A Protocol for Extraction of Infective Viromes Suitable for Metagenomics Sequencing from Low Volume Fecal Samples

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Storage
2.2. Virus Stock Production
2.3. Spiking of Fecal Samples with Known Phages
2.4. Plaque Assay
2.5. Virome Isolation from Feces
2.6. Nucleic Acid Extraction of Virome from Feces
2.7. Virus Quantification by Quantitative Real-Time PCR (qPCR)
2.8. Sequencing of Fecal Virome Nucleic Acids
2.9. Processing and Analysis of the Sequencing Results
3. Results and Discussion
3.1. Design of the Experiments
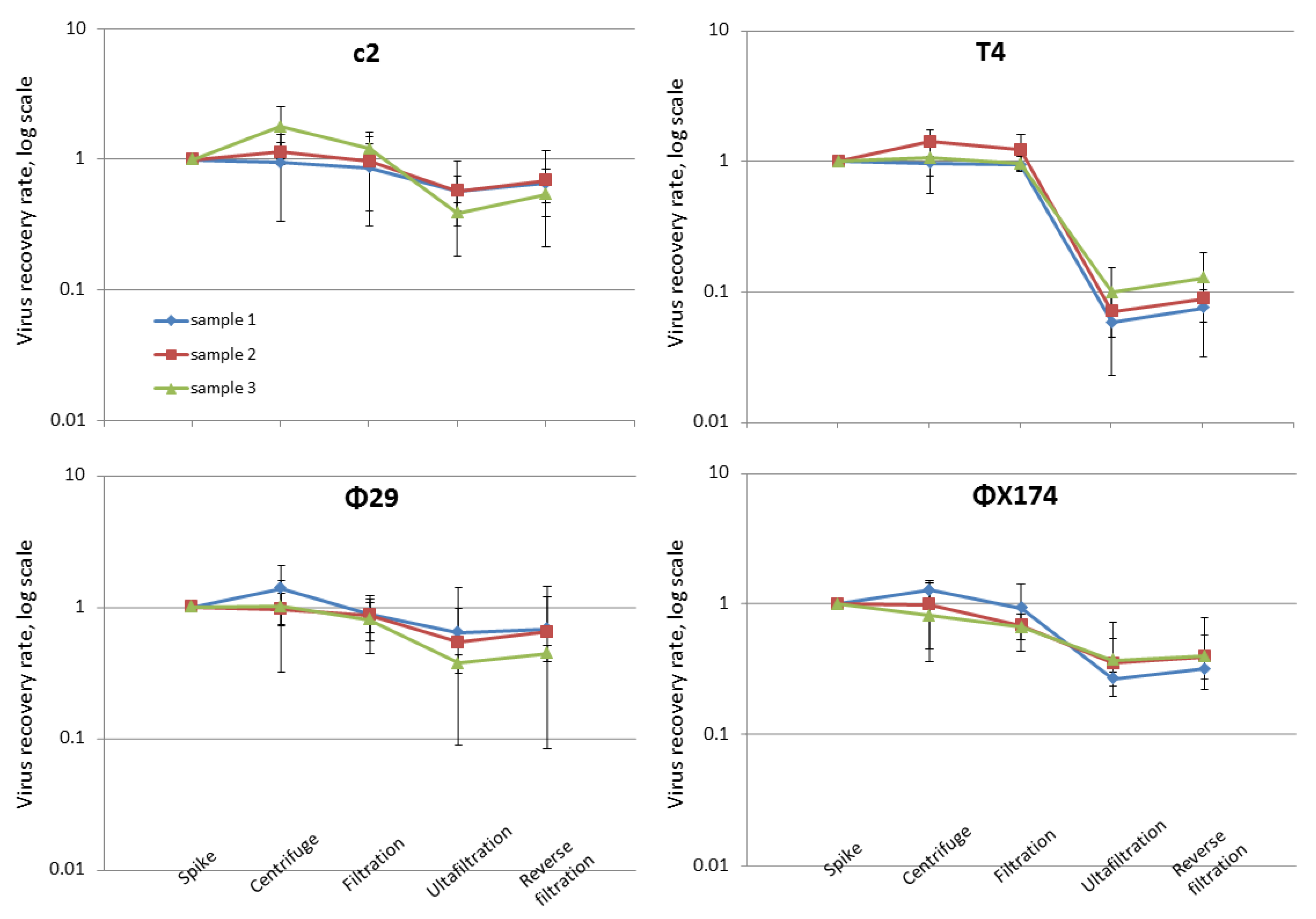
3.2. Assessing the Protocol Design by Recovery Rates of Spiked Phages
3.3. Determination of T4 Genome Recovery Rate by qPCR
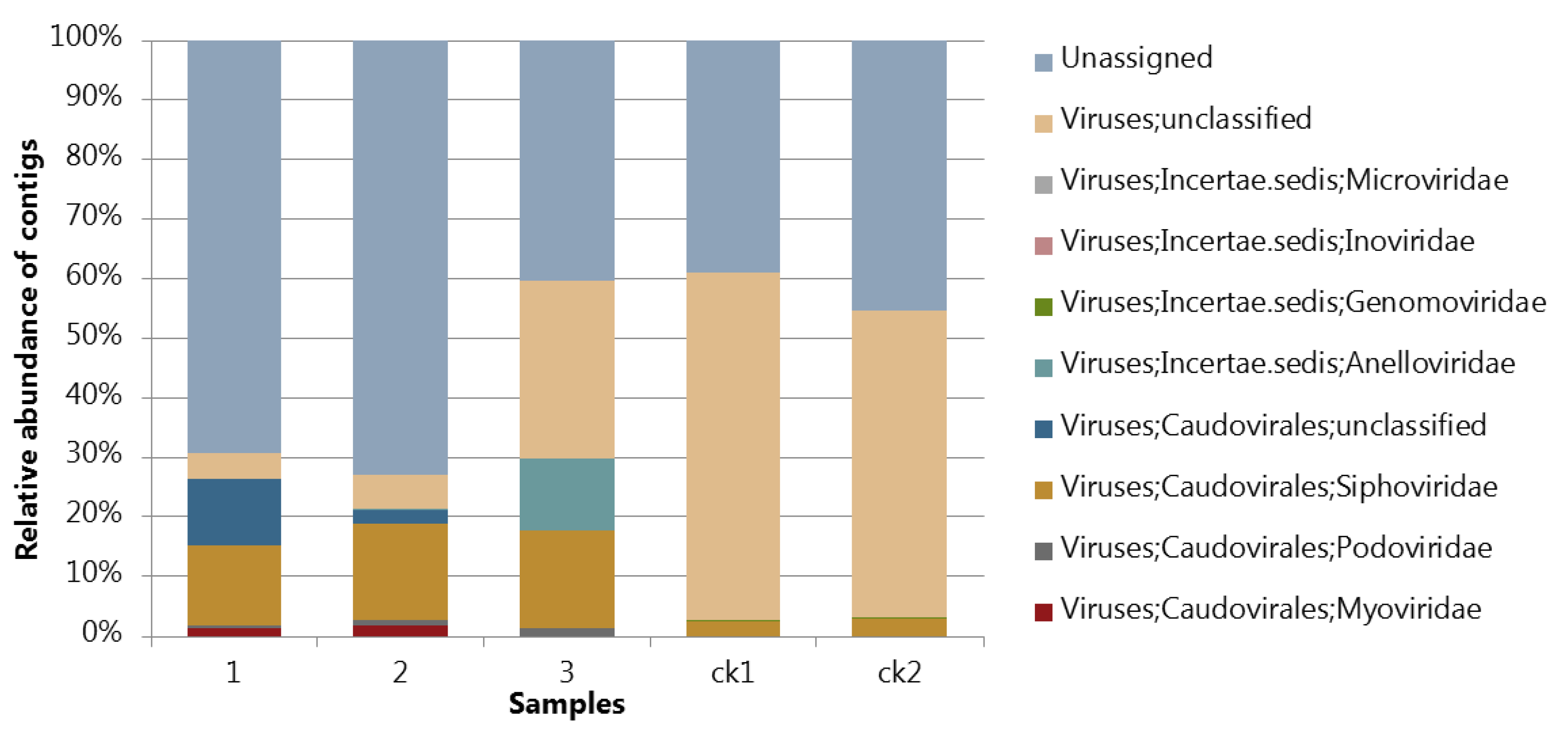
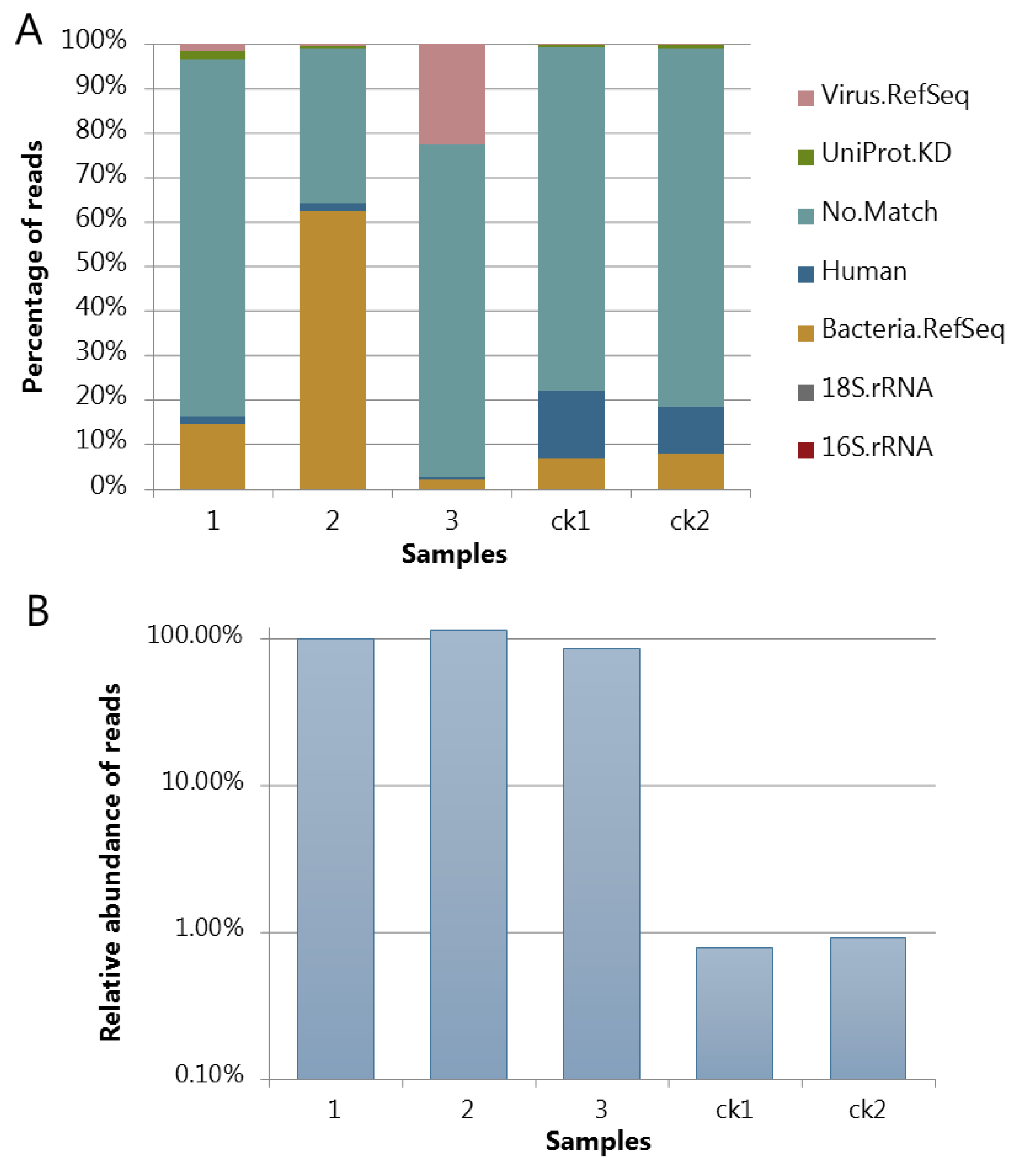
3.4. Assessing the Protocol by Sequencing and Bioinfomatic Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Andersen, L.O.; Vedel Nielsen, H.; Stensvold, C.R. Waiting for the human intestinal Eukaryotome. Isme. J. 2013, 7, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Gaci, N.; Borrel, G.; Tottey, W.; O’Toole, P.W.; Brugere, J.F. Archaea and the human gut: New beginning of an old story. World J. Gastroenterol. 2014, 20, 16062–16078. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, E.; Ianiro, G.; Attili, F.; Bassanelli, C.; De Santis, A.; Gasbarrini, A. The human gut microbiota and virome: Potential therapeutic implications. Dig. Liver Dis. 2015, 47, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.A.; Jones, B.V. The human gut virome: A multifaceted majority. Front. Microbiol. 2015, 6, 918. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nature reviews. Microbiology 2012, 10, 607–617. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nature reviews. Microbiology 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; U’Ren, J.M.; Youens-Clark, K. Computational prospecting the great viral unknown. Fems Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef]
- Castro-Mejia, J.L.; Muhammed, M.K.; Kot, W.; Neve, H.; Franz, C.M.; Hansen, L.H.; Vogensen, F.K.; Nielsen, D.S. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 2015, 3, 64. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Conceicao-Neto, N.; Zeller, M.; Lefrere, H.; De Bruyn, P.; Beller, L.; Deboutte, W.; Yinda, C.K.; Lavigne, R.; Maes, P.; Van Ranst, M.; et al. Modular approach to customise sample preparation procedures for viral metagenomics: A reproducible protocol for virome analysis. Sci. Rep. 2015, 5, 16532. [Google Scholar] [CrossRef]
- Muhammed, M.K.; Krych, L.; Nielsen, D.S.; Vogensen, F.K. A high-throughput qPCR system for simultaneous quantitative detection of dairy Lactococcus lactis and Leuconostoc bacteriophages. PLoS ONE 2017, 12, e0174223. [Google Scholar] [CrossRef]
- Rasmussen, T.S.; de Vries, L.; Kot, W.; Hansen, L.H.; Castro-Mejía, J.L.; Vogensen, F.K.; Hansen, A.K.; Nielsen, D.S. Mouse vendor influence on the bacterial and viral gut composition exceeds the effect of diet. Viruses 2019, 11, 435. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Reyes, A.; Blanton, L.V.; Cao, S.; Zhao, G.; Manary, M.; Trehan, I.; Smith, M.I.; Wang, D.; Virgin, H.W.; Rohwer, F. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 2015, 112, 11941–11946. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Cambuy, D.D.; Coutinho, F.H.; Dutilh, B.E. Contig annotation tool CAT robustly classifies assembled metagenomic contigs and long sequences. bioRxiv 2016, 072868. [Google Scholar] [CrossRef]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470. [Google Scholar] [CrossRef]
- Vajda, B. Concentration and purification of viruses and bacteriophages with polyethylene glycol. Folia Microbiol. 1978, 23, 88–96. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Ryan, F.J.; Draper, L.A.; Forde, A.; Stockdale, S.R.; Daly, K.M.; McDonnell, S.A.; Nolan, J.A.; Sutton, T.D.S.; Dalmasso, M.; et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 2018, 6, 68. [Google Scholar] [CrossRef]
- Conceicao-Neto, N.; Yinda, K.C.; Van Ranst, M.; Matthijnssens, J. NetoVIR: Modular Approach to Customize Sample Preparation Procedures for Viral Metagenomics. Methods Mol. Biol. 2018, 1838, 85–95. [Google Scholar] [CrossRef]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenck, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbart, M.; Sullivan, M.B. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016, 4, e2777. [Google Scholar] [CrossRef]
- Kleiner, M.; Hooper, L.V.; Duerkop, B.A. Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. Bmc Genom. 2015, 16, 7. [Google Scholar] [CrossRef]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Bakir, M.A.; Sakamoto, M.; Kitahara, M.; Matsumoto, M.; Benno, Y. Bacteroides dorei sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2006, 56, 1639–1643. [Google Scholar] [CrossRef]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage-host relationships. Fems Microbiol. Rev. 2016, 40, 258–272. [Google Scholar] [CrossRef]
- Villarroel, J.; Kleinheinz, K.A.; Jurtz, V.I.; Zschach, H.; Lund, O.; Nielsen, M.; Larsen, M.V. HostPhinder: A Phage Host Prediction Tool. Viruses 2016, 8. [Google Scholar] [CrossRef]
- Galiez, C.; Siebert, M.; Enault, F.; Vincent, J.; Soding, J. WIsH: Who is the host? Predicting prokaryotic hosts from metagenomic phage contigs. Bioinformatics 2017, 33, 3113–3114. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Strain | Phage (Family) | Growth Media | Source |
|---|---|---|---|
| Bacillus subtilis DSM 5547 | Φ29 (Podoviridae) | TSB | Lab.stock |
| Escherichia coli DSM 613 | T4 (Myoviridae) | LB medium | Lab.stock |
| Escherichia coli ATTC 13706 | ΦX174 (Microviridae) | BHI Broth | Félix d’Hérelle Reference Center |
| Lactococcus lactis MG1363 | c2 (Siphoviridae) | M17 | Lab. Stock |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, L.; Silins, R.; Castro-Mejía, J.L.; Kot, W.; Jessen, L.; Thorsen, J.; Shah, S.; Stokholm, J.; Bisgaard, H.; Moineau, S.; et al. A Protocol for Extraction of Infective Viromes Suitable for Metagenomics Sequencing from Low Volume Fecal Samples. Viruses 2019, 11, 667. https://doi.org/10.3390/v11070667
Deng L, Silins R, Castro-Mejía JL, Kot W, Jessen L, Thorsen J, Shah S, Stokholm J, Bisgaard H, Moineau S, et al. A Protocol for Extraction of Infective Viromes Suitable for Metagenomics Sequencing from Low Volume Fecal Samples. Viruses. 2019; 11(7):667. https://doi.org/10.3390/v11070667
Chicago/Turabian StyleDeng, Ling, Ronalds Silins, Josué L. Castro-Mejía, Witold Kot, Leon Jessen, Jonathan Thorsen, Shiraz Shah, Jakob Stokholm, Hans Bisgaard, Sylvain Moineau, and et al. 2019. "A Protocol for Extraction of Infective Viromes Suitable for Metagenomics Sequencing from Low Volume Fecal Samples" Viruses 11, no. 7: 667. https://doi.org/10.3390/v11070667
APA StyleDeng, L., Silins, R., Castro-Mejía, J. L., Kot, W., Jessen, L., Thorsen, J., Shah, S., Stokholm, J., Bisgaard, H., Moineau, S., & Nielsen, D. S. (2019). A Protocol for Extraction of Infective Viromes Suitable for Metagenomics Sequencing from Low Volume Fecal Samples. Viruses, 11(7), 667. https://doi.org/10.3390/v11070667