Viral Strain Determines Disease Symptoms, Pathology, and Immune Response in Neonatal Rats with Lymphocytic Choriomeningitis Virus Infection

Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Virus
2.3. Infections
2.4. Perfusions and Pathological Analysis
2.5. Quantifying Cross-Sectional Area of the Cerebellum
2.6. Immunohistochemistry
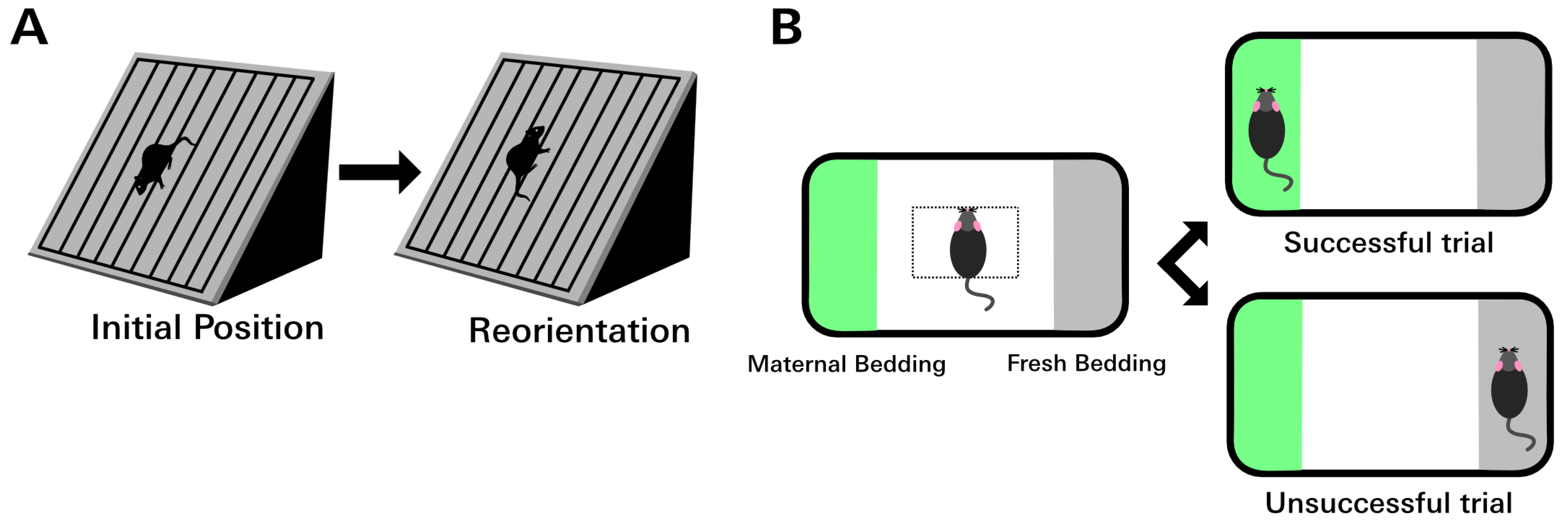
2.7. Negative Geotaxis Assay
2.8. Olfaction Discrimination Assay
2.9. Isolating Leukocytes from Brain Tissue
2.10. Gene Expression Analysis
2.11. Statistical Analyses
3. Results
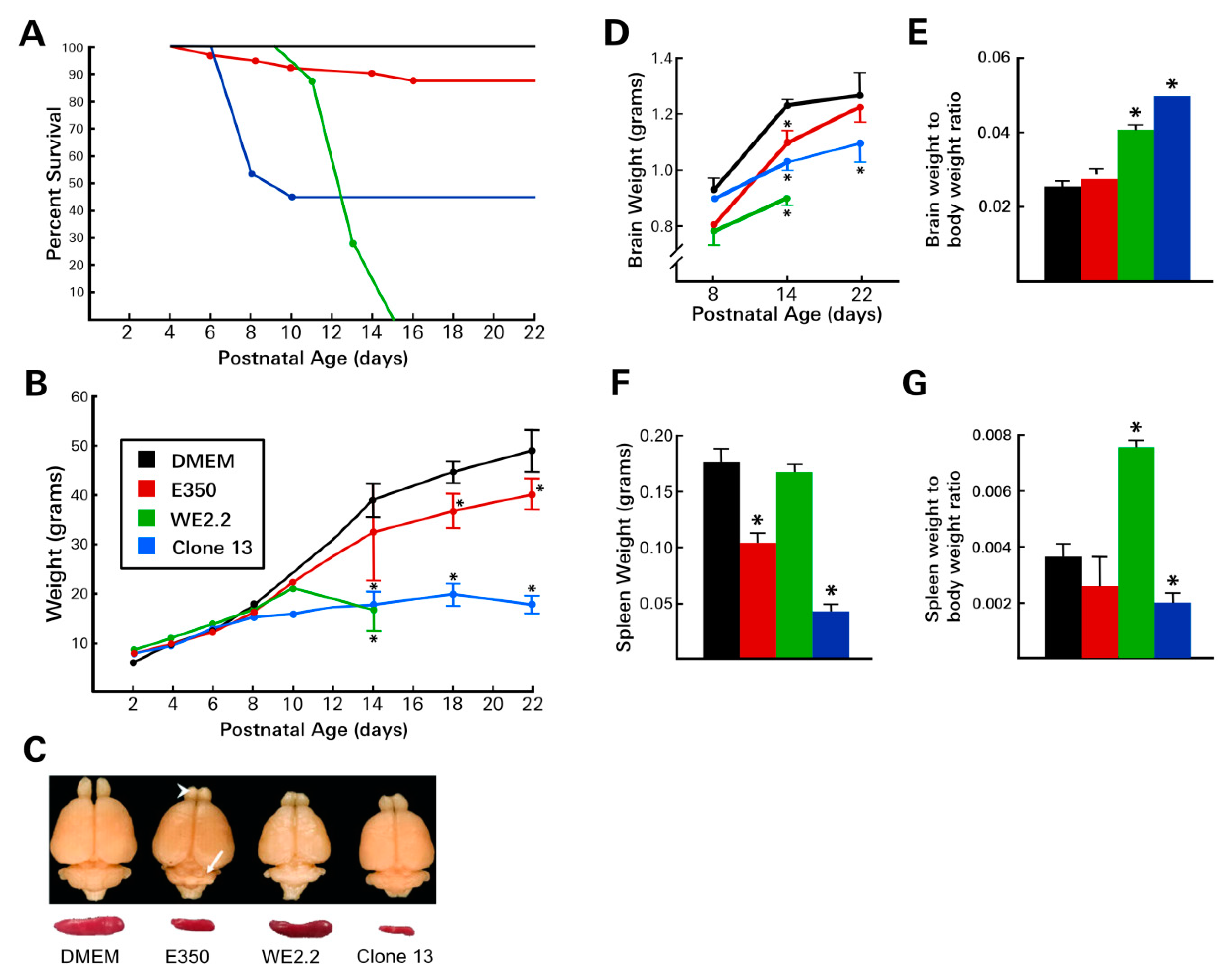
3.1. Different LCMV Strains Produced Different Diseases
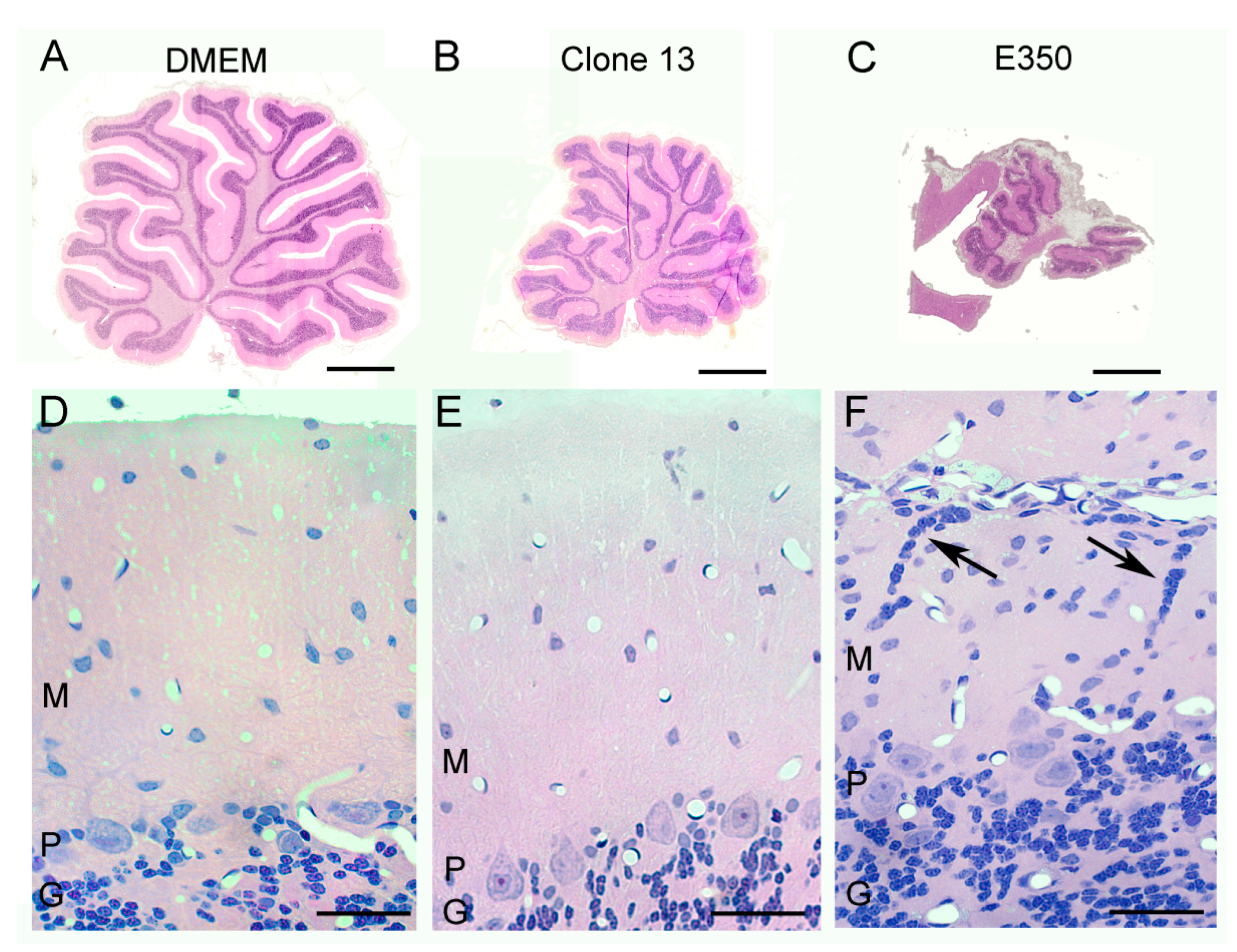
3.2. Different LCMV Strains Induced Different Patterns of Growth Disturbance and Pathology in Spleen and Brain
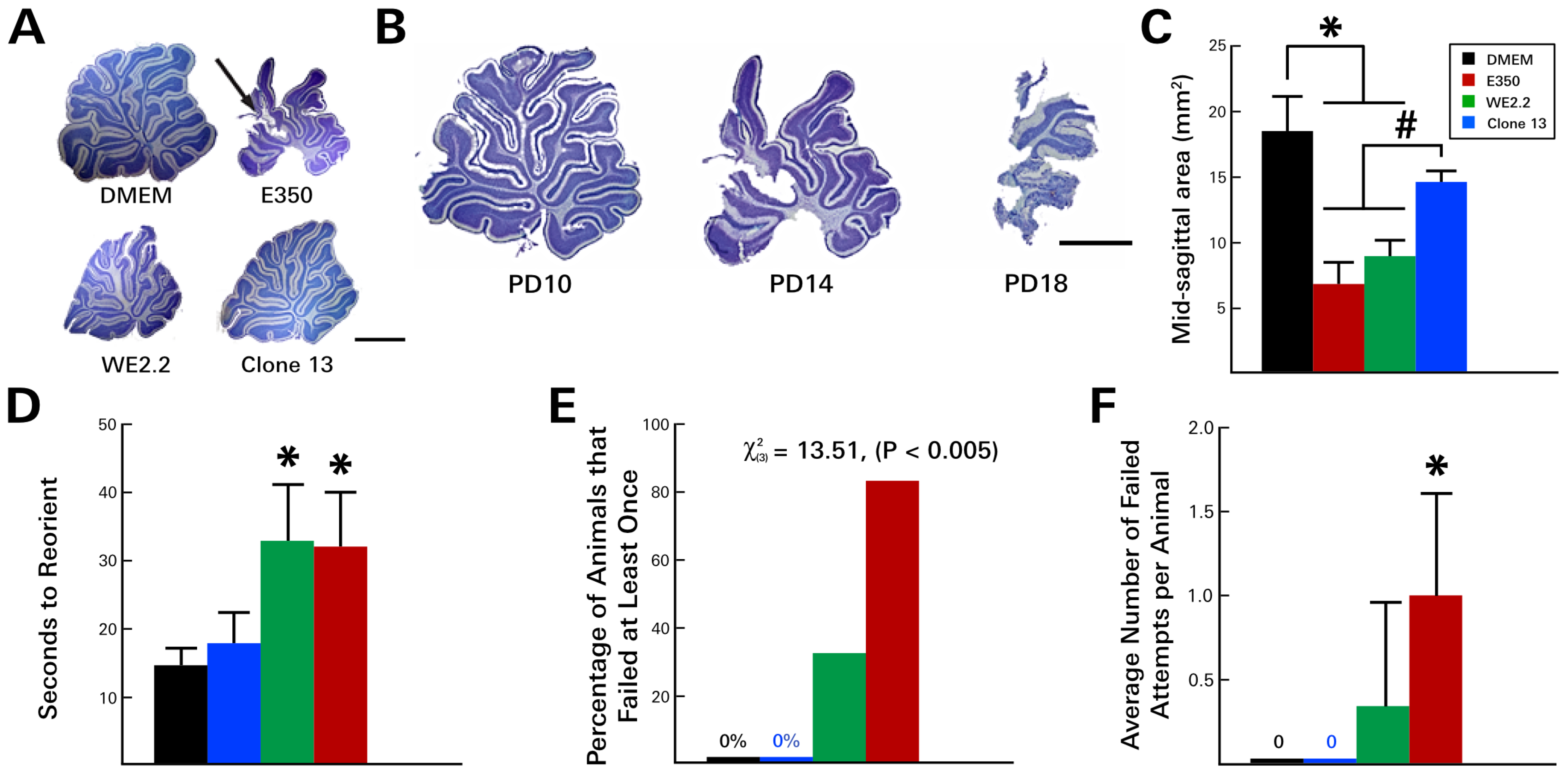
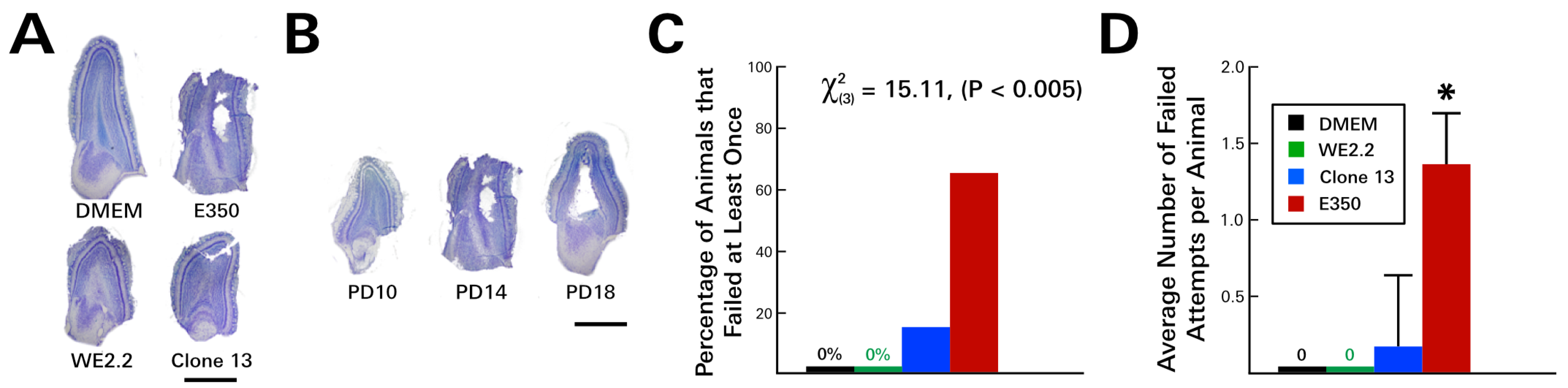
3.3. Different LCMV Strains Induced Different Behavioral Deficits
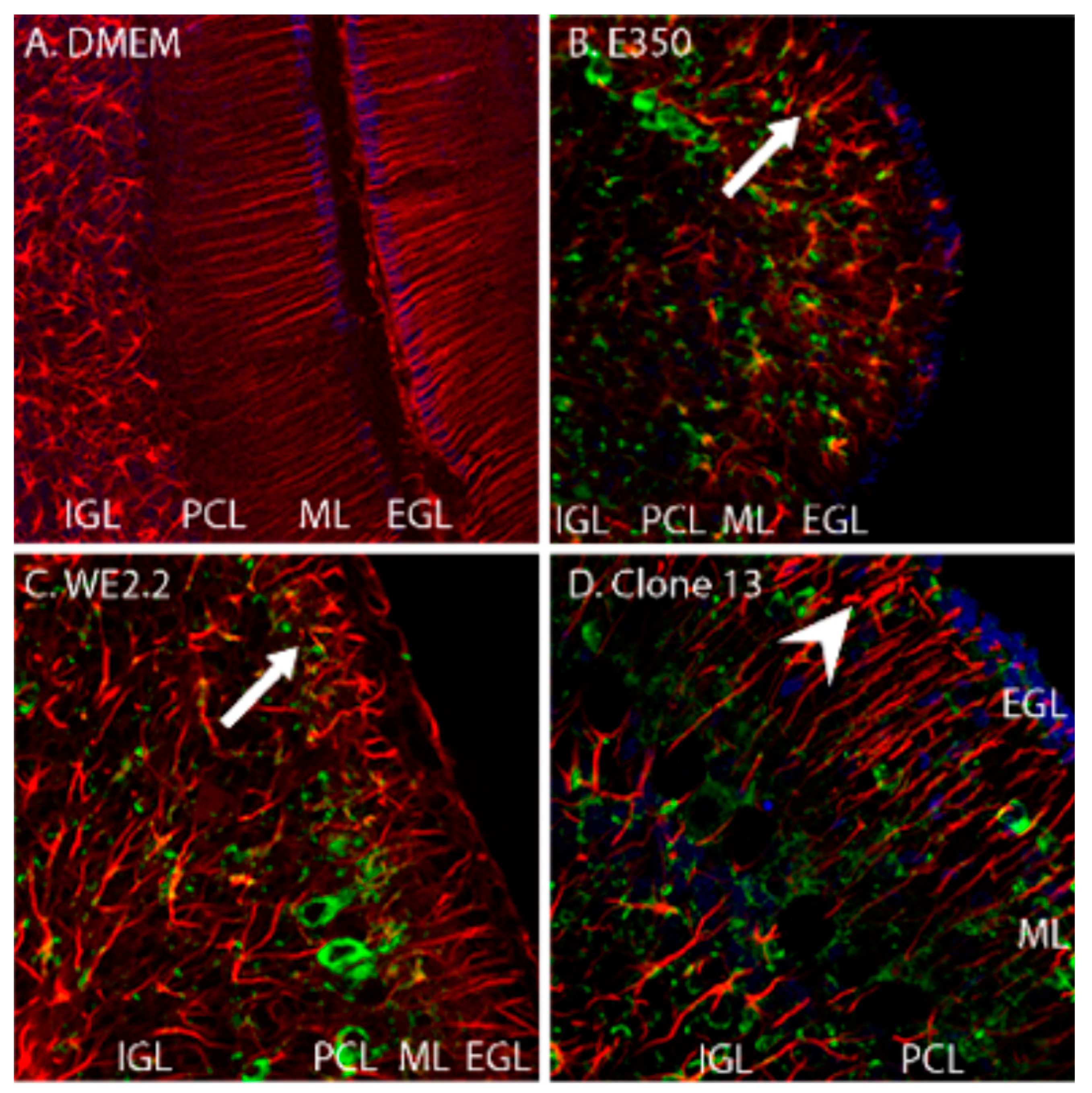
3.4. Different LCMV Strains Induced Different Forms and Degree of Neuropathology within the Cerebellum
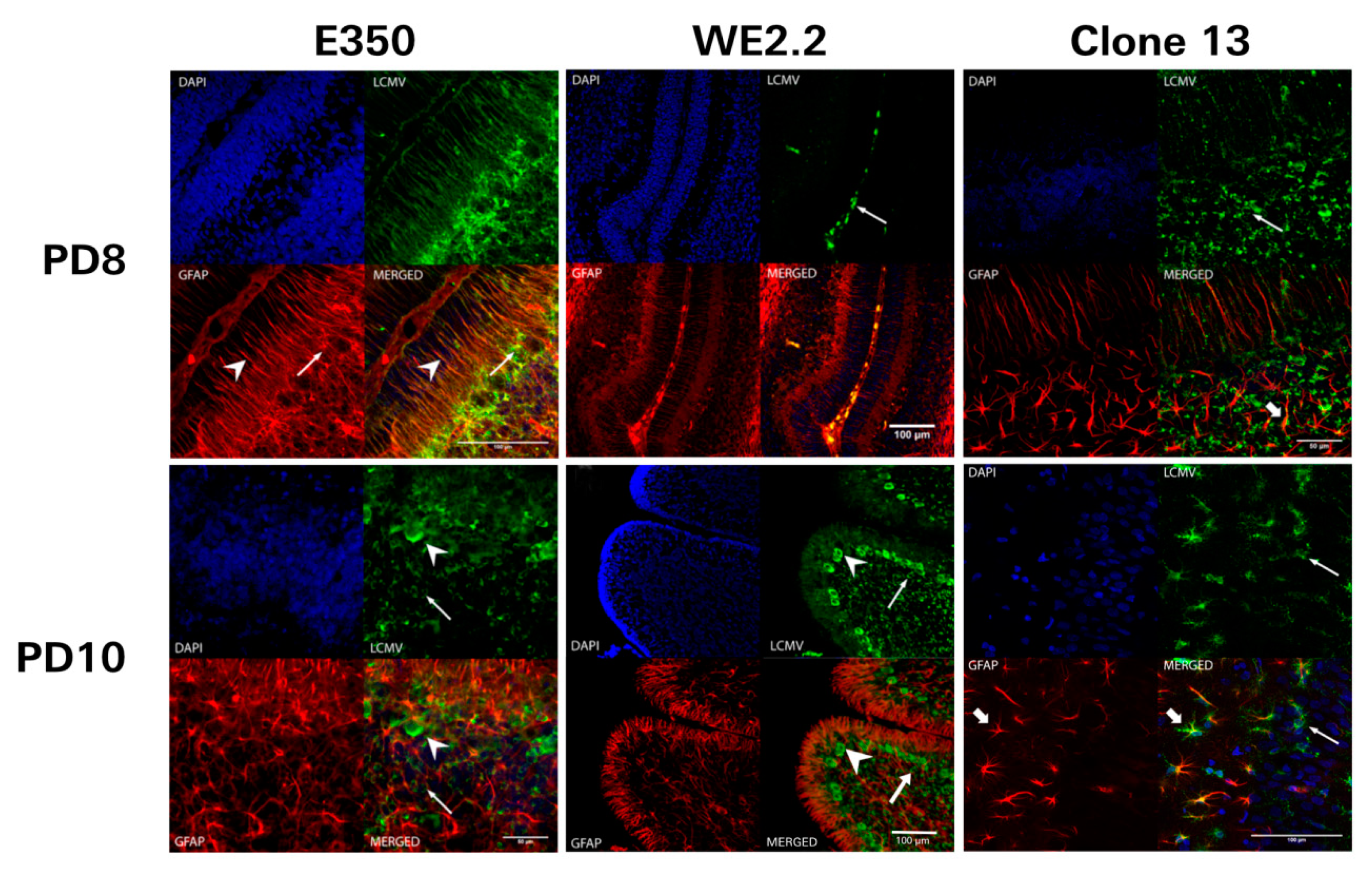
3.5. Different LCMV Strains Had Different Cellular Targets of Infection
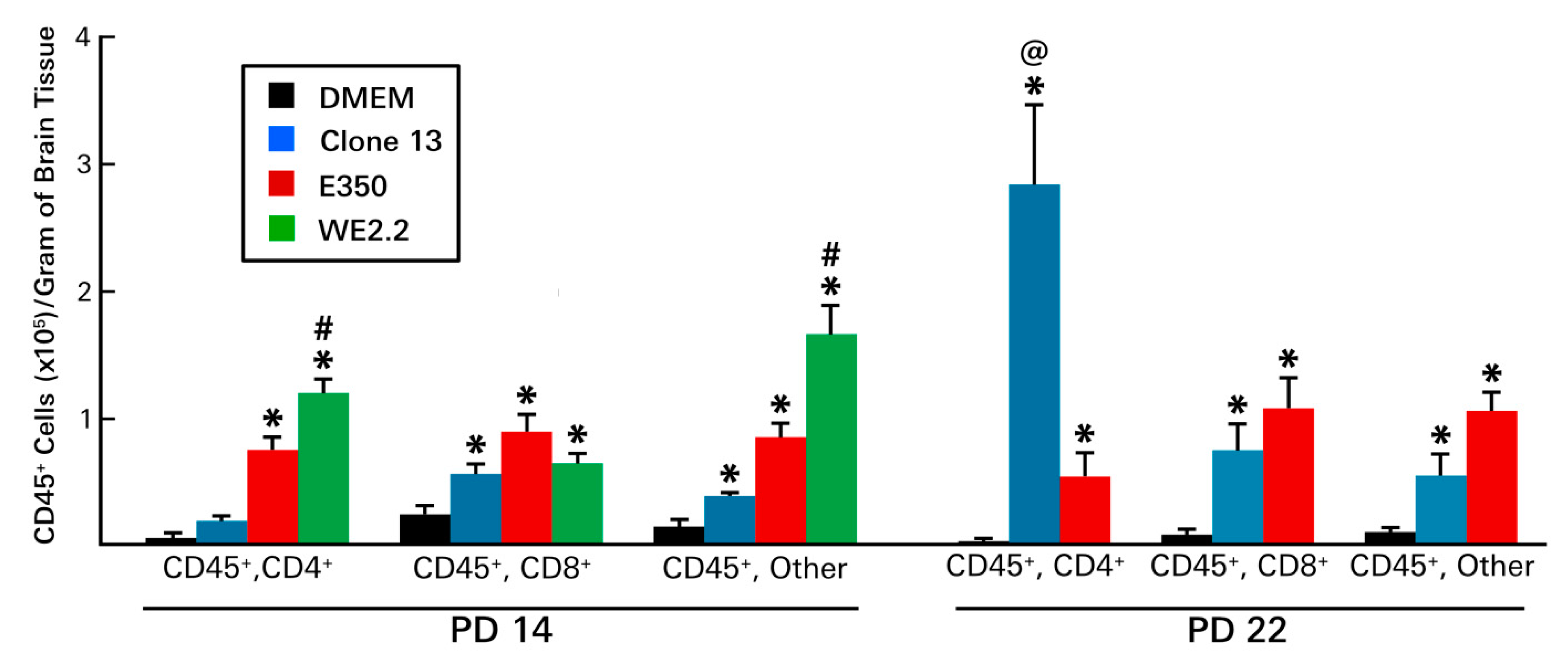
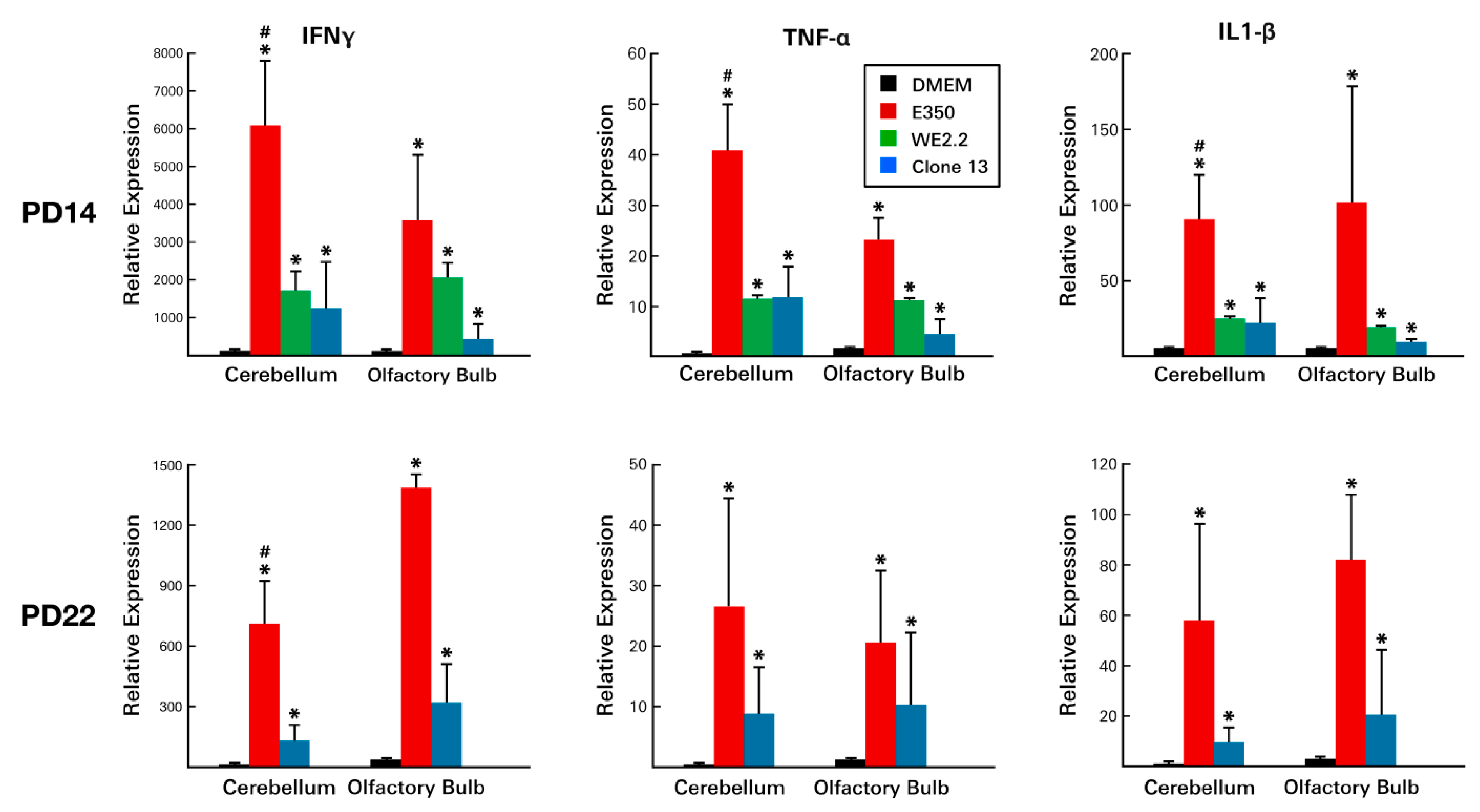
3.6. Different LCMV Strains Elicited Different Host Immune Responses to Infection
4. Discussion
4.1. Different Strains of LCMV Differed in Their Lethality
4.2. Different Strains of LCMV Differed in their Cellular Targets of Infection
4.3. Different Strains of LCMV Differed in their Sites and Forms of Pathology
4.4. Strengths and Limitations of This Study
4.5. Neonatal Rat Infected with LCMV is a Powerful Model System of Human Congenital LCMV Infection
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Stephensen, C.B.; Blount, S.R.; Lanford, R.E.; Holmes, K.V.; Montali, R.J.; Fleenor, M.E.; Shaw, J.F. Prevalence of serum antibodies against lymphocytic choriomeningitis virus in selected populations from two U.S. cities. J. Med. Virol. 1992, 38, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, A.M.; Feuillade, M.R.; Gamboa, G.S.; Maiztegui, J.I. Prevalence of lymphocytic choriomeningitis virus infection in a human population of Argentina. Am. J. Trop. Med. Hyg. 1994, 50, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Childs, J.E.; Glass, G.E.; Ksiazek, T.G.; Rossi, C.A.; Oro, J.G.B.; Leduc, J.W. Human-rodent contact and infection with lymphocytic choriomeningitis and Seoul viruses in an inner-city population. Am. J. Trop. Med. Hyg. 1991, 44, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Fevola, C.; Kuivanen, S.; Smura, T.; Vaheri, A.; Kallio-Kokko, H.; Hauffe, H.C.; Vapalahti, O.; Jääskeläinen, A.J. Seroprevalence of lymphocytic choriomeningitis virus and Ljungan virus virus in Finnish patients with suspected neurological infections. J. Med. Virol. 2017, 90, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Childs, J.E.; Glass, G.E.; Korch, G.W.; Ksiazek, T.G.; Leduc, J.W. Lymphocytic choriomeningitis virus infection and house mouse (mus musculus) distribution in urban Baltimore. Am. J. Trop. Med. Hyg. 1992, 47, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, P.B.; Peters, C.J. Lymphocytic choriomeningitis virus: A neglected pathogen of man. Arch. Pathol. Lab. Med. 1992, 116, 486–488. [Google Scholar]
- Peters, C.J. Lymphocytic choriomeningitis virus—An old enemy up to new tricks. N. Engl. J. Med. 2006, 354, 2208–2211. [Google Scholar] [CrossRef]
- Bonthius, D.J. Lymphocytic choriomeningitis virus: An under-recognized cause of neurologic disease in the fetus, child, and adult. Semin. Pediatric Neurol. 2012, 19, 89–95. [Google Scholar] [CrossRef]
- Enders, G.; Varho-Göbel, M.; Löhler, J.; Terletskaia-Ladwig, E.; Eggers, M. Congenital lymphocytic choriomeningitis virus infection: An underdiagnosed disease. Pediatric Infect. Dis. J. 1999, 18, 652–655. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Wright, R.; Tseng, B.; Barton, L.; Marco, E.; Karacay, B.; Larsen, P.D. Congenital lymphocytic choriomeningitis virus infection: Spectrum of disease. Ann. Neurol. 2007, 62, 347–355. [Google Scholar] [CrossRef]
- Wright, R.; Johnson, D.; Neumann, M.; Ksiazek, T.G.; Rollin, P.; Keech, R.V.; Bonthius, D.J.; Hitchon, P.; Grose, C.F.; Bell, W.E.; et al. Congenital lymphocytic choriomeningitis virus syndrome: A disease that mimics congenital toxoplasmosis or cytomegalovirus infection. Pediatrics 1997, 100, E9. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.L.; Peters, C.J.; Ksiazek, T.G. Lymphocytic choriomeningitis virus: An unrecognized teratogenic pathogen. Emerg. Infect. Dis. 1995, 1, 152–153. [Google Scholar] [CrossRef] [PubMed]
- Delaine, M.; Weingertner, A.S.; Nougairede, A.; Lepiller, Q.; Fafi-Kremer, S.; Favre, R.; Charrel, R. Microcephaly caused by lymphocytic choriomeningitis virus. Emerg. Infect. Dis. 2017, 23, 1548–1550. [Google Scholar] [CrossRef] [PubMed]
- Mets, M.B.; Barton, L.L.; Khan, A.S.; Ksiazek, T.G. Lymphocytic choriomeningitis virus: An underdiagnosed cause of congenital chorioretinitis. Am. J. Ophthalmol. 2000, 130, 209–215. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Nichols, B.; Harb, H.; Mahoney, J.; Karacay, B. Lymphocytic choriomeningitis virus infection of the developing brain: Critical role of host age. Ann. Neurol. 2007, 62, 356–374. [Google Scholar] [CrossRef]
- Albariño, C.G.; Palacios, G.; Khristova, M.L.; Erickson, B.R.; Carroll, S.A.; Comer, J.A.; Hui, J.; Briese, T.; St George, K.; Ksiazek, T.G.; et al. High diversity and ancient common ancestry of lymphocytic choriomeningitis virus. Emerg. Infect. Dis. 2010, 16, 1093–1100. [Google Scholar] [CrossRef]
- Djavani, M.M.; Crasta, O.R.; Zapata, J.C.; Fei, Z.; Folkerts, O.; Sobral, B.; Swindells, M.; Bryant, J.; Davis, H.; Pauza, C.D.; et al. Early blood profiles of virus infection in a monkey model for Lassa fever. J. Virol. 2007, 81, 7960–7973. [Google Scholar] [CrossRef]
- Lukashevich, I.S.; Rodas, J.D.; Tikhonov, I.I.; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. LCMV-mediated hepatitis in rhesus macaques: WE but not ARM strain activates hepatocytes and induces liver regeneration. Arch. Virol. 2004, 149, 2319–2336. [Google Scholar] [CrossRef]
- Smelt, S.C.; Borrow, P.; Kunz, S.; Cao, W.; Tishon, A.; Lewicki, H.; Campbell, K.P.; Oldstone, M.B. Differences in affinity of binding of lymphocytic choriomeningitis virus strains to the cellular receptor alpha-dystroglycan correlate with viral tropism and disease kinetics. J. Virol. 2001, 75, 448–457. [Google Scholar] [CrossRef]
- Dobbing, J. The later development of the brain and its vulnerability. In Scientific Foundations of Paediatrics; Davis, J.A., Dobbing, J., Eds.; Heinemann: London, UK, 1981; pp. 331–336. [Google Scholar]
- Dobbing, J.; Sands, J. Quantitative growth and development of the human brain. Arch. Dis. Child. 1973, 48, 757–767. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 89–93. [Google Scholar] [CrossRef]
- Bonthius, D.J., Jr.; Winters, Z.; Karacay, B.; Bousquet, S.L.; Bonthius, D.J. Importance of genetics in fetal alcohol effects: Null mutation of the nNOS gene worsens alcohol-induced cerebellar neuronal losses and behavioral deficits. Neurotoxicology 2015, 46, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Goodlett, C.R.; Lundahl, K.R. Temporal determinants of neonatal alcohol-induced cerebellar damage and motor performance deficits. Pharmacol. Biochem. Behav. 1996, 55, 531–540. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Perlman, S. Congenital viral infections of the brain: Lessons learned from lymphocytic choriomeningitis virus in the neonatal rat. PLoS Pathog. 2007, 3, e149. [Google Scholar] [CrossRef] [PubMed]
- Monjan, A.A.; Cole, G.A.; Nathanson, N. Pathogenesis of LCM disease in the rat. In Lymphocytic Choriomeningitis Virus and Other Arenaviruses; Lehmann, F., Ed.; Springer: Berlin, Germany, 1973; pp. 195–206. [Google Scholar]
- Bonthius, D.J.; Mahoney, J.; Buchmeier, M.J.; Karacay, B.; Taggard, D. Critical role for glial cells in the propagation and spread of lymphocytic choriomeningitis virus in the developing rat brain. J. Virol. 2002, 76, 6618–6635. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.; Rabe, G.K.; Karacay, B.; Bonthius, D.J. T-cells underlie some, but not all, of the cerebellar pathology in a neonatal rat model of congenital lymphocytic choriomeningitis virus infection. J. Neuropathol. Exp. Neurol. 2016, 75, 1031–1047. [Google Scholar] [CrossRef]
- Pearce, B.D.; Steffensen, S.C.; Paoletti, A.D.; Henriksen, S.J.; Buchmeier, M.J. Persistent dentate granule cell hyperexcitability after neonatal infection with lymphocytic choriomeningitis virus. J. Neurosci. 1996, 16, 220–228. [Google Scholar] [CrossRef]
- Monjan, A.A.; Cole, G.A.; Nathanson, H. Pathogenesis of cerebellar hypoplasia produced by lymphocytic choriomeningitis virus infection of neonatal rats: Protective effect of immunosuppression with anti-lymphoid serum. Infect. Immun. 1974, 10, 499–502. [Google Scholar]
- Armstrong, C.; Lillie, R.D. Experimental lymphocytic choriomeningitis of monkeys and mice produced by a virus encountered in studies of the 1933 St. Louis encephalitis epidemic. Public Health Rep. 1934, 49, 1019–1022. [Google Scholar] [CrossRef]
- Bergthaler, A.; Flatz, L.; Hegazy, A.N.; Johnson, S.; Horvath, E.; Löhning, M.; Pinschewer, D.D. Viral replicative capacity is the primary determinant of lymphocytic choriomeningitis virus persistence and immunosuppression. Proc. Natl. Acad. Sci. USA 2010, 107, 21641–21646. [Google Scholar] [CrossRef]
- Sevilla, N.; Kunz, S.; Holz, A.; Lewicki, H.; Homann, D.; Yamada, H.; Campbell, K.P.; de la Torre, J.C.; Oldstone, M.B. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 2000, 192, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Sevilla, N.; Rojek, J.M.; Oldstone, M.B.A. Use of alternative receptors different than a-dystroglycan by selected isolates of lymphocytic choriomeningitis virus. Virology 2004, 325, 432–445. [Google Scholar] [CrossRef]
- Dutko, F.J.; Oldstone, M.B.A. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J. Gen. Virol. 1983, 64, 1689–1698. [Google Scholar] [CrossRef]
- Motz, B.A.; Alberts, J.R. The validity and utility of geotaxis in young rodents. Neurotoxicol. Teratol. 2005, 27, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Smart, J.L.; Dobbing, J. Vulnerability of the developing brain. II. Effects of early nutritional deprivation on reflex ontogeny and development of behavior in the rat. Brain Res. 1971, 28, 85–95. [Google Scholar] [CrossRef]
- Carr, W.J.; Marasco, E.; Landauer, M.R. Responses by rat pups to their own nest versus a strange conspecific nest. Physiol. Behav. 1979, 23, 1149–1151. [Google Scholar] [CrossRef]
- Lehmann-Grube, F. Portraits of viruses: Arenaviruses. Intervirology 1984, 22, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Buchmeier, M.J.; Zajac, A.J. Lymphocytic choriomeningitis virus. In Persistent Viral Infections; Ahmed, R., Chen, I., Eds.; Wiley: New York, NY, USA, 1999; pp. 575–605. [Google Scholar]
- Holland, J.J.D.; De La Torre, J.C.; Steinhauer, D.A. RNA virus populations as quasispecies. In Genetic Diversity of RNA Viruses; Springer: Berlin/Heidelberg, Germany, 1992; Volume 176, pp. 1–20. [Google Scholar]
- Ahmed, R.; Simon, R.S.; Matloubian, M.; Kolhekar, S.R.; Southern, P.J.; Freedman, D.M. Genetic analysis of in vivo-selected viral variants causing chronic infection: Importance of mutation in the L RNA segment of lymphocytic choriomeningitis virus. J. Virol. 1988, 62, 3301–3308. [Google Scholar]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B.A. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef]
- Welsh, R.M.; Lampert, P.W.; Oldstone, M.B.A. Prevention of virus-induced cerebellar disease by defective-interfering lymphocytic choriomeningitis virus. J. Infect. Dis. 1977, 136, 391–399. [Google Scholar] [CrossRef]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C. Innate immune responses to LCMV infections: Natural killer cells and cytokines. Curr. Top. Microbiol. Immunol. 2002, 263, 7–27. [Google Scholar] [PubMed]
- Suprunenko, T.; Hofer, M.J. Complexities of type I interferon biology: Lessons learned from LCMV. Viruses 2019, 11, 172. [Google Scholar] [CrossRef] [PubMed]
- Monjan, A.A.; Silverstein, A.M.; Cole, G.A. Lymphocytic choriomeningitis virus-induced retinopathy in newborn rats. Investig. Ophthalmol. 1972, 11, 850–856. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect in Humans | Comparable Effect in Rats | LCMV Strain | Infection Day |
|---|---|---|---|
| Fetal Demise | Early Death | WE 2.2 | PD4 |
| Microencephaly | Microencephaly | WE 2.2, Clone 13 | PD4 |
| Chorioretinitis | Chorioretinitis | Arm-4 | PD1 |
| Hydrocephalus | Hydrocephalus | Arm-4 | PD10-PD60 |
| Lissencephaly (Neuronal Migration Disturbance) | Neuronal Migration Disturbance | E350 | PD4 |
| Arm-4 | PD4-PD6 | ||
| Porencephalic Cyst | Porencephalic Cyst (Olfactory Bulb) | Arm-4 | PD10-PD60 |
| E350 | PD4 | ||
| Cerebellar Hypoplasia | Cerebellar Hypoplasia | Arm-4 | PD1 |
| WE 2.2, Clone 13 | PD4 | ||
| Encephalomalacia | Encephalomalacia (Cerebellum) | Arm-4 | PD4-PD6 |
| E350 | PD4 | ||
| Encephalomalacia (Dentate Gyrus) | Arm-4 | PD6 | |
| Periventricular Cyst | Periventricular Cyst (Occipital Horn) | Arm-4 | PD10-PD60 |
| Periventricular Calcification | Periventricular Infection | Arm-4 | PD1-PD10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plume, J.M.; Todd, D.; Bonthius, D.J. Viral Strain Determines Disease Symptoms, Pathology, and Immune Response in Neonatal Rats with Lymphocytic Choriomeningitis Virus Infection. Viruses 2019, 11, 552. https://doi.org/10.3390/v11060552
Plume JM, Todd D, Bonthius DJ. Viral Strain Determines Disease Symptoms, Pathology, and Immune Response in Neonatal Rats with Lymphocytic Choriomeningitis Virus Infection. Viruses. 2019; 11(6):552. https://doi.org/10.3390/v11060552
Chicago/Turabian StylePlume, Jeffrey M., Dylan Todd, and Daniel J. Bonthius. 2019. "Viral Strain Determines Disease Symptoms, Pathology, and Immune Response in Neonatal Rats with Lymphocytic Choriomeningitis Virus Infection" Viruses 11, no. 6: 552. https://doi.org/10.3390/v11060552
APA StylePlume, J. M., Todd, D., & Bonthius, D. J. (2019). Viral Strain Determines Disease Symptoms, Pathology, and Immune Response in Neonatal Rats with Lymphocytic Choriomeningitis Virus Infection. Viruses, 11(6), 552. https://doi.org/10.3390/v11060552