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH like gene profile. Nat. Immunol. 2012, 13, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, K.J.; Read, K.A.; Gilbertson, S.E.; Hough, K.P.; McDonald, P.W.; Krishnamoorthy, V.; Weinmann, A.S. Bcl-6 directly represses the gene program of the glycolysis pathway. Nat. Immunol. 2014, 15, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Pipkin, M.E.; Sacks, J.A.; Cruz-Guilloty, F.; Lichtenheld, M.G.; Bevan, M.J.; Rao, A. Interleukin-2 and Inflammation Induce Distinct Transcriptional Programs that Promote the Differentiation of Effector Cytolytic T Cells. Immunity 2010, 32, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Beima, K.M.; Miazgowicz, M.M.; Lewis, M.D.; Yan, P.S.; Huang, T.H.-M.; Weinmann, A.S. T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J. Biol. Chem. 2006, 281, 11992–12000. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Huang, A.C.; Miazgowicz, M.M.; Brassil, M.M.; Weinmann, A.S. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev. 2008, 22, 2980–2993. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A Novel Transcription Factor, T-bet, Directs Th1 Lineage Commitment. Cell 2000, 100, 655–669. [Google Scholar]
- Sullivan, B.M.; Stemmann, C.; Satoskar, A.R.; Sleckman, B.P.; Glimcher, L.H. Distinct Effects of T-bet in TH1 Lineage Commitment and IFN-γ Production in CD4 and CD8 T Cells. Science 2002, 295, 338–342. [Google Scholar]
- Yoon, J.; Feng, X.; Kim, Y.-S.; Shin, D.-M.; Hatzi, K.; Wang, H.; Morse, H.C. Interferon regulatory factor 8 (IRF8) interacts with the B cell lymphoma 6 (BCL6) corepressor BCOR. J. Biol. Chem. 2014, 289, 34250–34257. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tsuzuki, S.; Tsuzuki, M.; Handa, K.; Inaguma, Y.; Emi, N. BCOR as a novel fusion partner of retinoic acid receptor alpha in a t(X;17)(p11;q12) variant of acute promyelocytic leukemia. Blood 2010, 116, 4274–4283. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Van der Windt, G.J.W.; Everts, B.; Chang, C.-H.; Freitas, T.C.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef]
- Van der Windt, G.J.W.; O’Sullivan, D.; Everts, B.; Huang, S.C.-C.; Buck, M.D.; Chang, C.-H.; Smith, A.M.; Ai, T.; Faubert, B.; Jones, R.G.; et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc. Natl. Acad. Sci. USA 2013, 110, 14336–14341. [Google Scholar] [CrossRef]
- Trautmann, L.; Mbitikon-Kobo, F.-M.; Goulet, J.-P.; Peretz, Y.; Shi, Y.; Van Grevenynghe, J.; Procopio, F.A.; Boulassel, M.R.; Routy, J.-P.; Chomont, N.; et al. Profound metabolic, functional, and cytolytic differences characterize HIV-specific CD8 T cells in primary and chronic HIV infection. Blood 2012, 120, 3466–3477. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Fisicaro, P.; Barili, V.; Montanini, B.; Acerbi, G.; Ferracin, M.; Guerrieri, F.; Salerno, D.; Boni, C.; Massari, M.; Cavallo, M.C.; et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017, 57, 167–336. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Berkers, C.R.; Maddocks, O.D.K.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic regulation by p53 family members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef]
- Verma, N.K.; Kelleher, D. Not Just an Adhesion Molecule: LFA-1 Contact Tunes the T Lymphocyte Program. J. Immunol. 2017, 199, 1213–1221. [Google Scholar] [CrossRef]
- Gerlach, C.; Moseman, E.A.; Loughhead, S.M.; Alvarez, D.; Zwijnenburg, A.J.; Waanders, L.; Garg, R.; de la Torre, J.C.; von Andrian, U.H. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 2016, 45, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Crompton, J.G.; Clever, D.; Vizcardo, R.; Rao, M.; Restifo, N.P. Reprogramming antitumor immunity. Trends Immunol. 2014, 35, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Themeli, M.; Rivière, I.; Sadelain, M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell 2015, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Abdelsamed, H.A.; Moustaki, A.; Fan, Y.; Dogra, P.; Ghoneim, H.E.; Zebley, C.C.; Triplett, B.M.; Sekaly, R.-P.; Youngblood, B. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J. Exp. Med. 2017, 214, 1593–1606. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.A.; Crawford, A.; Angelosanto, J.M.; Paley, M.A.; Ziegler, C.G.; Wherry, E.J. Network Analysis Reveals Centrally Connected Genes and Pathways Involved in CD8+ T Cell Exhaustion versus Memory. Immunity 2012, 37, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Kurachi, M.; Barnitz, R.A.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The epigenetic landscape of T cell exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Scott-Browne, J.P.; López-Moyado, I.F.; Trifari, S.; Wong, V.; Chavez, L.; Rao, A.; Pereira, R.M. Dynamic Changes in Chromatin Accessibility Occur in CD8+ T Cells Responding to Viral Infection. Immunity 2016, 45, 1327–1340. [Google Scholar] [CrossRef]
- Shin, H.; Wherry, E.J. CD8 T cell dysfunction during chronic viral infection. Curr. Opin. Immunol. 2007, 19, 408–415. [Google Scholar] [CrossRef]
- Gallimore, A.; Gallimore, A.; Glithero, A.; Glithero, A.; Godkin, A.; Godkin, A.; Tissot, A.C.; Tissot, A.C.; Plückthun, A.; Plückthun, A.; et al. Induction and Exhaustion of Lymphocytic Choriomeningitis Virus–specific Cytotoxic T Lymphocytes Visualized Using Soluble Tetrameric Major Histocompatibility Complex Class I–Peptide Complexes. J. Exp. Med. 1998, 187, 1383–1393. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Blattman, J.N.; Murali-Krishna, K.; Murali-Krishna, K.; Sourdive, D.J.; Sourdive, D.J.D.; Suresh, M.; Altman, J.D.; et al. Viral Immune Evasion Due to Persistence of Activated T Cells Without Effector Function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Freeman, G.J.; Wherry, E.J. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc. Natl. Acad. Sci. USA 2008, 105, 15016–15021. [Google Scholar] [CrossRef]
- He, R.; Hou, S.; Liu, C.; Zhang, A.; Bai, Q.; Han, M.; Yang, Y.; Wei, G.; Shen, T.; Yang, X.; et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature 2016, 537, 412–428. [Google Scholar] [CrossRef]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Kissick, H.T.; Burger, M.C.; Lee, J.; Nasti, T.H.; Sharpe, A.H.; Freeman, G.J.; Germain, R.N.; et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef]
- Wu, T.; Ji, Y.; Moseman, E.A.; Xu, H.C.; Manglani, M.; Kirby, M.; Anderson, S.M.; Handon, R.; Kenyon, E.; Elkahloun, A.; et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 2016, 1, eaai8593. [Google Scholar] [CrossRef]
- Carey, B.W.; Finley, L.W.S. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef]
- TeSlaa, T.; Chaikovsky, A.C.; Lipchina, I.; Escobar, S.L.; Hochedlinger, K.; Huang, J.; Graeber, T.G.; Braas, D.; Teitell, M.A. α-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016, 24, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Macias, D.; Lee, K.L.; Veliça, P.; You, J.; Chia, G.S.; Sim, J.; Doedens, A.; Abelanet, A.; Evans, C.E.; Griffiths, J.R.; et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 2016, 540, 236–241. [Google Scholar]
- Jasper, H.; Ho, T.T. Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol. 2016, 18, 823–832. [Google Scholar]
- Kaelin, W.G.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Mentch, S.J. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef]
- Huang, Z.; Cai, L. Dietary control of chromatin. Curr. Opin. Cell Biol. 2015, 34, 69–74. [Google Scholar] [CrossRef][Green Version]
- Wellen, K.E. A two-way street: Reciprocal regulation of metabolism and signalling. Nat. Rev. Mol. Cell. Biol. 2012, 13, 270–276. [Google Scholar] [CrossRef]
- Ryall, J.G.; Cliff, T.; Dalton, S.; Sartorelli, V. Metabolic Reprogramming of Stem Cell Epigenetics. Cell Stem Cell 2015, 17, 651–662. [Google Scholar] [CrossRef]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Pavlova, N.N. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Losman, J.-A.; Kaelin, W.G. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013, 27, 836–852. [Google Scholar] [CrossRef]
- Hörmanseder, E.; Simeone, A.; Allen, G.E.; Bradshaw, C.R.; Figlmüller, M.; Gurdon, J.; Jullien, J. H3K4 Methylation-Dependent Memory of Somatic Cell Identity Inhibits Reprogramming and Development of Nuclear Transfer Embryos. Cell Stem Cell 2017, 21, 135–143. [Google Scholar] [CrossRef]
- Matoba, S.; Liu, Y.; Lu, F.; Iwabuchi, K.A.; Inoue, A.; Zhang, Y. Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell 2014, 159, 884–895. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Consortium; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Phan, A.T.; Goldrath, A.W.; Glass, C.K. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity 2017, 46, 714–729. [Google Scholar] [CrossRef]
- Rosenberg, B.R.; Depla, M.; Freije, C.A.; Gaucher, D.; Mazouz, S.; Boisvert, M.; Bédard, N.; Bruneau, J.; Rice, C.M.; Shoukry, N.H. Longitudinal transcriptomic characterization of the immune response to acute hepatitis C virus infection in patients with spontaneous viral clearance. PLoS Pathog. 2018, 14, e1007290. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolski, D.; Lauer, G.M. Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection. Viruses 2019, 11, 683. https://doi.org/10.3390/v11080683
Wolski D, Lauer GM. Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection. Viruses. 2019; 11(8):683. https://doi.org/10.3390/v11080683
Chicago/Turabian StyleWolski, David, and Georg M. Lauer. 2019. "Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection" Viruses 11, no. 8: 683. https://doi.org/10.3390/v11080683
APA StyleWolski, D., & Lauer, G. M. (2019). Hepatitis C Virus as a Unique Human Model Disease to Define Differences in the Transcriptional Landscape of T Cells in Acute versus Chronic Infection. Viruses, 11(8), 683. https://doi.org/10.3390/v11080683