Molecular Analysis of the Complete Genome of a Simian Foamy Virus Infecting Hylobates pileatus (pileated gibbon) Reveals Ancient Co-Evolution with Lesser Apes



Abstract
1. Introduction
2. Materials and Methods
2.1. Blood Sample Processing, Co-Culture, and PCR Identification of a Novel Divergent SFV in Gibbons
2.2. Next Generation Sequencing and SFVhpi_SAM106 Genome Assembly
2.3. Sanger Sequencing of LTR Region
2.4. Complete SFVhpi_SAM106 Genome Sequence Analysis
2.5. Comparison of FV tRNA Binding Motifs
2.6. Nucleotide Sequence Accession Number
3. Results
3.1. SFVhpi_SAM106 Genome Assembly
3.2. Organization of the SFVhpi_SAM106 Genome and Comparison with Other Ape SFVs
3.3. Absence of Evidence of Genetic Recombination in the SFVhpi_SAM106 Genome
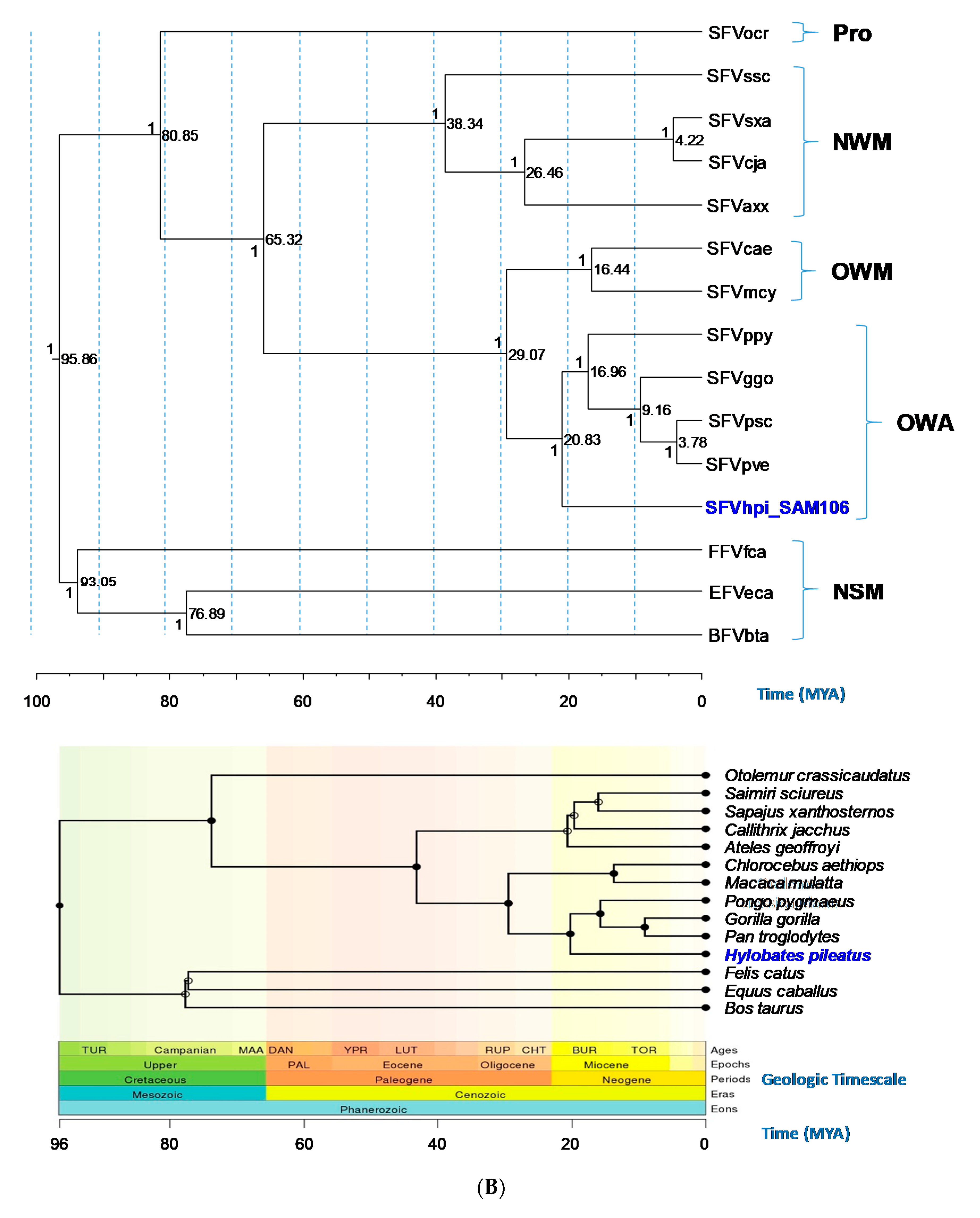
3.4. Evolutionary Relationships and Divergence Dating of SFVhpi_SAM106 and Other FVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Linial, M.L. Foamy viruses are unconventional retroviruses. J. Virol. 1999, 73, 1747–1755. [Google Scholar] [PubMed]
- Rethwilm, A. Molecular biology of foamy viruses. Med. Microbiol. Immunol. 2010, 199, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Santini, D.M.; Stenbak, C.R.; Linial, M.L. Foamy virus zoonotic infections. Retrovirology 2017, 14, 55. [Google Scholar] [CrossRef] [PubMed]
- Ghersi, B.M.; Jia, H.; Aiewsakun, P.; Katzourakis, A.; Mendoza, P.; Bausch, D.G.; Kasper, M.R.; Montgomery, J.M.; Switzer, W.M. Wide distribution and ancient evolutionary history of simian foamy viruses in new world primates. Retrovirology 2015, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Katzourakis, A.; Aiewsakun, P.; Jia, H.; Wolfe, N.D.; LeBreton, M.; Yoder, A.D.; Switzer, W.M. Discovery of prosimian and afrotherian foamy viruses and potential cross species transmissions amidst stable and ancient mammalian co-evolution. Retrovirology 2014, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.E.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Betsem, E.; Rua, R.; Tortevoye, P.; Froment, A.; Gessain, A. Frequent and recent human acquisition of simian foamy viruses through apes’ bites in central africa. PLoS Pathog. 2011, 7, e1002306. [Google Scholar] [CrossRef] [PubMed]
- Calattini, S.; Nerrienet, E.; Mauclere, P.; Georges-Courbot, M.C.; Saib, A.; Gessain, A. Natural simian foamy virus infection in wild-caught gorillas, mandrills and drills from cameroon and gabon. J. Gen. Virol. 2004, 85, 3313–3317. [Google Scholar] [CrossRef]
- Calattini, S.; Nerrienet, E.; Mauclere, P.; Georges-Courbot, M.C.; Saib, A.; Gessain, A. Detection and molecular characterization of foamy viruses in central african chimpanzees of the pan troglodytes troglodytes and pan troglodytes vellerosus subspecies. J. Med. Primatol. 2006, 35, 59–66. [Google Scholar] [CrossRef]
- Mouinga-Ondeme, A.; Betsem, E.; Caron, M.; Makuwa, M.; Salle, B.; Renault, N.; Saib, A.; Telfer, P.; Marx, P.; Gessain, A.; et al. Two distinct variants of simian foamy virus in naturally infected mandrills (Mandrillus sphinx) and cross-species transmission to humans. Retrovirology 2010, 7, 105. [Google Scholar] [CrossRef]
- Schulze, A.; Lemey, P.; Schubert, J.; McClure, M.O.; Rethwilm, A.; Bodem, J. Complete nucleotide sequence and evolutionary analysis of a gorilla foamy virus. J. Gen. Virol. 2011, 92, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Switzer, W.M.; Heneine, W. Foamy virus infection of humans. In Molecular Detection of Human Viral Pathogens; Liu, D., Ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2011; Volume 1, pp. 131–146. [Google Scholar]
- Troncoso, L.L.; Muniz, C.P.; Siqueira, J.D.; Curty, G.; Schrago, C.G.; Augusto, A.; Fedullo, L.; Soares, M.A.; Santos, A.F. Characterization and comparative analysis of a simian foamy virus complete genome isolated from brazilian capuchin monkeys. Virus Res. 2015, 208, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Bodem, J.; Buseyne, F.; Gessain, A.; Johnson, W.; Kuhn, J.H.; Kuzmak, J.; Lindemann, D.; Linial, M.L.; Lochelt, M.; et al. Spumaretroviruses: Updated taxonomy and nomenclature. Virology 2018, 516, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Rethwilm, A.; Bodem, J. Evolution of foamy viruses: The most ancient of all retroviruses. Viruses 2013, 5, 2349–2374. [Google Scholar] [CrossRef] [PubMed]
- Leendertz, F.H.; Zirkel, F.; Couacy-Hymann, E.; Ellerbrok, H.; Morozov, V.A.; Pauli, G.; Hedemann, C.; Formenty, P.; Jensen, S.A.; Boesch, C.; et al. Interspecies transmission of simian foamy virus in a natural predator-prey system. J. Virol. 2008, 82, 7741–7744. [Google Scholar] [CrossRef]
- Leendertz, S.A.; Junglen, S.; Hedemann, C.; Goffe, A.; Calvignac, S.; Boesch, C.; Leendertz, F.H. High prevalence, coinfection rate, and genetic diversity of retroviruses in wild red colobus monkeys (Piliocolobus badius badius) in Tai National Park, Cote D’ivoire. J. Virol. 2010, 84, 7427–7436. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Worobey, M.; Li, Y.; Keele, B.F.; Bibollet-Ruche, F.; Guo, Y.; Goepfert, P.A.; Santiago, M.L.; Ndjango, J.B.; Neel, C.; et al. Molecular ecology and natural history of simian foamy virus infection in wild-living chimpanzees. PLoS Pathog. 2008, 4, e1000097. [Google Scholar] [CrossRef]
- Richard, L.; Rua, R.; Betsem, E.; Mouinga-Ondeme, A.; Kazanji, M.; Leroy, E.; Njouom, R.; Buseyne, F.; Afonso, P.V.; Gessain, A. Cocirculation of two env molecular variants, of possible recombinant origin, in gorilla and chimpanzee simian foamy virus strains from central Africa. J. Virol. 2015, 89, 12480–12491. [Google Scholar] [CrossRef]
- Heneine, W.; Switzer, W.M.; Sandstrom, P.; Brown, J.; Vedapuri, S.; Schable, C.A.; Khan, A.S.; Lerche, N.W.; Schweizer, M.; Neumann-Haefelin, D.; et al. Identification of a human population infected with simian foamy viruses. Nat. Med. 1998, 4, 403–407. [Google Scholar] [CrossRef]
- Switzer, W.M.; Bhullar, V.; Shanmugam, V.; Cong, M.E.; Parekh, B.; Lerche, N.W.; Yee, J.L.; Ely, J.J.; Boneva, R.; Chapman, L.E.; et al. Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J. Virol. 2004, 78, 2780–2789. [Google Scholar] [CrossRef]
- Khan, A.S. Simian foamy virus infection in humans: Prevalence and management. Expert Rev. Anti-Infect. Ther. 2009, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Murphy, H.W.; Miller, M.; Ramer, J.; Travis, D.; Barbiers, R.; Wolfe, N.D.; Switzer, W.M. Implications of simian retroviruses for captive primate population management and the occupational safety of primate handlers. J. Zoo Wildl. Med. 2006, 37, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Rua, R.; Betsem, E.; Turpin, J.; Mahieux, R. Htlv-3/4 and simian foamy retroviruses in humans: Discovery, epidemiology, cross-species transmission and molecular virology. Virology 2013, 435, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.; Anthony, S.J.; Switzer, W.M.; Epstein, J.H.; Seimon, T.; Jia, H.; Sanchez, M.D.; Huynh, T.T.; Galland, G.G.; Shapiro, S.E.; et al. Zoonotic viruses associated with illegally imported wildlife products. PLoS ONE 2012, 7, e29505. [Google Scholar] [CrossRef] [PubMed]
- Switzer, W.M.; Tang, S.; Ahuka-Mundeke, S.; Shankar, A.; Hanson, D.L.; Zheng, H.; Ayouba, A.; Wolfe, N.D.; LeBreton, M.; Djoko, C.F.; et al. Novel simian foamy virus infections from multiple monkey species in women from the Democratic Republic of Congo. Retrovirology 2012, 9, 100. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, N.D.; Switzer, W.M.; Carr, J.K.; Bhullar, V.B.; Shanmugam, V.; Tamoufe, U.; Prosser, A.T.; Torimiro, J.N.; Wright, A.; Mpoudi-Ngole, E.; et al. Naturally acquired simian retrovirus infections in central African hunters. Lancet 2004, 363, 932–937. [Google Scholar] [CrossRef]
- Muniz, C.P.; Cavalcante, L.T.F.; Jia, H.; Zheng, H.; Tang, S.; Augusto, A.M.; Pissinatti, A.; Fedullo, L.P.; Santos, A.F.; Soares, M.A.; et al. Zoonotic infection of Brazilian primate workers with new world simian foamy virus. PLoS ONE 2017, 12, e0184502. [Google Scholar] [CrossRef]
- Feeroz, M.M.; Soliven, K.; Small, C.T.; Engel, G.A.; Andreina Pacheco, M.; Yee, J.L.; Wang, X.; Kamrul Hasan, M.; Oh, G.; Levine, K.L.; et al. Population dynamics of rhesus macaques and associated foamy virus in Bangladesh. Emerg. Microbes Infect. 2013, 2, 1–14. [Google Scholar] [CrossRef]
- Mergia, A.; Blackwell, J.; Papadi, G.; Johnson, C. Simian foamy virus type 1 (sfv-1) induces apoptosis. Virus Res. 1997, 50, 129–137. [Google Scholar] [CrossRef]
- Boneva, R.S.; Switzer, W.M.; Spira, T.J.; Bhullar, V.B.; Shanmugam, V.; Cong, M.E.; Lam, L.; Heneine, W.; Folks, T.M.; Chapman, L.E. Clinical and virological characterization of persistent human infection with simian foamy viruses. AIDS Res. Hum. Retroviruses 2007, 23, 1330–1337. [Google Scholar] [CrossRef]
- Buseyne, F.; Betsem, E.; Montange, T.; Njouom, R.; Bilounga Ndongo, C.; Hermine, O.; Gessain, A. Clinical signs and blood test results among humans infected with zoonotic simian foamy virus: A case-control study. J. Infect. Dis. 2018, 218, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Linial, M. Why aren’t foamy viruses pathogenic? Trends Microbiol. 2000, 8, 284–289. [Google Scholar] [CrossRef]
- Murray, S.M.; Linial, M.L. Foamy virus infection in primates. J. Med. Primatol. 2006, 35, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Vieira, I.C.; Toledo, P.M.; Silva, J.M.; Higuchi, H. Deforestation and threats to the biodiversity of Amazonia. Braz. J. Biol. 2008, 68, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, N.D.; Eitel, M.N.; Gockowski, J.; Muchaal, P.K.; Nolte, C.; Prosser, A.T.; Torimiro, J.N.; Weise, S.; Burke, D.S. Deforestation, hunting and the ecology of microbial emergence. Glob. Chang. Hum. Health 2000, 1, 10–25. [Google Scholar] [CrossRef]
- Cunningham, C.; Mootnick, A. Gibbons. Curr. Biol. 2009, 19, R543–R544. [Google Scholar] [CrossRef]
- Thinh, V.N.; Rawson, B.; Hallam, C.; Kenyon, M.; Nadler, T.; Walter, L.; Roos, C. Phylogeny and distribution of crested gibbons (genus Nomascus) based on mitochondrial cytochrome b gene sequence data. Am. J. Primatol. 2010, 72, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Carbone, L.; Becquet, C.; Mootnick, A.R.; Li, D.J.; de Jong, P.J.; Wall, J.D. Patterns of genetic variation within and between gibbon species. Mol. Biol. Evol. 2011, 28, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Bartlett, T.Q. Overlooked small apes need more attention! Am. J. Primatol. 2017, 79, e22658. [Google Scholar] [CrossRef]
- Ayouba, A.; Duval, L.; Liegeois, F.; Ngin, S.; Ahuka-Mundeke, S.; Switzer, W.M.; Delaporte, E.; Ariey, F.; Peeters, M.; Nerrienet, E. Nonhuman primate retroviruses from cambodia: High simian foamy virus prevalence, identification of divergent stlv-1 strains and no evidence of siv infection. Infect. Genet. Evol. 2013, 18, 325–334. [Google Scholar] [CrossRef]
- Hussain, A.I.; Shanmugam, V.; Bhullar, V.B.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; Wolfe, N.D.; Karesh, W.B.; Kilbourn, A.M.; Tooze, Z.; et al. Screening for simian foamy virus infection by using a combined antigen western blot assay: Evidence for a wide distribution among old world primates and identification of four new divergent viruses. Virology 2003, 309, 248–257. [Google Scholar] [CrossRef]
- Lauck, M.; Hyeroba, D.; Tumukunde, A.; Weny, G.; Lank, S.M.; Chapman, C.A.; O’Connor, D.H.; Friedrich, T.C.; Goldberg, T.L. Novel, divergent simian hemorrhagic fever viruses in a wild ugandan red colobus monkey discovered using direct pyrosequencing. PLoS ONE 2011, 6, e19056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Gaschen, B.; Blay, W.; Foley, B.; Haigwood, N.; Kuiken, C.; Korber, B. Tracking global patterns of n-linked glycosylation site variation in highly variable viral glycoproteins: Hiv, siv, and hcv envelopes and influenza hemagglutinin. Glycobiology 2004, 14, 1229–1246. [Google Scholar] [CrossRef] [PubMed]
- Calattini, S.; Betsem, E.; Froment, A.; Bassot, S.; Chevalier, S.A.; Mahieux, R.; Gessain, A. Identification and complete sequence analysis of a new htlv-3 strain from South Cameroon. In Proceedings of the 13th International Conference on Human Retrovirology: HTLV and Related Viruses, Hakone, Japan, 21–25 May 2007. [Google Scholar]
- Switzer, W.M.; Qari, S.H.; Wolfe, N.D.; Burke, D.S.; Folks, T.M.; Heneine, W. Ancient origin and molecular features of the novel human t-lymphotropic virus type 3 revealed by complete genome analysis. J. Virol. 2006, 80, 7427–7438. [Google Scholar] [CrossRef] [PubMed]
- Switzer, W.M.; Salemi, M.; Qari, S.H.; Jia, H.; Gray, R.R.; Katzourakis, A.; Marriott, S.J.; Pryor, K.N.; Wolfe, N.D.; Burke, D.S.; et al. Ancient, independent evolution and distinct molecular features of the novel human T-lymphotropic virus type 4. Retrovirology 2009, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Galvin, T.A.; Ahmed, I.A.; Shahabuddin, M.; Bryan, T.; Khan, A.S. Identification of recombination in the envelope gene of simian foamy virus serotype 2 isolated from Macaca cyclopis. J. Virol. 2013, 87, 8792–8797. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype c-infected seroconverters in india, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. Iq-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. Beast: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Perelman, P.; Johnson, W.E.; Roos, C.; Seuanez, H.N.; Horvath, J.E.; Moreira, M.A.; Kessing, B.; Pontius, J.; Roelke, M.; Rumpler, Y.; et al. A molecular phylogeny of living primates. PLoS Genet. 2011, 7, e1001342. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Rua, R.; Betsem, E.; Calattini, S.; Saib, A.; Gessain, A. Genetic characterization of simian foamy viruses infecting humans. J. Virol. 2012, 86, 13350–13359. [Google Scholar] [CrossRef] [PubMed]
- Kehl, T.; Tan, J.; Materniak, M. Non-simian foamy viruses: Molecular virology, tropism and prevalence and zoonotic/interspecies transmission. Viruses 2013, 5, 2169–2209. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.F.; Edelmann, K.; Strong, R.K.; Moebes, A.; Rethwilm, A.; Linial, M.L. The carboxyl terminus of the human foamy virus gag protein contains separable nucleic acid binding and nuclear transport domains. J. Virol. 1996, 70, 8255–8262. [Google Scholar]
- Renault, N.; Tobaly-Tapiero, J.; Paris, J.; Giron, M.L.; Coiffic, A.; Roingeard, P.; Saib, A. A nuclear export signal within the structural gag protein is required for prototype foamy virus replication. Retrovirology 2011, 8, 6. [Google Scholar] [CrossRef]
- Wang, G.; Mulligan, M.J. Comparative sequence analysis and predictions for the envelope glycoproteins of foamy viruses. J. Gen. Virol. 1999, 80, 245–254. [Google Scholar] [CrossRef]
- Lecellier, C.H.; Vermeulen, W.; Bachelerie, F.; Giron, M.L.; Saib, A. Intra- and intercellular trafficking of the foamy virus auxiliary bet protein. J. Virol. 2002, 76, 3388–3394. [Google Scholar] [CrossRef]
- Xia, X.; Xie, Z.; Salemi, M.; Chen, L.; Wang, Y. An index of substitution saturation and its application. Mol. Phylogenet. Evol. 2003, 26, 1–7. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Katzourakis, A. Marine origin of retroviruses in the early Palaeozoic Era. Nat. Commun. 2017, 8, 13954. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, L.; Hodgson, J.A.; Burrell, A.S.; Sterner, K.N.; Raaum, R.L.; Disotell, T.R. Primate phylogenetic relationships and divergence dates inferred from complete mitochondrial genomes. Mol. Phylogenet. Evol. 2014, 75, 165–183. [Google Scholar] [CrossRef]
- dos Reis, M.; Inoue, J.; Hasegawa, M.; Asher, R.J.; Donoghue, P.C.; Yang, Z. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc. Biol. Sci. 2012, 279, 3491–3500. [Google Scholar] [CrossRef]
- Nandakumar, S.; Bae, E.H.; Khan, A.S. Complete genome sequence of a naturally occurring simian foamy virus isolate from rhesus macaque (sfvmmu_k3t). Genome Announc. 2017, 5. [Google Scholar] [CrossRef]
- Nandakumar, S.; Bae, E.H.; Khan, A.S. Complete genome sequence of the African green monkey simian foamy virus serotype 3 strain fv2014 (sfvcae_fv2014). Genome Announc. 2018, 6. [Google Scholar] [CrossRef]
- Schmidt, M.; Herchenroder, O.; Heeney, J.; Rethwilm, A. Long terminal repeat u3 length polymorphism of human foamy virus. Virology 1997, 230, 167–178. [Google Scholar] [CrossRef] [PubMed]
- van Opijnen, T.; Kamoschinski, J.; Jeeninga, R.E.; Berkhout, B. The human immunodeficiency virus type 1 promoter contains a cata box instead of a tata box for optimal transcription and replication. J. Virol. 2004, 78, 6883–6890. [Google Scholar] [CrossRef]
- Matsudaira, K.; Ishida, T. Phylogenetic relationships and divergence dates of the whole mitochondrial genome sequences among three gibbon genera. Mol. Phylogenet. Evol. 2010, 55, 454–459. [Google Scholar] [CrossRef]
- Chan, Y.C.; Roos, C.; Inoue-Murayama, M.; Inoue, E.; Shih, C.C.; Pei, K.J.; Vigilant, L. Mitochondrial genome sequences effectively reveal the phylogeny of Hylobates gibbons. PLoS ONE 2010, 5, e14419. [Google Scholar] [CrossRef]
- Bergsten, J. A review of long-branch attraction. Cladistics 2005, 21, 163–193. [Google Scholar] [CrossRef]
- Kumar, S.; Subramanian, S. Mutation rates in mammalian genomes. Proc. Natl. Acad. Sci. USA 2002, 99, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Coffin, J.M. Constructing primate phylogenies from ancient retrovirus sequences. Proc. Natl. Acad. Sci. USA 1999, 96, 10254–10260. [Google Scholar] [CrossRef] [PubMed]
- Rawson, B.M.; Insua-Cao, P.; Ha, N.M.; Thinh, V.N.; Duc, H.M.; Mahood, S.; Geissmann, T.; Roos, C. The Conservation Status of Gibbons in Vietnam; Fauna & Flora International Vietnam Programme: Hanoi, Vietnam, 2011; p. 155. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Foamy Virus | Mammalian Host | Host Scientific Name | Family | GenBank Accession Number |
|---|---|---|---|---|
| SFVhpi_SAM106 | Pileated gibbon | Hylobates pileatus | Hylobatidae | M621235 |
| SFVpve | Western chimpanzee | Pan troglodytes verus | Hominidae | U04327 |
| SFVpsc | Eastern chimpanzee | Pan troglodytes schweinfurthii | Hominidae | Y07725 |
| SFVppy | Bornean orangutan | Pongo pygmaeus | Hominidae | AJ544579 |
| SFVggo | Lowland gorilla | Gorilla gorilla gorilla | Hominidae | NC_039029 |
| SFVcae | African green monkey | Cercopithecus aethiops | Cercopithecidae | M74895 |
| SFVmcy | Formosan rock macaque | Macaca cyclopsis | Cercopithecidae | X54482 |
| SFVcja | Common marmoset | Callithrix jacchus | Callitrichinae | GU356395 |
| SFVsxa | Yellow-breasted capuchin | Sapajus xanthosternos | Cebinae | KP143760 |
| SFVaxx | Spider monkey | Ateles species | Atelinae | EU010385 |
| SFVssc | Common squirrel monkey | Saimiri sciureus | Saimirinae | GU356394 |
| SFVocr | Brown greater galago | Otolemur crassicaudatus | Galagidae | KM233624 |
| EFVeca | Horse | Equus caballus | Equidae | AF201902 |
| BFVbta | Cow | Bos taurus | Bovidae | U94514 |
| FFVfca | Cat | Felis catus | Felidae | Y08851 |
| Virus 1 | LTR | gag | pol | env | tas | bet | Genome |
|---|---|---|---|---|---|---|---|
| SFVhpi_SAM106 | 2071 | 1974 | 3420 | 2966 | 897 | 1449 | 13,885 |
| SFVpve | 1760 | 1959 | 3438 | 2964 | 900 | 1470 | 13,246 |
| SFVpsc | 1767 | 1944 | 3431 | 2964 | 900 | 1446 | 13,242 |
| SFVggo | 1283 | 1964 | 3154 | 2963 | 897 | 1443 | 12,258 |
| SFVppy | 1621 | 1987 | 3236 | 2965 | 834 | 1392 | 12,823 |
| Virus 1 | gag/Gag | pol/Pol | env/Env | tas/Tas | bet/Bet | Concatamer 2 |
|---|---|---|---|---|---|---|
| SFVpve | 49.2/38.9 | 74.3/75.9 | 67.0/66.2 | 49.2/31.3 | 49.2/27.9 | 65.6/63.4 |
| SFVpsc | 49.6/38.3 | 73.4/75.2 | 67.3/67.1 | 49.7/38.5 | 50.3/29.6 | 65.7/63.5 |
| SFVggo | 48.0/40.2 | 74.1/76.6 | 67.8/67.5 | 48.3/29.7 | 48.0/30.7 | 65.7/64.7 |
| SFVppy | 48.7/40.6 | 73.2/76.1 | 65.8/63.8 | 48.0/32.7 | 41.3/22.2 | 64.7/63.3 |
| FV 2, Host or Analysis Parameter | FV gag-pol-env | FV gag-pol-env (12cdp)3 | FV Pol 4 | Host 5 |
|---|---|---|---|---|
| SFVhpi_SAM106/SFVgreat ape split (Crown Hominoidea) | 20.69 (19.13–22.19) | 20.83 (19.32–22.45) | N/A 6 | 22.32 (20.54–23.85 |
| SFV OWM/ape split (Crown Catarrhini) | 29.13 (27.28–30.09) | 29.07 (27.37–30.96) | 31.60 (25.36–37.85) | 32.12 (29.44–33.82) |
| SFVpve, SFVpsc/SFVggo split (Crown Homininae) | 9.18 (8.54–9.8) | 9.16 (8.53–9.8) | 8.29 (6.52–10.05) | 10.63 (10.02–11.68) |
| SFV OWM (Crown Cercopithecinae) | 18.05 (11.29–24.77) | 16.44 (10.18–22.81) | N/A | 14.09 (12.24–15.82) |
| Prosimian SFV (Crown Primates) | 82.53 (64.77–96.82) | 80.85 (65.25–95.16) | 86.92 (76.34–97.22) | 74.11 (68.17–81.20) |
| Non-simian FV (horse/cat split) | 92.4 (85.03–98.69) | 93.05 (86.2–99.12) | N/A | 80.6 (63.0–115.4) |
| BFV/EFV split (cow/horse split) | 77.2 (73.26–80.85) | 76.89 (73.11–80.61) | N/A | 105.7 (74.1–130.8) |
| Root FV placental mammals (Crown Boreotheria) | 95.46 (90.08–101.19) | 95.86 (90.47–101.36) | 98.59 (95.94–100.78) | 120.5 (97.8–136.6) |
| Mean rate 7 | 8.68 × 10−9 (7.88 × 10−9–9.48 × 10−9) | 5.05 × 10−9 (4.62 × 10−9–5.51 × 10−9) | N/A | N/A9 |
| ucld.stdev 8 | 0.5524 (0.4084, 0.7252) | 0.435 (0.2931–0.5911) | N/A | N/A |
| α-parameter (Γ-distribution) 9 | 1.079 (0.9704, 1.1858) | 1.064 (0.8954, 1.2586) | N/A | N/A |
| ESS 10 | >258 | >1063 | N/A | N/A |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shankar, A.; Sibley, S.D.; Goldberg, T.L.; Switzer, W.M. Molecular Analysis of the Complete Genome of a Simian Foamy Virus Infecting Hylobates pileatus (pileated gibbon) Reveals Ancient Co-Evolution with Lesser Apes. Viruses 2019, 11, 605. https://doi.org/10.3390/v11070605
Shankar A, Sibley SD, Goldberg TL, Switzer WM. Molecular Analysis of the Complete Genome of a Simian Foamy Virus Infecting Hylobates pileatus (pileated gibbon) Reveals Ancient Co-Evolution with Lesser Apes. Viruses. 2019; 11(7):605. https://doi.org/10.3390/v11070605
Chicago/Turabian StyleShankar, Anupama, Samuel D. Sibley, Tony L. Goldberg, and William M. Switzer. 2019. "Molecular Analysis of the Complete Genome of a Simian Foamy Virus Infecting Hylobates pileatus (pileated gibbon) Reveals Ancient Co-Evolution with Lesser Apes" Viruses 11, no. 7: 605. https://doi.org/10.3390/v11070605
APA StyleShankar, A., Sibley, S. D., Goldberg, T. L., & Switzer, W. M. (2019). Molecular Analysis of the Complete Genome of a Simian Foamy Virus Infecting Hylobates pileatus (pileated gibbon) Reveals Ancient Co-Evolution with Lesser Apes. Viruses, 11(7), 605. https://doi.org/10.3390/v11070605