Bacillus Phage vB_BtS_B83 Previously Designated as a Plasmid May Represent a New Siphoviridae Genus

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Phage Isolation and Propagation
2.3. Host Range Determination
2.4. Transmission Electron Microscopy
2.5. Phage DNA Sequencing and Analyzing
2.6. Comparative Genomics
2.7. Accession Number
3. Results
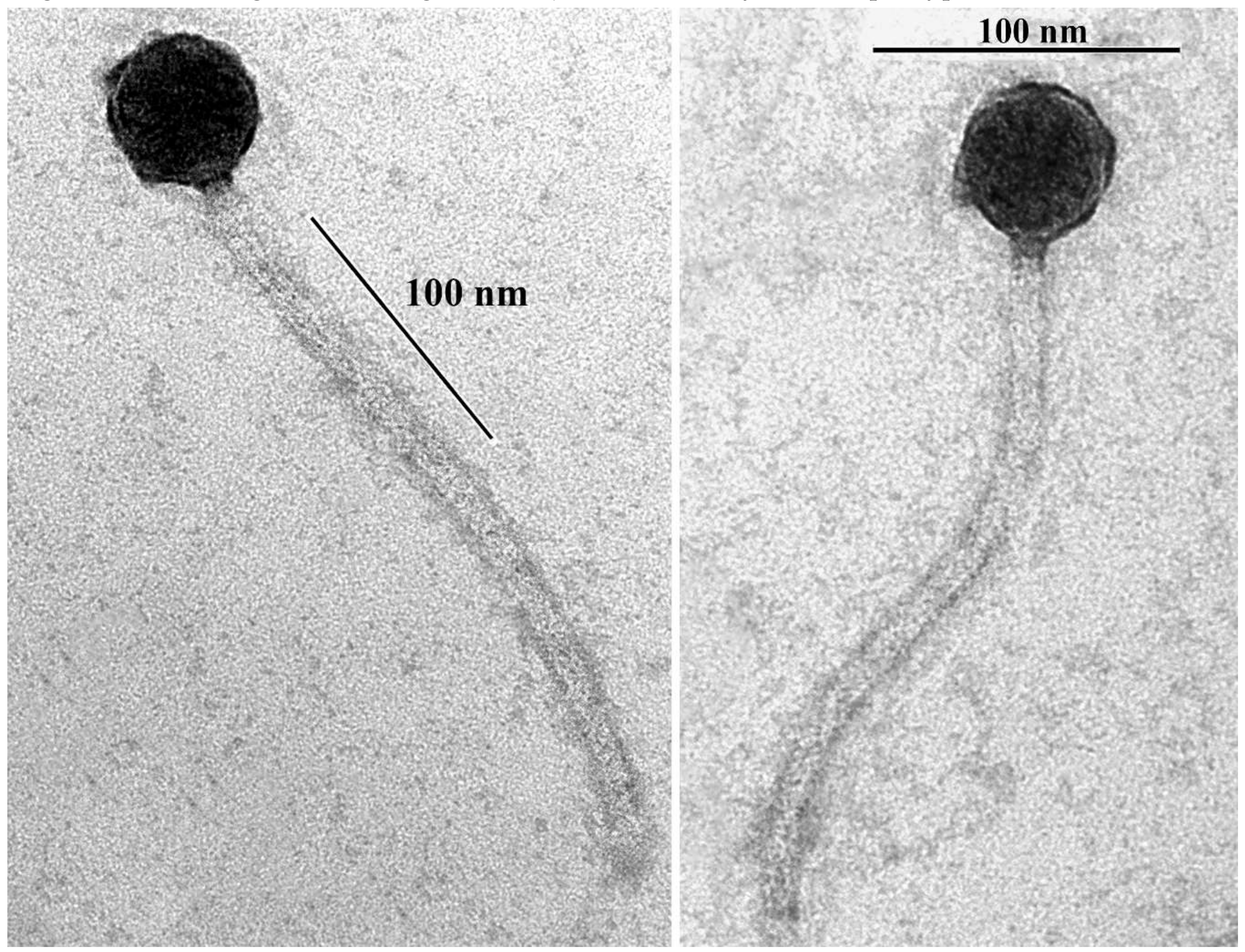
3.1. Isolation, Host Range and Morphology
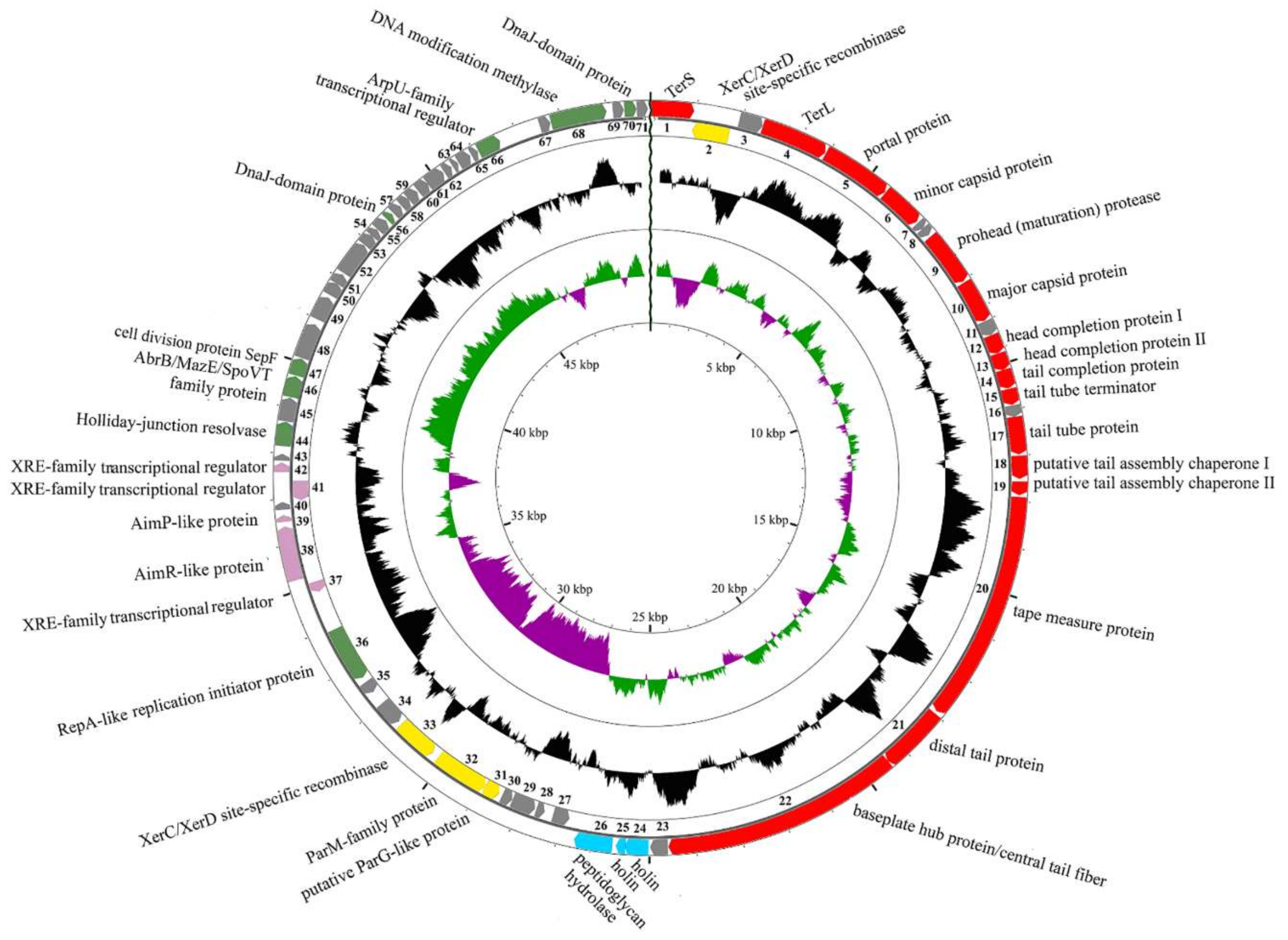
3.2. General Genome Organization of B83
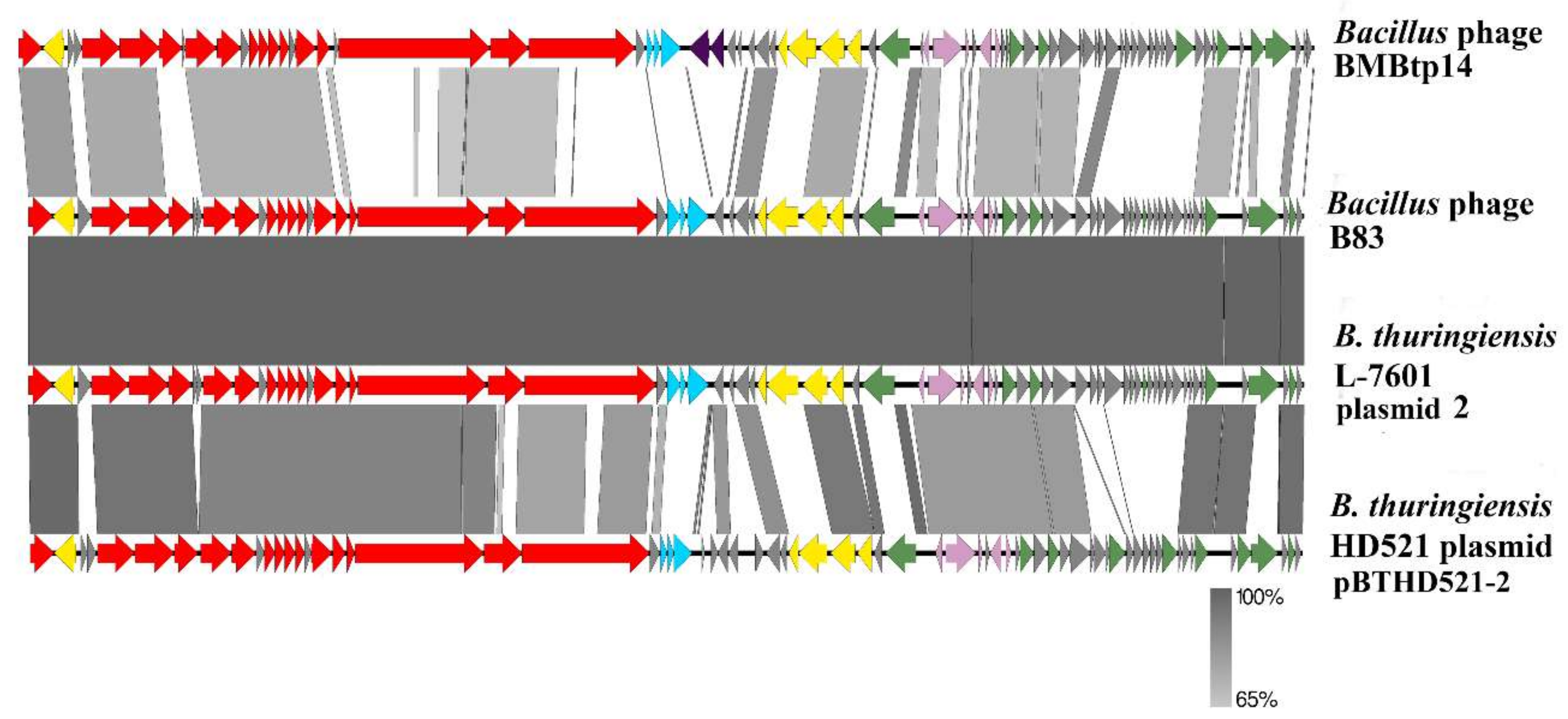
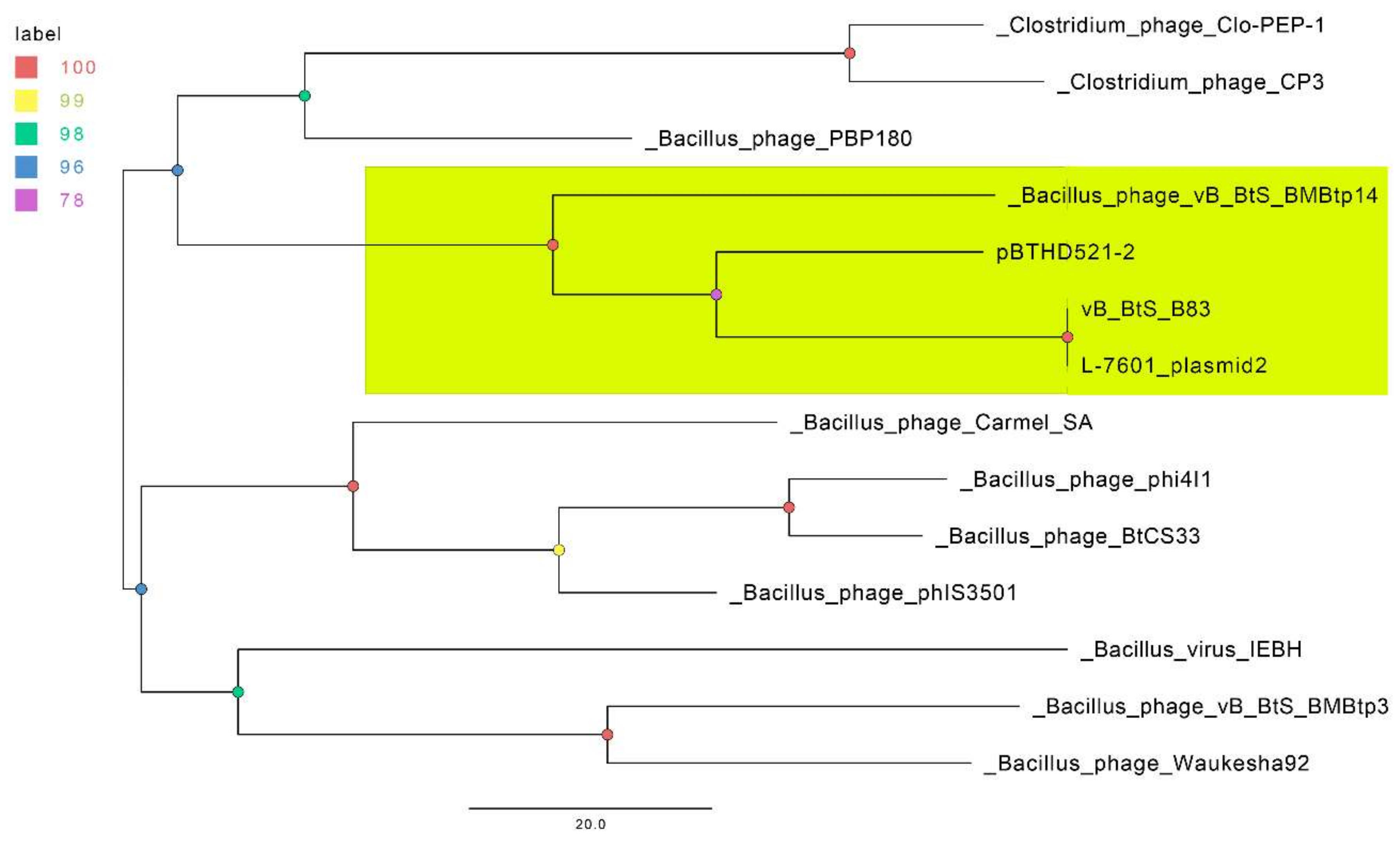
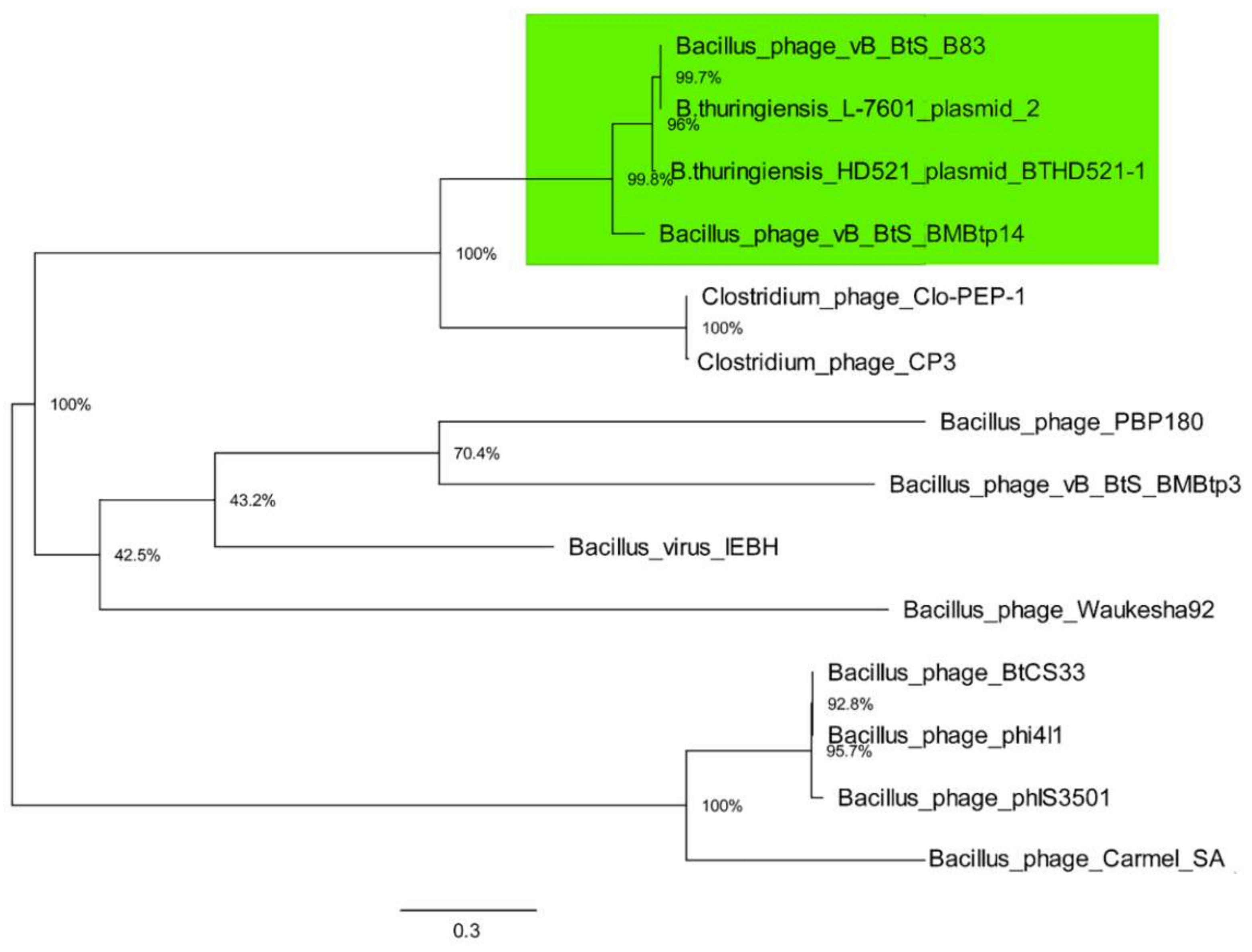
3.3. Comparative Genomics
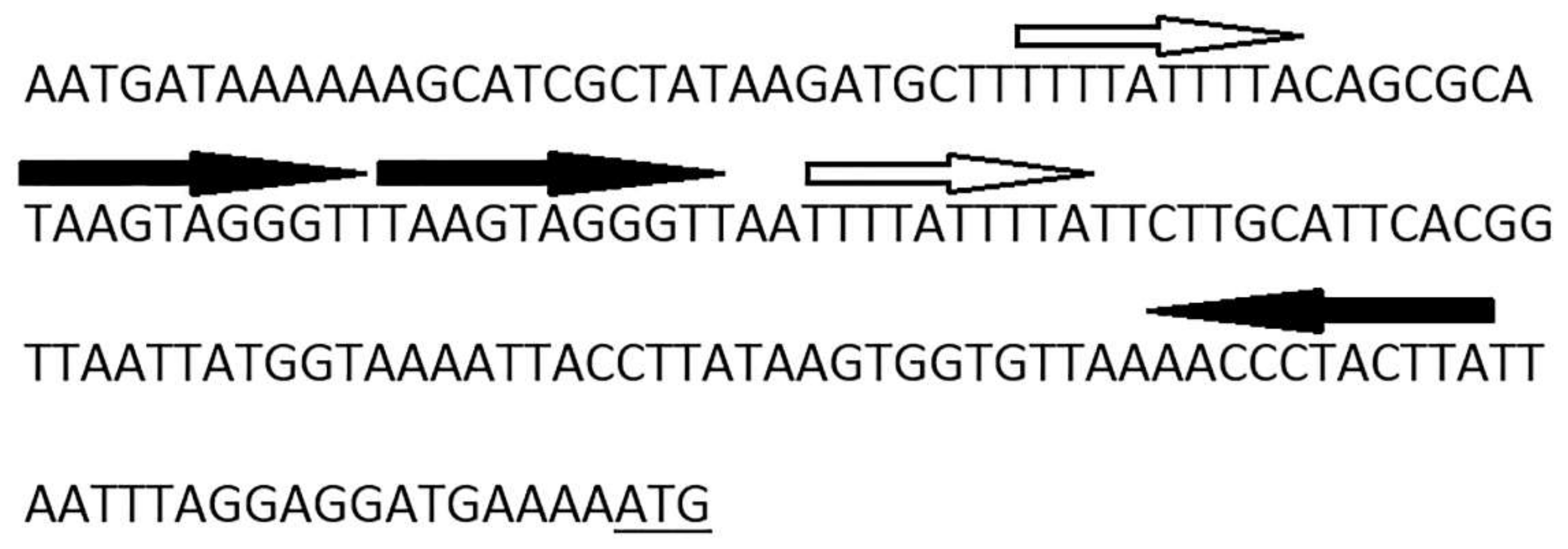
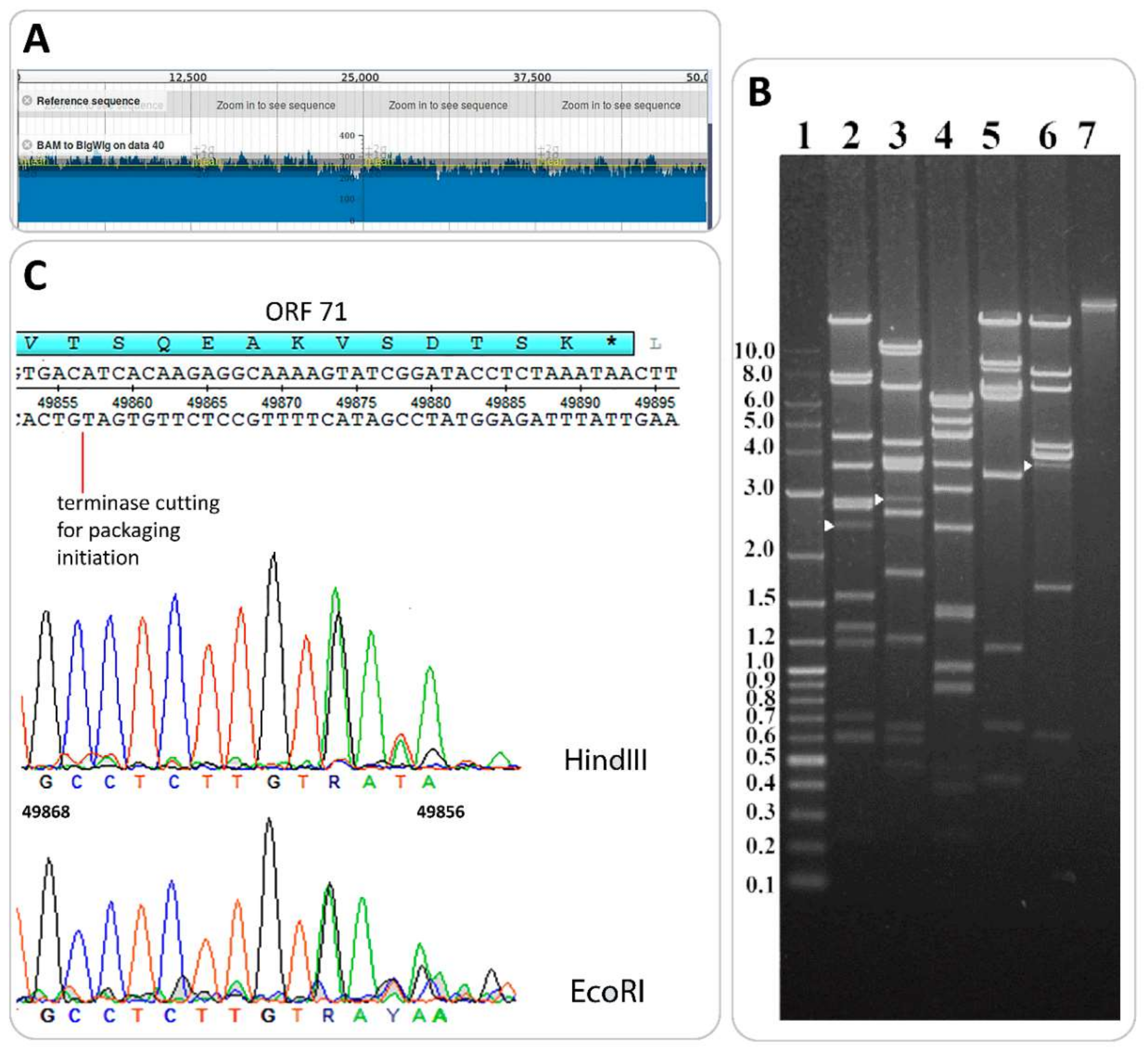
3.4. Determination of Packaging Strategy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Adriaenssens, E.M.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.E. The structure of coliphages. Microbiology 1963, 31, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Bradley, D.E. The fluorescent staining of bacteriophage nucleic acids. Microbiology 1966, 44, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Tolstoy, I.; Kropinski, A.M.; Brister, J.R. Bacteriophage taxonomy: An evolving discipline. Methods Mol. Biol. 2018, 57–71. [Google Scholar]
- Gillis, A.; Mahillon, J. Phages preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: Past, present and future. Viruses 2014, 6, 2623–2672. [Google Scholar] [CrossRef] [PubMed]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis–lysogeny decisions. Nature 2017, 541, 488. [Google Scholar] [CrossRef]
- Stokar-Avihail, A.; Tal, N.; Erez, Z.; Lopatina, A.; Sorek, R. Widespread utilization of peptide communication in phages infecting soil and pathogenic bacteria. Cell Host Microbe 2019, 25, 746–755. [Google Scholar] [CrossRef]
- Moumen, B.; Sorokin, A. Sequence analysis of inducible prophage phIS3501 integrated into the haemolysin II gene of Bacillus thuringiensis var israelensis ATCC35646. Genet. Res. Int. 2012. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Pile-up Analysis Using Starts & Ends. Available online: https://cpt.tamu.edu/computer-resources/pause/ (accessed on 29 May 2019).
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 10, 8365. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- BLAST®. Basic Local Alignment Search Tool. Available online: http://www.ncbi.nlm.nih.gov/blast/Blast.cgi (accessed on 29 May 2019).
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2004, 21, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics. 2011, 7, 1009–1010. [Google Scholar] [CrossRef]
- Zafar, N.; Mazumder, R.; Seto, D. CoreGenes: A computational tool for identifying and cataloging “core” genes in a set of small genomes. BMC Bioinform. 2002, 3, 12. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic virus orthologous groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2016, 45, 491–498. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.M.; Kannan, L.; Coleman, M.K.; Wolf, Y.I.; Sorokin, A.; Koonin, E.V.; Mushegian, A. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 2010, 26, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, P.; Ochoa-Sánchez, L.E.; Contreras-Moreira, B. GET_PHYLOMARKERS, a software package to select optimal orthologous clusters for phylogenomics and inferring pan-genome phylogenies, used for a critical geno-taxonomic revision of the genus Stenotrophomonas. Front. Microbiol. 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree 1.4.A Graphical Viewer of Phylogenetic Trees and a Program for Producing Publication-Ready Figures. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 29 May 2019).
- Shadrin, A.M.; Shapyrina, E.V.; Siunov, A.V.; Severinov, K.V.; Solonin, A.S. Bacillus cereus pore-forming toxins hemolysin II and cytotoxin K: Polymorphism and distribution of genes among representatives of the cereus group. Microbiology 2007, 76, 405–412. [Google Scholar] [CrossRef]
- Ackermann, H.W. Frequency of morphological phage descriptions in the year 2000. Arch. Virol. 2001, 146, 843–857. [Google Scholar] [CrossRef]
- Weigel, C.; Seitz, H. Bacteriophage replication modules. FEMS Microbiol. Rev. 2006, 30, 321–381. [Google Scholar] [CrossRef]
- Ikeda, H.; Tomizawa, J. Prophage P1, an extrachromosomal replication unit. Cold Spring Harb. Symp. Quant. Biol. 1968, 33, 791–798. [Google Scholar] [CrossRef]
- Jensen, R.B.; Gerdes, K. Mechanism of DNA segregation in prokaryotes: ParM partitioning protein of plasmid R1 co-localizes with its replicon during the cell cycle. EMBO J. 1999, 18, 4076–4084. [Google Scholar] [CrossRef] [PubMed]
- Salje, J.; Gayathri, P.; Löwe, J. The ParMRC system: Molecular mechanisms of plasmid segregation by actin-like filaments. Nat. Rev. Microbiol. 2010, 8, 683. [Google Scholar] [CrossRef] [PubMed]
- Golovanov, A.P.; Barillà, D.; Golovanova, M.; Hayes, F.; Lian, L.Y. ParG, a protein required for active partition of bacterial plasmids, has a dimeric ribbon–helix–helix structure. Mol. Microbiol. 2003, 50, 1141–1153. [Google Scholar] [CrossRef] [PubMed]
- Barillà, D.; Hayes, F. Architecture of the ParF• ParG protein complex involved in prokaryotic DNA segregation. Mol. Microbiol. 2003, 49, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.R.; Cardarelli, L.; Pell, L.G.; Radford, D.R.; Maxwell, K.L. Long noncontractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012, 115–142. [Google Scholar]
- Cao, Z.L.; Tan, T.T.; Jiang, K.; Mei, S.Q.; Hou, X.Y.; Cai, J. Complete genome sequence of Bacillus thuringiensis L-7601, a wild strain with high production of melanin. J. Biotechnol. 2018, 275, 40–43. [Google Scholar] [CrossRef]
- Li, Q.; Xu, L.Z.; Zou, T.; Ai, P.; Huang, G.H.; Li, P.; Zheng, A.P. Complete genome sequence of Bacillus thuringiensis strain HD521. Stand. Genomic Sci. 2015, 10, 62. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Edwards, R.; Nash, J.H.; Mahadevan, P.; Seto, D.; Ackermann, H.W.; Lavigne, R.; Kropinski, A.M. Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology. 2015, 477, 144–154. [Google Scholar] [CrossRef]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Russell, D.A. Sequencing, assembling, and finishing complete bacteriophage genomes. Methods Mol. Biol. 2018, 3, 109–125. [Google Scholar]
- Colloms, S.D.; Sykora, P.; Szatmari, G.; Sherratt, D.J. Recombination at ColE1 cer requires the Escherichia coli xerC gene product, a member of the lambda integrase family of site-specific recombinases. J. Bacteriol. 1990, 172, 6973–6980. [Google Scholar] [CrossRef]
- Blakely, G.; May, G.; McCulloch, R.; Arciszewska, L.K.; Burke, M.; Lovett, S.T.; Sherratt, D.J. Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 1993, 75, 351–361. [Google Scholar] [CrossRef]
- Jensen, R.B.; Gerdes, K. Partitioning of plasmid RThe ParM protein exhibits ATPase activity and interacts with the centromere-like ParR-parC complex. J. Mol. Biol. 1997, 269, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Ruhfel, R.E.; Thorne, C.B. Physical and genetic characterisation of the Bacillus thuringiensis subsp. kurstaki HD-1 extrachromosomal temperate phage TP-21.00A0. In Proceedings of the 88th Annual Meeting of the American Social Microbiology Abstracts, Washington, DC, USA, 8–13 May 1988. [Google Scholar]
- Kanda, K.; Ohderaotoshi, T.; Shimojyo, A.; Kato, F.; Murata, A. An extrachromosomal prophage naturally associated with Bacillus thuringiensis serovar israelensis. Lett. Appl. Microbiol. 1999, 28, 305–308. [Google Scholar] [CrossRef]
- Smeesters, P.R.; Drèze, P.A.; Bousbata, S.; Parikka, K.J.; Timmery, S.; Hu, X.; Perez-Morga, D.; Deghorain, M.; Toussaint, A.; Mahillon, J.; et al. Characterization of a novel temperate phage originating from a cereulide-producing Bacillus cereus strain. Res. Microbiol. 2011, 162, 446–459. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Peng, Q.; Wu, D.; Kou, Z.; Wu, Y.; Liu, P.; Gao, M. Effects of actin-like proteins encoded by two Bacillus pumilus phages on unstable lysogeny, revealed by genomic analysis. Appl. Environ. Microbiol. 2015, 81, 339–350. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Species | Strain | Lysis * |
|---|---|---|---|
| 1 | B. cereus | VKM B-13 | − |
| 2 | B. cereus | VKM B-370 | + |
| 3 | B. cereus | VKM B-373 | + |
| 4 | B. cereus | VKM B-383 | − |
| 5 | B. cereus | VKM B-445 | − |
| 6 | B. cereus | VKM B-473 | + |
| 7 | B. cereus | VKM B-491 | − |
| 8 | B. cereus | VKM B-504T | − |
| 9 | B. cereus | VKM B-682 | − |
| 10 | B. cereus | VKM B-683 | − |
| 11 | B. cereus | VKM B-684 | − |
| 12 | B. cereus | VKM B-686 | − |
| 13 | B. cereus | VKM B-688 | − |
| 14 | B. cereus | VKM B-771 | − |
| 15 | B. cereus | VKM B-810 | − |
| 16 | B. cereus | VKM B-812 | − |
| 17 | B. cereus | ATCC 4342 | − |
| 18 | B. cereus | ATCC 14893 | − |
| 19 | B. thuringiensis | VKM B-83 | − |
| 20 | B. thuringiensis | VKM B-84 | − |
| 21 | B. thuringiensis | VKM B-85 | − |
| 22 | B. thuringiensis | VKM B-440 | − |
| 23 | B. thuringiensis | VKM B-446 | − |
| 24 | B. thuringiensis | VKM B-450 | − |
| 25 | B. thuringiensis | VKM B-453 | − |
| 26 | B. thuringiensis | VKM B-454 | − |
| 27 | B. thuringiensis | VKM B-1555 | − |
| 28 | B. thuringiensis | VKM B-1557 | − |
| 29 | B. thuringiensis | ATCC 35646 | − |
| 30 | B. weihenstephanensis | KBAB4 | − |
| Name | GenBank Accession No. | Genome Length (kb) | G+C-Content, % | ORFs | tRNA | Total DNA Sequence Identity *, % | Homologous Proteins **, % (Number) |
|---|---|---|---|---|---|---|---|
| B83 | MK759918.1 | 49,952 | 35.79 | 71 | 0 | 100 | 100 |
| plasmid unnamed 2 | CP020004.1 | 49,952 | 35.79 | 62 | 0 | 100 | 100 (62) |
| plasmid pBTHD521-2 | CP010108.1 | 49,838 | 35.91 | 70 | 0 | 64.7 | 60.0 (42) |
| BMBtp14 | KX190833.1 | 50,740 | 36.76 | 75 | 0 | 37.0 | 46.7 (35) |
| No. | Product | B83 Protein id | B83 Locus Tag | pVOG |
|---|---|---|---|---|
| 1 | terminase small subunit | QCQ57781.1 | B83_gp01 | - |
| 2 | XerC/XerD site-specific recombinase | QCQ57782.1 | B83_gp02 | VOG0275 |
| 3 | terminase large subunit | QCQ57785.1 | B83_gp04 | VOG4544 |
| 4 | portal protein | QCQ57783.1 | B83_gp05 | VOG4773 |
| 5 | putative phage head morphogenesis protein (minor capsid protein) | QCQ57786.1 | B83_gp06 | VOG4688 |
| 6 | phage prohead (maturation) protease | QCQ57790.1 | B83_gp09 | VOG4685 |
| 7 | major capsid protein | QCQ57789.1 | B83_gp10 | VOG0749 |
| 8 | hypothetical protein | QCQ57791.1 | B83_gp11 | - |
| 9 | phage head completion protein (neck protein) I | QCQ57792.1 | B83_gp12 | - |
| 10 | phage head completion protein (neck protein) II | QCQ57793.1 | B83_gp13 | - |
| 11 | tail completion protein | QCQ57794.1 | B83_gp14 | - |
| 12 | tail tube terminator | QCQ57795.1 | B83_gp15 | - |
| 13 | hypothetical protein | QCQ57796.1 | B83_gp16 | - |
| 14 | tail tube protein | QCQ57797.1 | B83_gp17 | - |
| 15 | putative tail assembly chaperone | QCQ57798.1 | B83_gp18 | - |
| 16 | tail tape measure protein | QCQ57800.1 | B83_gp20 | - |
| 17 | distal tail protein | QCQ57801.1 | B83_gp21 | VOG4605 |
| 18 | baseplate hub protein/central tail fiber | QCQ57802.1 | B83_gp22 | VOG4599 |
| 19 | hypothetical protein | QCQ57809.1 | B83_gp29 | - |
| 20 | XerC/XerD site-specific recombinase | QCQ57813.1 | B83_gp33 | VOG0275 |
| 21 | MerR family domain-containing protein | QCQ57835.1 | B83_gp34 | - |
| 22 | XRE family transcriptional regulator | QCQ57816.1 | B83_gp37 | - |
| 23 | AimR-like protein | QCQ57817.1 | B83_gp38 | VOG1563 |
| 24 | XRE family transcriptionalregulator | QCQ57820.1 | B83_gp41 | - |
| 25 | Holliday junction resolvase | QCQ57823.1 | B83_gp44 | VOG5617 |
| 26 | hypothetical protein | QCQ57824.1 | B83_gp45 | - |
| 27 | AbrB/MazE/SpoVT family DNA-binding domain-containing protein | QCQ57825.1 | B83_gp46 | VOG6628 |
| 28 | cell division protein SepF | QCQ57826.1 | B83_gp47 | VOG9647 |
| 29 | hypothetical protein | QCQ57827.1 | B83_gp48 | - |
| 30 | hypothetical protein | QCQ57844.1 | B83_gp64 | - |
| 31 | ArpU family transcriptional regulator | QCQ57846.1 | B83_gp66 | VOG0198 |
| 32 | DNA methyltransferase | QCQ57848.1 | B83_gp68 | VOG4571 |
| 33 | hypothetical protein | QCQ57849.1 | B83_gp69 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piligrimova, E.G.; Kazantseva, O.A.; Nikulin, N.A.; Shadrin, A.M. Bacillus Phage vB_BtS_B83 Previously Designated as a Plasmid May Represent a New Siphoviridae Genus. Viruses 2019, 11, 624. https://doi.org/10.3390/v11070624
Piligrimova EG, Kazantseva OA, Nikulin NA, Shadrin AM. Bacillus Phage vB_BtS_B83 Previously Designated as a Plasmid May Represent a New Siphoviridae Genus. Viruses. 2019; 11(7):624. https://doi.org/10.3390/v11070624
Chicago/Turabian StylePiligrimova, Emma G., Olesya A. Kazantseva, Nikita A. Nikulin, and Andrey M. Shadrin. 2019. "Bacillus Phage vB_BtS_B83 Previously Designated as a Plasmid May Represent a New Siphoviridae Genus" Viruses 11, no. 7: 624. https://doi.org/10.3390/v11070624
APA StylePiligrimova, E. G., Kazantseva, O. A., Nikulin, N. A., & Shadrin, A. M. (2019). Bacillus Phage vB_BtS_B83 Previously Designated as a Plasmid May Represent a New Siphoviridae Genus. Viruses, 11(7), 624. https://doi.org/10.3390/v11070624