Ubiquitous Carbohydrate Binding Modules Decorate 936 Lactococcal Siphophage Virions

 ,
,  ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Hosts and Bacteriophages
2.2. Host CWPS Genotyping
2.3. Phage Host-Range Assays
2.4. Bioinformatic Analysis
2.5. CBM-Encoding Gene Cloning
2.6. Protein Production
2.7. Fluorescent Binding Assays
3. Results
3.1. Targets of the Bioinformatic Analyses
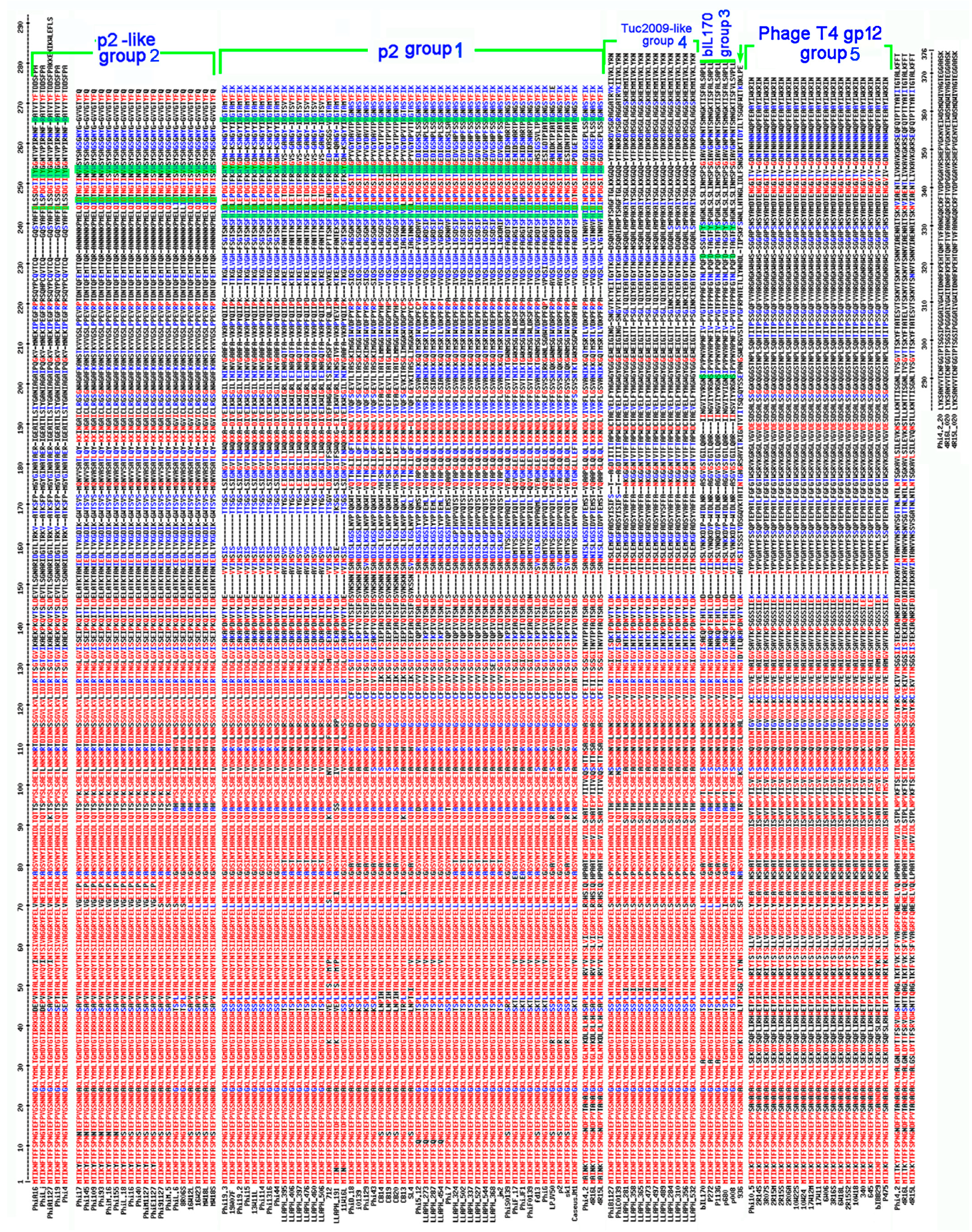
3.2. Bioinformatic Analysis of Receptor Binding Proteins (RBPs)
3.3. RBP Group vs. CWPS Type
3.4. Bioinformatic Analysis of the Major Tail Proteins
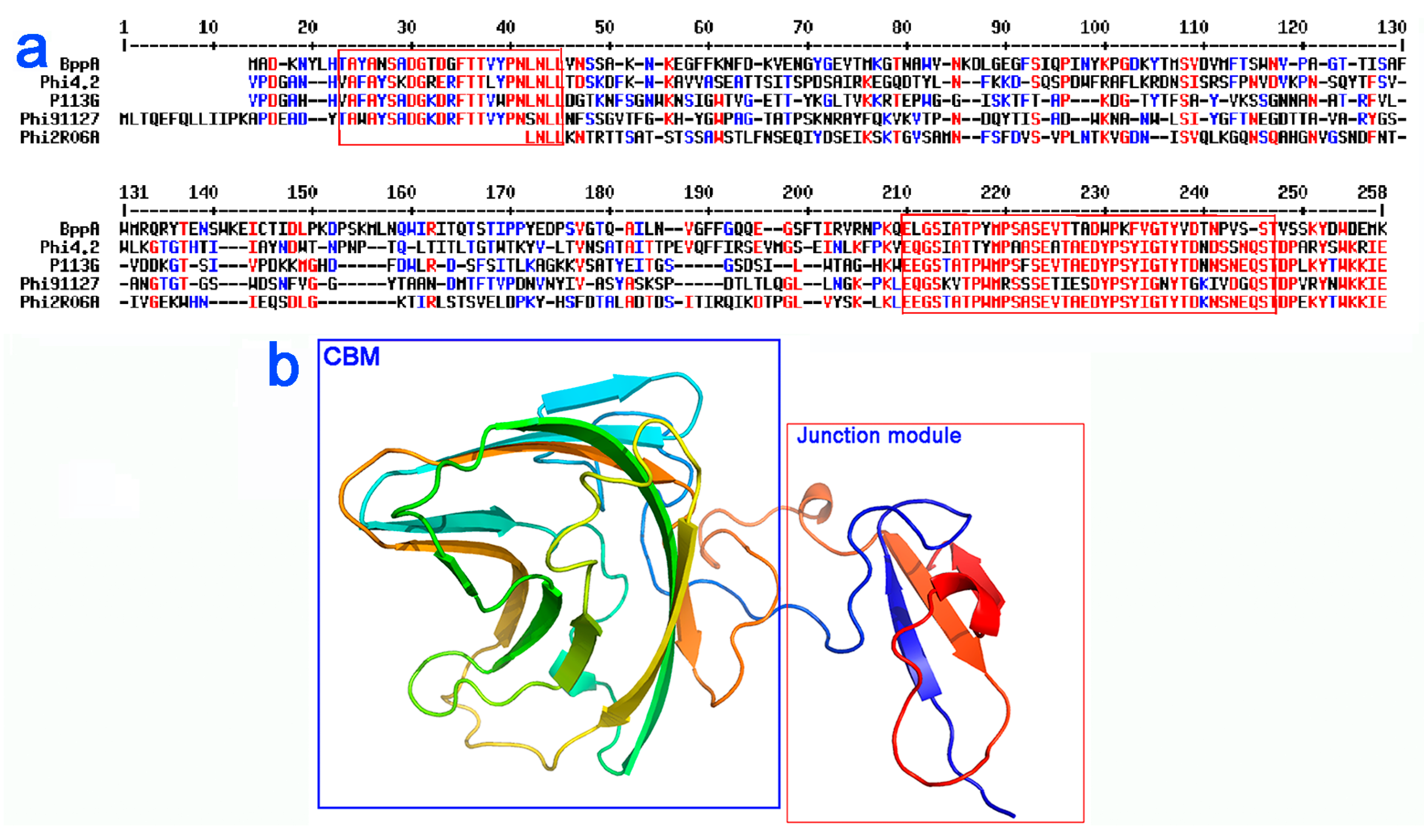
3.5. Bioinformatic Analysis of the Neck Passage Structure Proteins
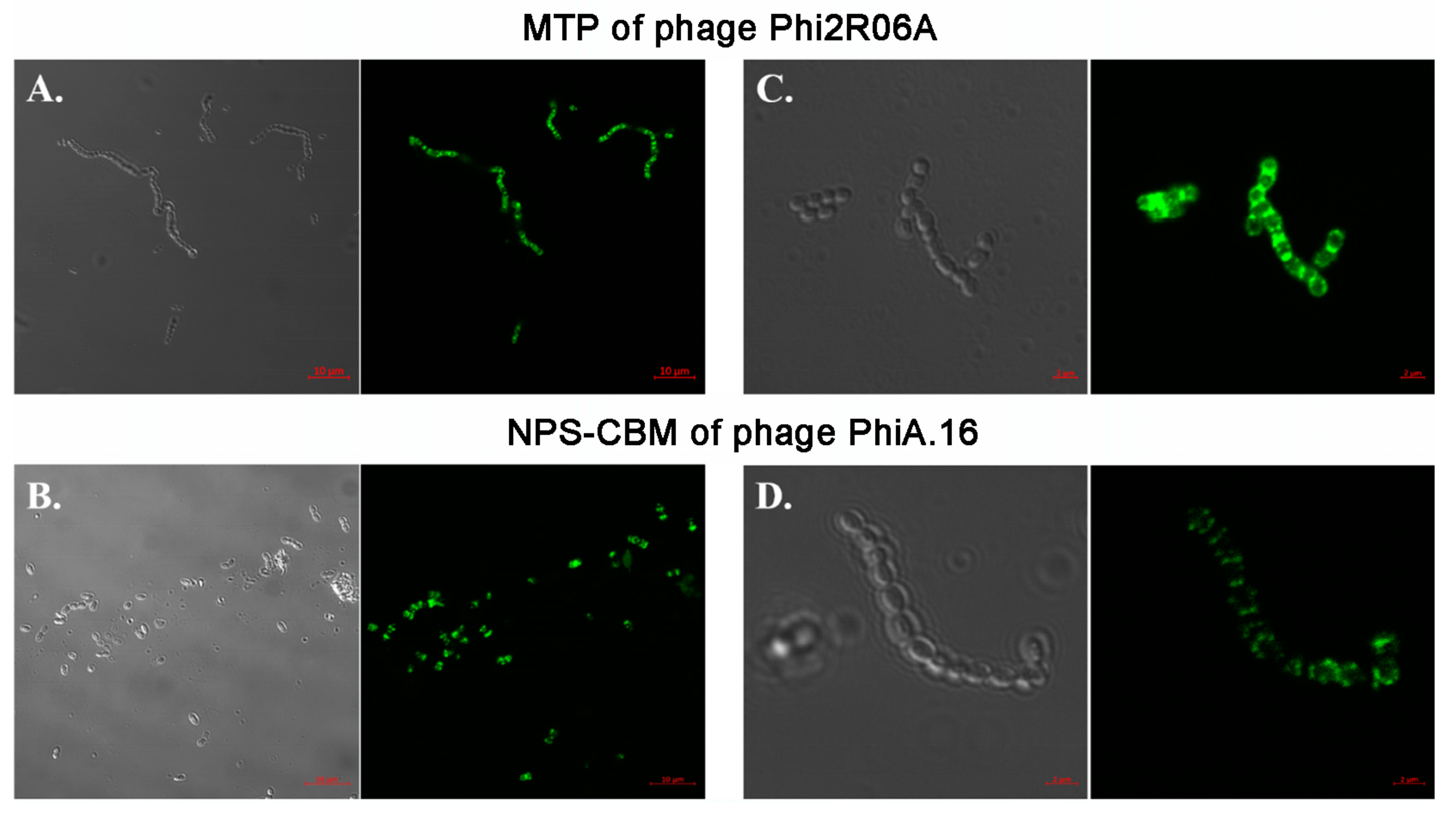
3.6. Fluorescent Binding Assays of NPS-CBM and MTP-TpeX Domains
3.7. Strain and CWPS Specificity
3.8. The Multiplicity of CBM Modules
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garneau, J.E.; Moineau, S. Bacteriophages of lactic acid bacteria and their impact on milk fermentations. Microb. Cell Factories 2011, 10, S20. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H. Population Genomics of Bacteriophages; Springer: New York, NY, USA, 2018; pp. 297–334. [Google Scholar]
- Mahony, J.; Murphy, J.; van Sinderen, D. Lactococcal 936-type phages and dairy fermentation problems: From detection to evolution and prevention. Front. Microbiol. 2012, 3, 335. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Cambillau, C. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. 2011, 75, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Campanacci, V.; Veesler, D.; Lichière, J.; Blangy, S.; Sciara, G.; Moineau, S.; Van Sinderen, D.; Bron, P.; Cambillau, C. Solution and electron microscopy characterization of lactococcal phage baseplates expressed in escherichia coli. J. Struct. Biol. 2010, 172, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, D.A.; Veesler, D.; Lichiere, J.; Ashcroft, A.E.; Cambillau, C. Unraveling lactococcal phages baseplate assembly by mass spectrometry. Mol. Cell. Proteomics 2011. [Google Scholar] [CrossRef] [PubMed]
- Veesler, D.; Spinelli, S.; Mahony, J.; Lichière, J.; Blangy, S.; Bricogne, G.; Legrand, P.; Ortiz-Lombardia, M.; Campanacci, V.; van Sinderen, D. Structure of the phage tp901-1 1.8 mda baseplate suggests an alternative host adhesion mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 8954–8958. [Google Scholar] [CrossRef]
- Sciara, G.; Bebeacua, C.; Bron, P.; Tremblay, D.; Ortiz-Lombardia, M.; Lichière, J.; Van Heel, M.; Campanacci, V.; Moineau, S.; Cambillau, C. Structure of lactococcal phage p2 baseplate and its mechanism of activation. Proc. Natl. Acad. Sci. USA 2010, 107, 6852–6857. [Google Scholar] [CrossRef]
- Hayes, S.; Duhoo, Y.; Neve, H.; Murphy, J.; Noben, J.-P.; Franz, C.; Cambillau, C.; Mahony, J.; Nauta, A.; van Sinderen, D. Identification of dual receptor binding protein systems in lactococcal 936 group phages. Viruses 2018, 10, 668. [Google Scholar] [CrossRef]
- Spinelli, S.; Desmyter, A.; Verrips, C.T.; de Haard, H.J.; Moineau, S.; Cambillau, C. Lactococcal bacteriophage p2 receptor-binding protein structure suggests a common ancestor gene with bacterial and mammalian viruses. Nat. Struct. Mol. Biol. 2006, 13, 85–89. [Google Scholar] [CrossRef]
- Tremblay, D.M.; Tegoni, M.; Spinelli, S.; Campanacci, V.; Blangy, S.; Huyghe, C.; Desmyter, A.; Labrie, S.; Moineau, S.; Cambillau, C. Receptor-binding protein of lactococcus lactis phages: Identification and characterization of the saccharide receptor-binding site. J. Bacteriol. 2006, 188, 2400–2410. [Google Scholar] [CrossRef]
- Ricagno, S.; Campanacci, V.; Blangy, S.; Spinelli, S.; Tremblay, D.; Moineau, S.; Tegoni, M.; Cambillau, C. Crystal structure of the receptor-binding protein head domain from lactococcus lactis phage bil170. J. Virol. 2006, 80, 9331–9335. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Veesler, D.; Bebeacua, C.; Cambillau, C. Structures and host-adhesion mechanisms of lactococcal siphophages. Front. Microbiol. 2014, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Mahony, J.; Kot, W.; Murphy, J.; Ainsworth, S.; Neve, H.; Hansen, L.H.; Heller, K.J.; Sørensen, S.J.; Hammer, K.; Cambillau, C. Investigation of the relationship between lactococcal host cell wall polysaccharide genotype and 936 phage receptor binding protein phylogeny. Appl. Environ. Microbiol. 2013, 79, 4385–4392. [Google Scholar] [CrossRef] [PubMed]
- Ainsworth, S.; Sadovskaya, I.; Vinogradov, E.; Courtin, P.; Guerardel, Y.; Mahony, J.; Grard, T.; Cambillau, C.; Chapot-Chartier, M.-P.; Van Sinderen, D. Differences in lactococcal cell wall polysaccharide structure are major determining factors in bacteriophage sensitivity. mBio 2014, 5, e00880-14. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Bottacini, F.; Mahony, J.; Kelleher, P.; Neve, H.; Zomer, A.; Nauta, A.; Van Sinderen, D. Comparative genomics and functional analysis of the 936 group of lactococcal siphoviridae phages. Sci. Rep. 2016, 6, 21345. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S.; Vincentelli, R.; Mahony, J.; Nauta, A.; Ramond, L.; Lugli, G.A.; Ventura, M.; van Sinderen, D.; Cambillau, C. Functional carbohydrate binding modules identified in evolved dits from siphophages infecting various gram-positive bacteria. Mol. Microbiol. 2018, 110, 777–795. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.; Bebeacua, C.; Mahony, J.; Blangy, S.; Douillard, F.P.; Veesler, D.; Cambillau, C.; van Sinderen, D. Structure and functional analysis of the host-recognition device of lactococcal phage tuc2009. J. Virol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Legrand, P.; Collins, B.; Blangy, S.; Murphy, J.; Spinelli, S.; Gutierrez, C.; Richet, N.; Kellenberger, C.; Desmyter, A.; Mahony, J. The atomic structure of the phage tuc2009 baseplate tripod suggests that host recognition involves two different carbohydrate binding modules. mBio 2016, 7, e01781-15. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.; Royer, B.; Mahony, J.; Hoyles, L.; Heller, K.; Neve, H.; Bonestroo, M.; Nauta, A.; van Sinderen, D. Biodiversity of lactococcal bacteriophages isolated from 3 gouda-type cheese-producing plants. J. Dairy Sci. 2013, 96, 4945–4957. [Google Scholar] [CrossRef]
- Ainsworth, S. Characterisation of Bacteriophage-Host Interactions in Lactococcus Lactis. Ph.D. Thesis, University College Cork, Cork, Ireland, 2014. [Google Scholar]
- Lillehaug, D. An improved plaque assay for poor plaque-producing temperate lactococcal bacteriophages. J. Appl. Microbiol. 1997, 83, 85–90. [Google Scholar] [CrossRef]
- Hildebrand, A.; Remmert, M.; Biegert, A.; Söding, J. Fast and accurate automatic structure prediction with hhpred. Proteins Struct. Funct. Bioinform. 2009, 77, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The hhpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The Pymol Molecular Graphics System. 2002. Available online: https://pymol.org/2/ (accessed on 27 May 2019).
- Turchetto, J.; Sequeira, A.F.; Ramond, L.; Peysson, F.; Brás, J.L.; Saez, N.J.; Duhoo, Y.; Blémont, M.; Guerreiro, C.I.; Quinton, L. High-throughput expression of animal venom toxins in escherichia coli to generate a large library of oxidized disulphide-reticulated peptides for drug discovery. Microb. Cell Factories 2017, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, M.E.; Spinelli, S.; Sadovskaya, I.; Piuri, M.; Cambillau, C. Evolved distal tail carbohydrate binding modules of l actobacillus phage j-1: A novel type of anti-receptor widespread among lactic acid bacteria phages. Mol. Microbiol. 2017, 104, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Bebeacua, C.; Tremblay, D.; Farenc, C.; Chapot-Chartier, M.-P.; Sadovskaya, I.; Van Heel, M.; Veesler, D.; Moineau, S.; Cambillau, C. Structure, adsorption to host, and infection mechanism of virulent lactococcal phage p2. J. Virol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Vegge, C.S.; Neve, H.; Brøndsted, L.; Heller, K.J.; Vogensen, F.K. Analysis of the collar-whisker structure of temperate lactococcal bacteriophage tp901-1. Appl. Environ. Microbiol. 2006, 72, 6815–6818. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leiman, P.G.; Shneider, M.M.; Mesyanzhinov, V.V.; Rossmann, M.G. Evolution of bacteriophage tails: Structure of t4 gene product 10. J. Mol. Biol. 2006, 358, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Bebeacua, C.; Lai, L.; Vegge, C.S.; Brøndsted, L.; van Heel, M.; Veesler, D.; Cambillau, C. Visualizing a complete siphoviridae member by single-particle electron microscopy: The structure of lactococcal phage tp901-1. J. Virol. 2013, 87, 1061–1068. [Google Scholar] [CrossRef]
- Pell, L.G.; Liu, A.; Edmonds, L.; Donaldson, L.W.; Howell, P.L.; Davidson, A.R. The x-ray crystal structure of the phage λ tail terminator protein reveals the biologically relevant hexameric ring structure and demonstrates a conserved mechanism of tail termination among diverse long-tailed phages. J. Mol. Biol. 2009, 389, 938–951. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.; Prokhorov, N.S.; Guerrero-Ferreira, R.C.; Shneider, M.M.; Browning, C.; Goldie, K.N.; Stahlberg, H.; Leiman, P.G. Structure of the t4 baseplate and its function in triggering sheath contraction. Nature 2016, 533, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, C.A.; Effantin, G.; Vivès, C.; Engilberge, S.; Bacia, M.; Boulanger, P.; Girard, E.; Schoehn, G.; Breyton, C. Bacteriophage T5 tail tube structure suggests a trigger mechanism for Siphoviridae DNA ejection. Nat. Commun. 2017, 8, 1953. [Google Scholar] [CrossRef] [PubMed]
- Sassi, M.; Bebeacua, C.; Drancourt, M.; Cambillau, C. The first structure of a mycobacteriophage, araucaria. J. Virol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Pell, L.G.; Gasmi-Seabrook, G.M.; Morais, M.; Neudecker, P.; Kanelis, V.; Bona, D.; Donaldson, L.W.; Edwards, A.M.; Howell, P.L.; Davidson, A.R. The solution structure of the c-terminal ig-like domain of the bacteriophage λ tail tube protein. J. Mol. Biol. 2010, 403, 468–479. [Google Scholar] [CrossRef]
- Fraser, J.S.; Maxwell, K.L.; Davidson, A.R. Immunoglobulin-like domains on bacteriophage: Weapons of modest damage? Curr. Opin. Microbiol. 2007, 10, 382–387. [Google Scholar] [CrossRef]
- Auzat, I.; Dröge, A.; Weise, F.; Lurz, R.; Tavares, P. Origin and function of the two major tail proteins of bacteriophage spp1. Mol. Microbiol. 2008, 70, 557–569. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Casjens, S.R.; Cingolani, G. Exploring the atomic structure and conformational flexibility of a 320 Å long engineered viral fiber using x-ray crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 342–353. [Google Scholar] [CrossRef]
- Montanier, C.; Money, V.A.; Pires, V.M.; Flint, J.E.; Pinheiro, B.A.; Goyal, A.; Prates, J.A.; Izumi, A.; Stålbrand, H.; Morland, C. The active site of a carbohydrate esterase displays divergent catalytic and noncatalytic binding functions. PLoS Biol. 2009, 7, e1000071. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, M.E.; Martin, J.F.; Durán, R.; Nemirovsky, S.I.; Rivas, C.S.; Bowman, C.; Russell, D.; Hatfull, G.F.; Cambillau, C.; Piuri, M. Characterization of prophages containing “evolved” dit/tal modules in the genome of lactobacillus casei bl23. Appl. Microbiol. Biotechnol. 2016, 100, 9201–9215. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | RBP Head | Dit † Class | NPS Group | TpeX Group | Phage | RBP Head | Dit † Class | NPS Group | TpeX Group |
|---|---|---|---|---|---|---|---|---|---|
| PhiA.16 | p2-like | A | ASCC502 | p2 | E | ||||
| PhiLj | p2-like | A | ASCC337 | p2 | E | ||||
| PhiA1127 | p2-like | A | ASCC527 | p2 | E | ||||
| Phi19 | p2-like | A | ASCC544 | p2 | E | ||||
| Phi4 | p2-like | A | ASCC368 | p2 | E | ||||
| Phi17 | p2-like | 1 | III | jm2 | p2 | D | |||
| Phi145 | p2-like | 1 | III | PhiS0139 | p2 | A | |||
| Phi109 | p2-like | 1 | III | PhiF.17 | p2 | A | |||
| Phi93 | p2-like | 1 | III | PhiJF1 | p2 | A | |||
| PhiM.16 | p2-like | 1 | A | PhiF0139 | p2 | A | |||
| Phi155 | p2-like | 1 | B | fd13 | p2 | A | |||
| PhiL.18 | p2-like | 1 | III | PhiG | p2 | A | |||
| Phi16 | p2-like | 1 | III | jj50 | p2 | ||||
| Phi40 | p2-like | 1 | III | p2 | p2 | ||||
| PhiM1127 | p2-like | 1 | III | sk1 | p2 | ||||
| PhiE1127 | p2-like | 1 | III | JM1 | p2 | B | |||
| Phi91127 | p2-like | 1 | III | Phi4.2 ‡ | p2 | II | |||
| PhiM.5 | p2-like | 1 | III | Phi4R16L ‡ | p2 | II | |||
| PhiL.6 | p2-like | 4 | III | Phi4R15L ‡ | p2 | II | |||
| Phi8R06S | p2-like | 4 | III | PhiB1127 | Tuc2009 | C | I | ||
| Phi16W12L | p2-like | 1 | III | PhiC0139 | Tuc2009 | C | I | ||
| Phi16W23 | p2-like | 1 | III | ASCC281 | Tuc2009 | G | |||
| PhiMW18L | p2-like | 1 | III | ASCC358 | Tuc2009 | G | |||
| PhiMW18S | p2-like | 1 | III | ASCC365 | Tuc2009 | G | |||
| MP1 | p2-like | 1 | A | III | ASCC473 | Tuc2009 | G | ||
| Phi19.3 | p2 | D | ASCC497 | Tuc2009 | G | ||||
| Phi19W07F | p2 | F | ASCC489 | Tuc2009 | G | ||||
| Phi19.2 | p2 | D | ASCC284 | Tuc2009 | G | ||||
| Phi15 | p2 | F | ASCC310 | Tuc2009 | G | ||||
| Phi13W11L | p2 | D | ASCC356 | Tuc2009 | G | ||||
| Phi114 | p2 | F | ASCC532 | Tuc2009 | G | ||||
| Phi13.16 | p2 | D | bIL170 | bIL170 | A | ||||
| Phi44 | p2 | F | P272 | bIL170 | I | ||||
| ASCC395 | p2 | E | P113G | bIL170 | I | ||||
| ASCC406 | p2 | E | p680 | bIL170 | B | ||||
| ASCC397 | p2 | E | p008 | bIL170 | G | ||||
| ASCC476 | p2 | E | 936 | bIL170 | |||||
| ASCC460 | p2 | E | Phi10.5 | T4 gp12 | 2 | ||||
| ASCC506 | p2 | E | Phi2R14S | T4 gp12 | 2 | ||||
| 712 | p2 | 3 | I | Phi3R07S | T4 gp12 | 2 | |||
| ASCC191 | p2 | E | Phi2R15M | T4 gp12 | 2 | ||||
| Phi11W16L | p2 | D | Phi2R15S | T4 gp12 | 2 | B | |||
| PhiD.18 | p2 | C | Phi2R06A | T4 gp12 | 2 | IV | |||
| i0139 | p2 | C | Phi10W22S | T4 gp12 | 2 | ||||
| Phi129 | p2 | C | Phi10W24 | T4 gp12 | 2 | ||||
| Phi43 | p2 | C | Phi17W12M | T4 gp12 | 2 | B | |||
| CB14 | p2 | E | Phi17W11 | T4 gp12 | 2 | ||||
| CB19 | p2 | A | Phi6W06 | T4 gp12 | 2 | B | |||
| CB20 | p2 | A | Phi3R16S | T4 gp12 | 2 | ||||
| CB13 | p2 | E | Phi6W18L | T4 gp12 | 2 | ||||
| SL4 | p2 | 3 | Phi2R15S2 | T4 gp12 | 2 | ||||
| Phi5.12 | p2 | IV | Phi10W18 | T4 gp12 | 2 | B | |||
| ASCC273 | p2 | E | 340 | T4 gp12 | F | ||||
| ASCC287 | p2 | E | 645 | T4 gp12 | F | ||||
| ASCC454 | p2 | E | bIBB29 | T4 gp12 | 2 | ||||
| Phi7 | p2 | G | I | P475 | T4 gp12 | I | |||
| ASCC324 | p2 | E |
| Subgroup | Lead Phage | HHpred Hit | Corresponding Protein | No. of Phages |
|---|---|---|---|---|
| I | P113G | 5e7t | Tuc2009, BppA | 7 |
| II | Phi4.2 | 5e7t | Tuc2009, BppA | 3 |
| III | Phi91127 | 5e7t | Tuc2009, BppA | 18 |
| IV | Phi2R06A | 5e7t | Tuc2009, BppA | 2 |
| Group | Lead Phage | HHpred Hit | Corresponding Protein | No. of Phages |
|---|---|---|---|---|
| Group A | PhiA.16 | 5e7t | Tuc2009, BppA | 16 |
| Group B | Phi155 | 5e7t | Tuc2009, BppA | 7 |
| Group C | PhiB1127 | 5e7t | Tuc2009, BppA | 6 |
| Group D | Phi19.3 | 1gq8, 2pyg | Pectin | 6 |
| Group E | ASCC365 | 2pyg, 4mxn | Alginate | 18 |
| Group F | Phi114 | 2waa | Carbohydrate esterase | 6 |
| Group G | ASCC191 | 2waa | Carbohydrate esterase | 12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayes, S.; Mahony, J.; Vincentelli, R.; Ramond, L.; Nauta, A.; van Sinderen, D.; Cambillau, C. Ubiquitous Carbohydrate Binding Modules Decorate 936 Lactococcal Siphophage Virions. Viruses 2019, 11, 631. https://doi.org/10.3390/v11070631
Hayes S, Mahony J, Vincentelli R, Ramond L, Nauta A, van Sinderen D, Cambillau C. Ubiquitous Carbohydrate Binding Modules Decorate 936 Lactococcal Siphophage Virions. Viruses. 2019; 11(7):631. https://doi.org/10.3390/v11070631
Chicago/Turabian StyleHayes, Stephen, Jennifer Mahony, Renaud Vincentelli, Laurie Ramond, Arjen Nauta, Douwe van Sinderen, and Christian Cambillau. 2019. "Ubiquitous Carbohydrate Binding Modules Decorate 936 Lactococcal Siphophage Virions" Viruses 11, no. 7: 631. https://doi.org/10.3390/v11070631
APA StyleHayes, S., Mahony, J., Vincentelli, R., Ramond, L., Nauta, A., van Sinderen, D., & Cambillau, C. (2019). Ubiquitous Carbohydrate Binding Modules Decorate 936 Lactococcal Siphophage Virions. Viruses, 11(7), 631. https://doi.org/10.3390/v11070631