Structure and Hierarchy of Influenza Virus Models Revealed by Reaction Network Analysis


 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
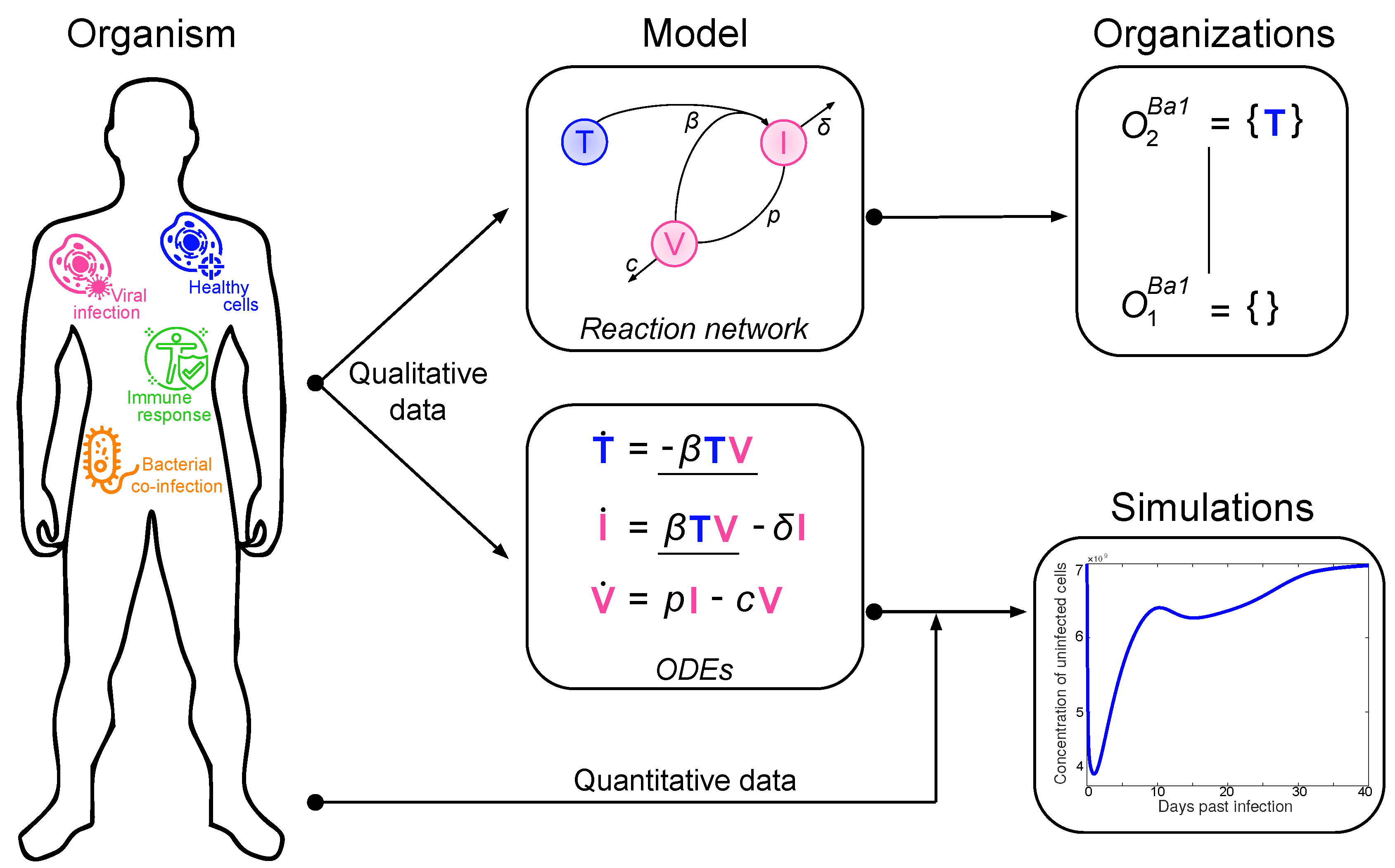
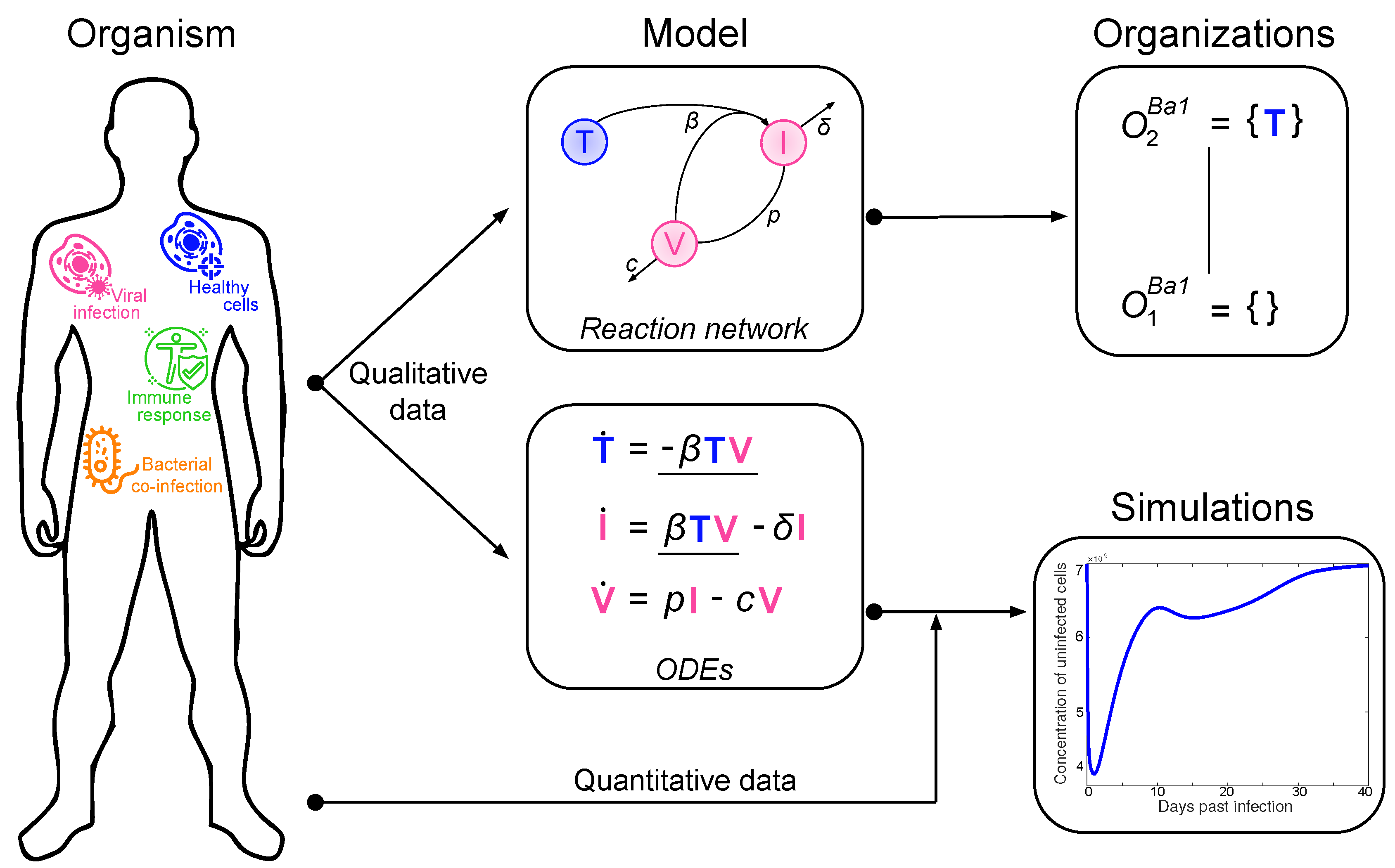
2. Materials and Methods: Procedure for the Organizational Analysis
- Uninfected (target) cells or those resistant/refractory to infection are marked in blue, e.g., T.
- Infected cells, partially or latently infected cells, and viruses are marked in magenta, e.g., I and V.
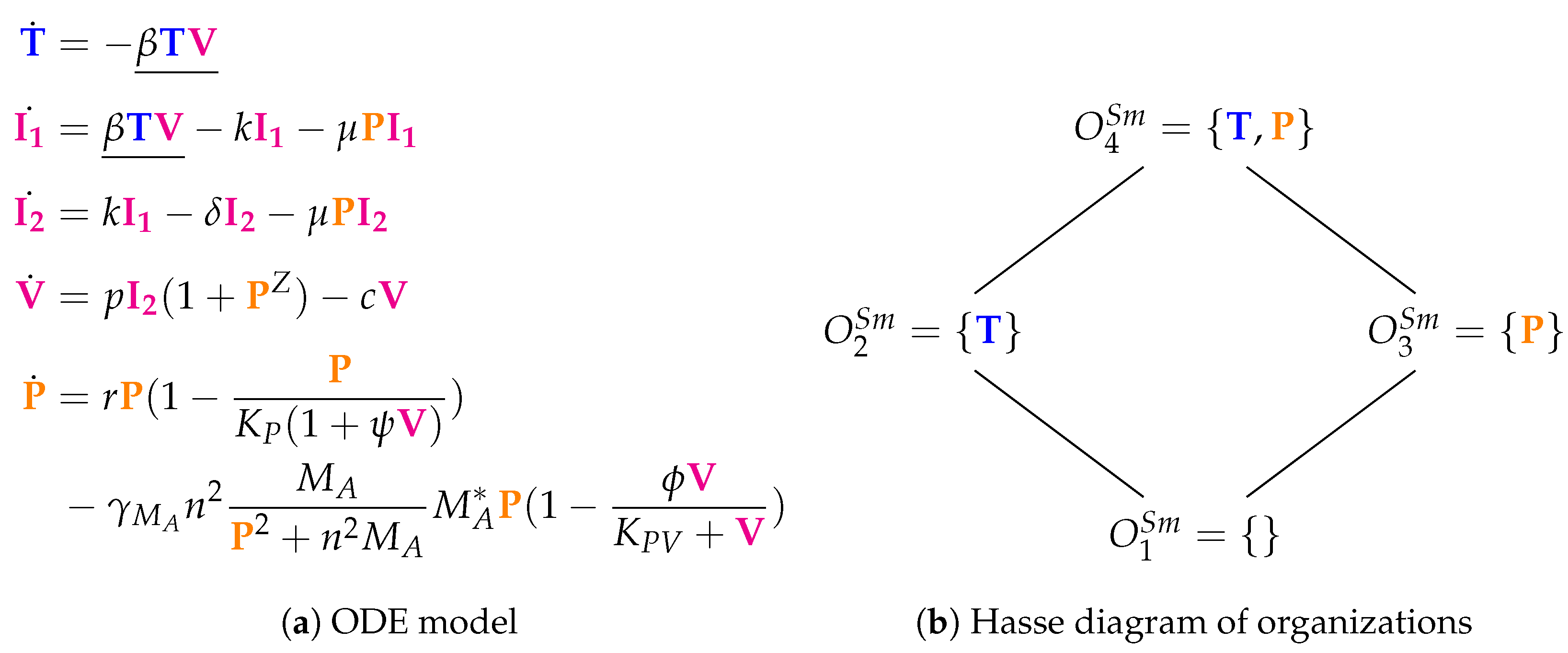
- Bacterial co-infection species are marked in orange. These species are only occurring in Smith’s model [15].
- Text referring to any other species is marked in black, e.g., transient target cell states, passive immune system, or dead cells.
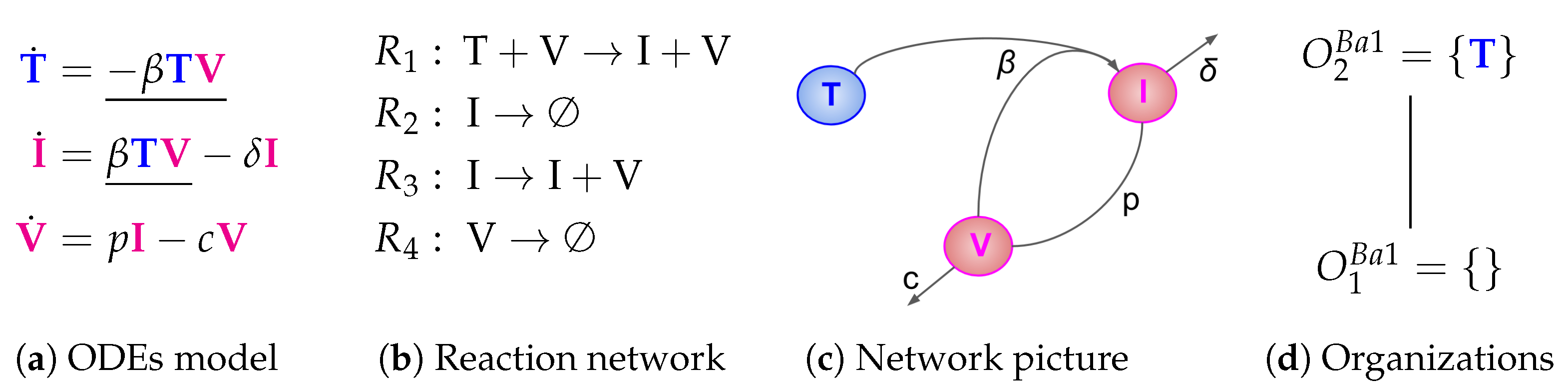
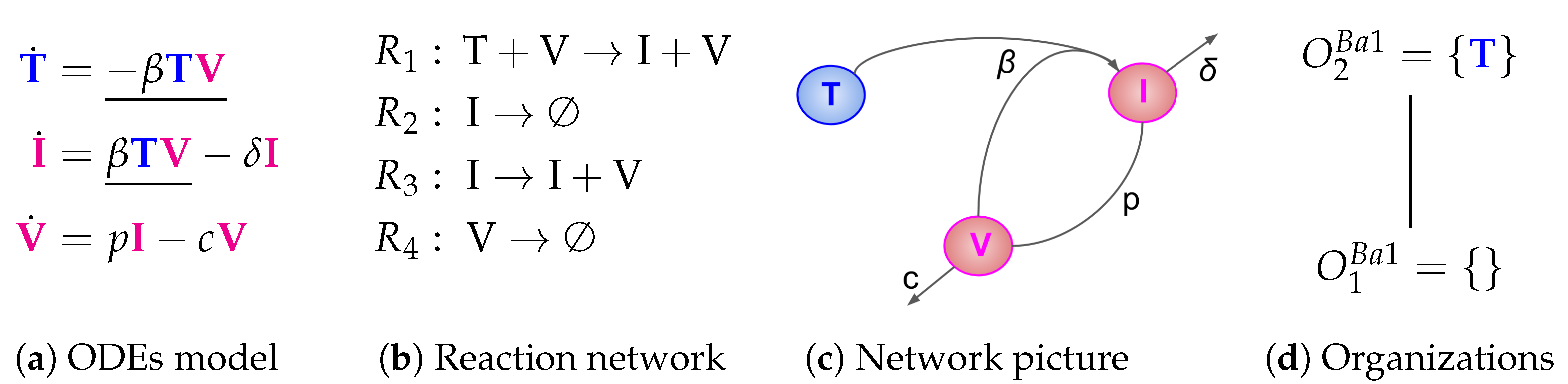
2.1. Deriving the Reaction Network from the ODE System
- The term represents the reaction , which in turn denotes the transformation of an uninfected target cell T to an infected cell I catalysed by the virus V.
- The terms and represent reactions and which are the outflow of infected cells I resp. virus V.
- The term represents the reaction which is the production of viruses V catalysed by infected cells I.
- Single underline for the transformation of uninfected cells into infected ones by the action of viruses.
- for kinetic terms involving interferon.
2.2. Computing the Organizations from the Reaction Network
2.3. The Role Organizations Play in the Dynamics
3. Results and Discussion
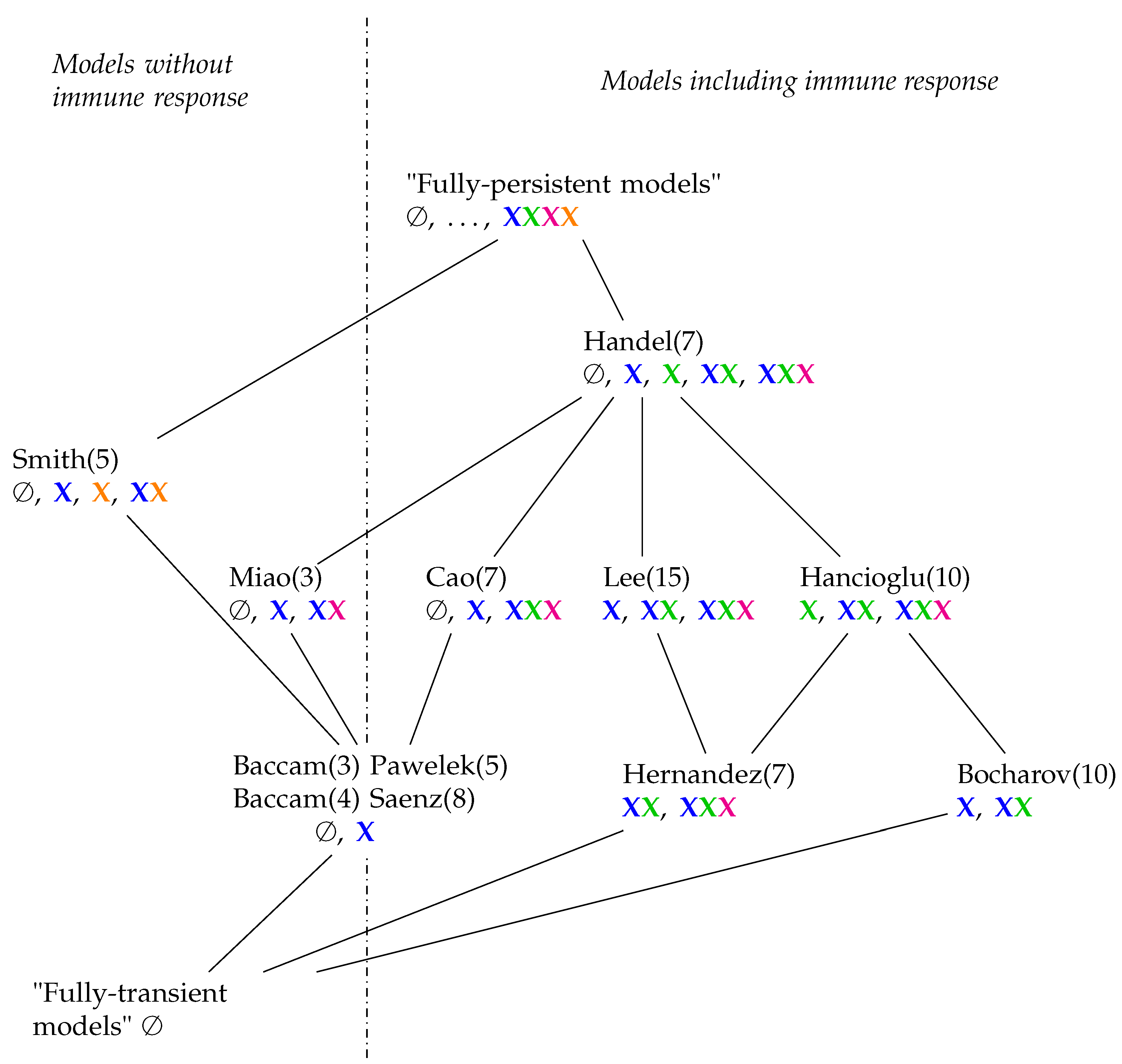
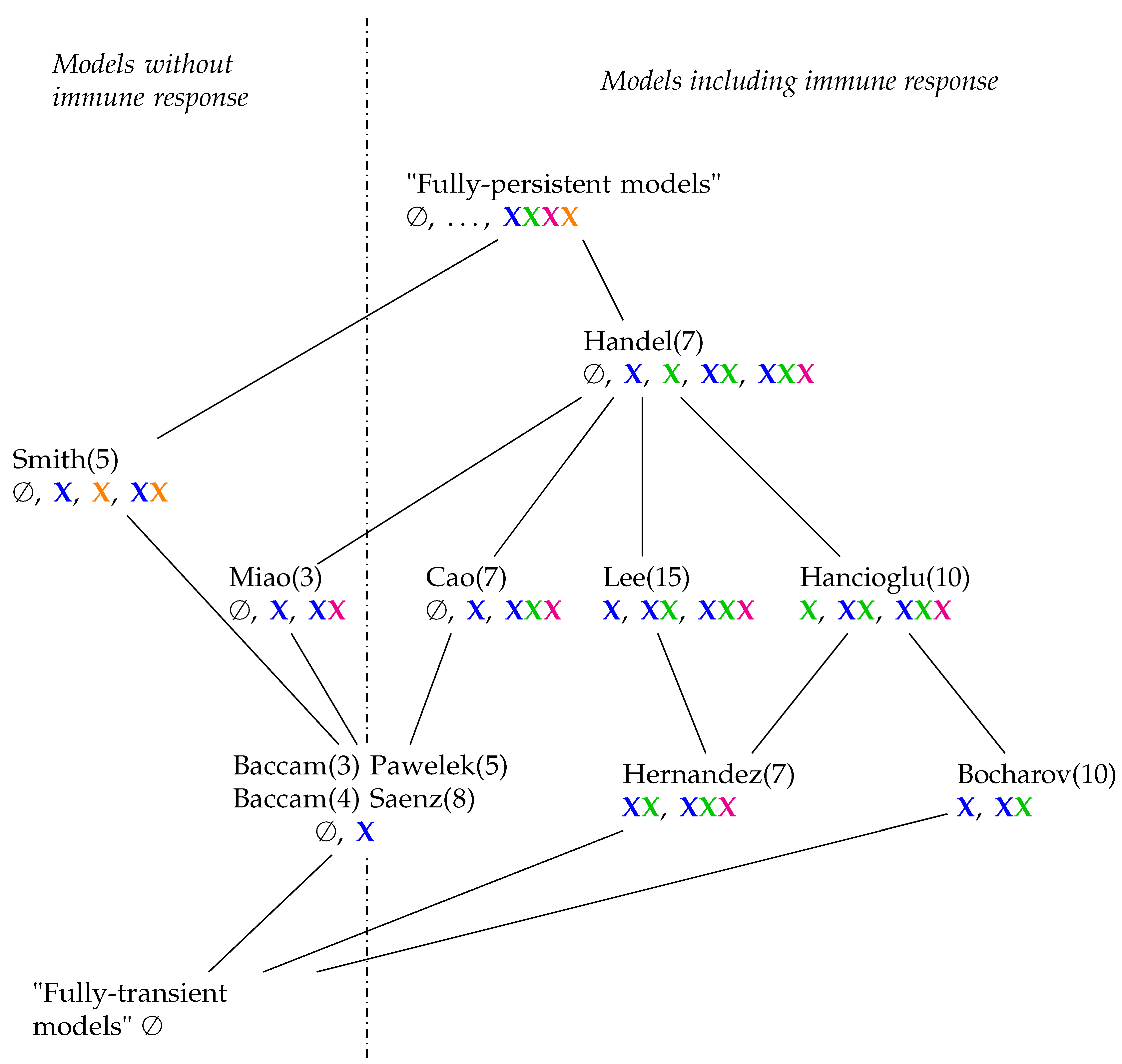
3.1. Target Cell Limited Model by Miao et al. (Miao Model, M, 2010)
3.2. Target Cell Limited Model with Delayed Virus Production (Baccam II Model, Ba2, 2006)
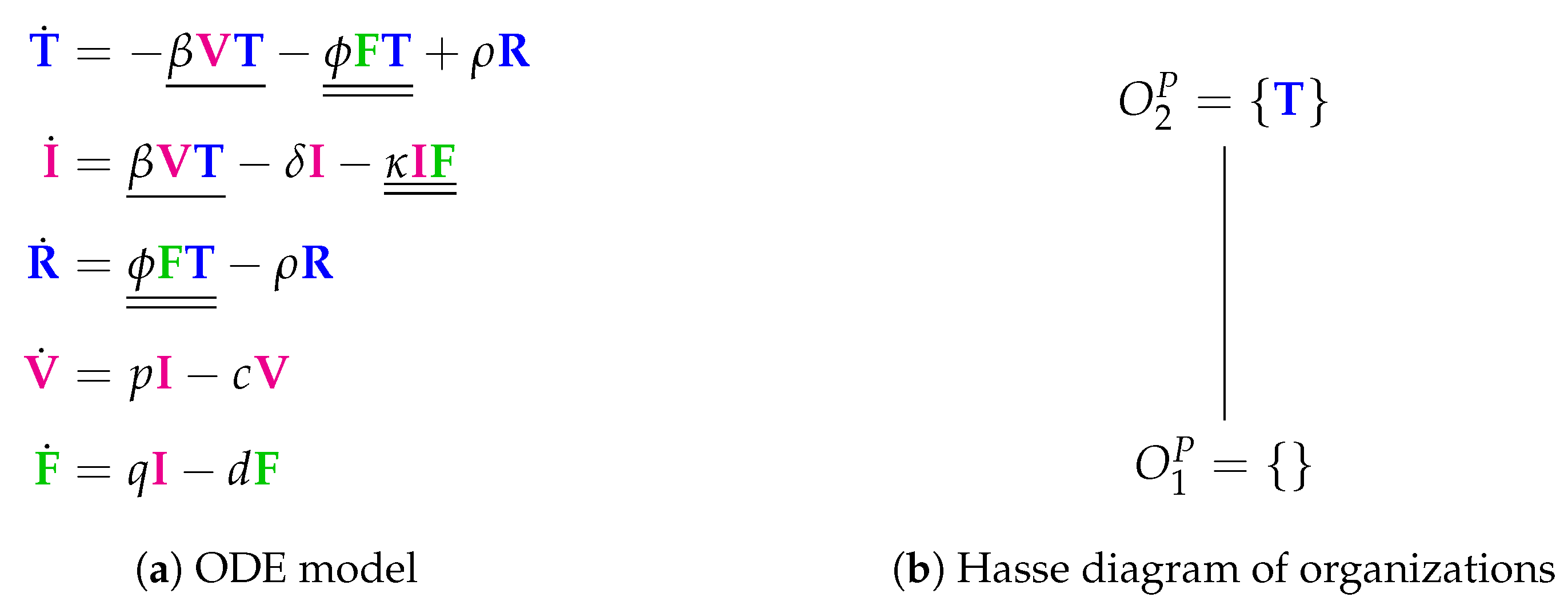
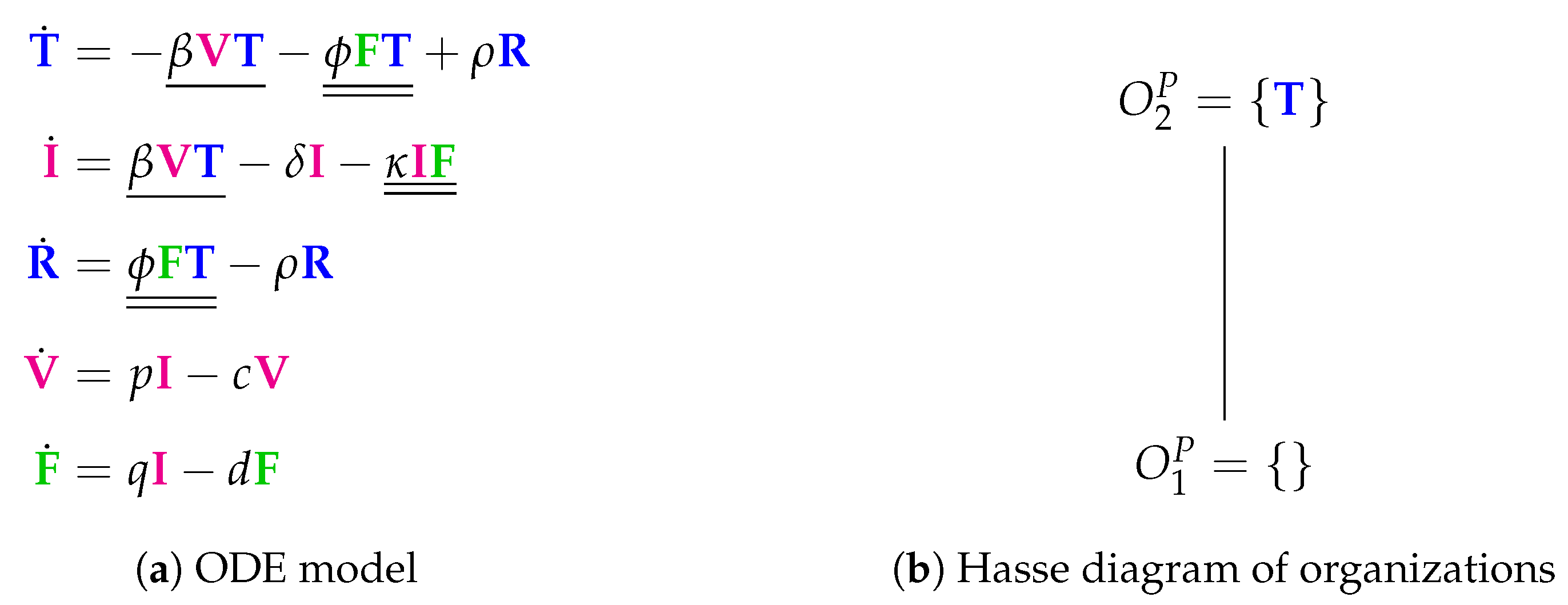
3.3. Innate and (Simple) Adaptive Immune Response (Pawelek Model, P, 2012)
- The rate term represents the transformation of uninfected target cells to refractory cells catalysed by interferon.
- The reverse shift back from refractory to simple uninfected cells is represented by the term .
- Furthermore, infected cells are deleted by the action of interferon at a rate .
- Interferon is produced in the presence of infected cells at a rate .
3.4. A Model Including Bacterial Co-Infection (Smith Model, Sm, 2016)
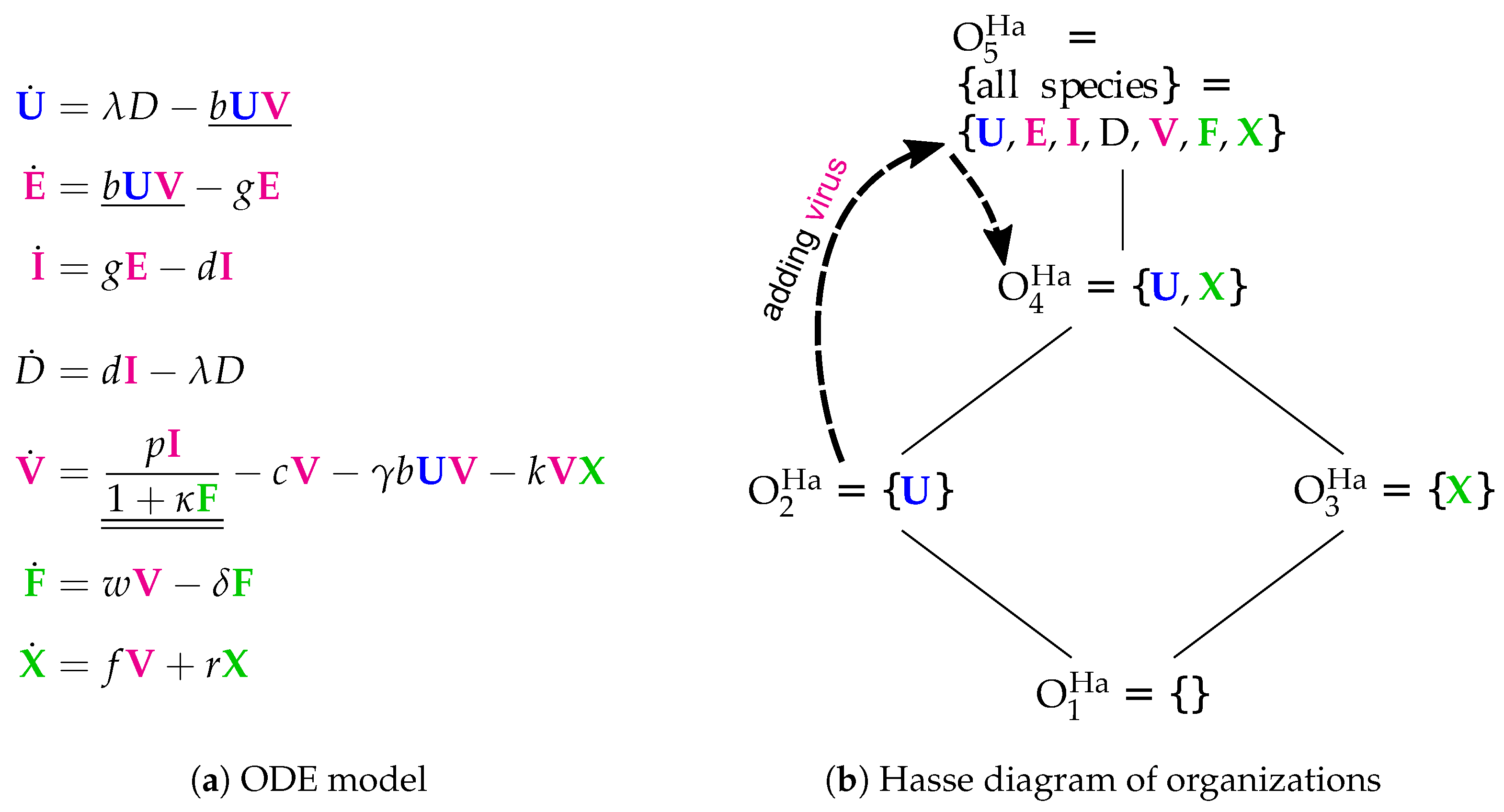
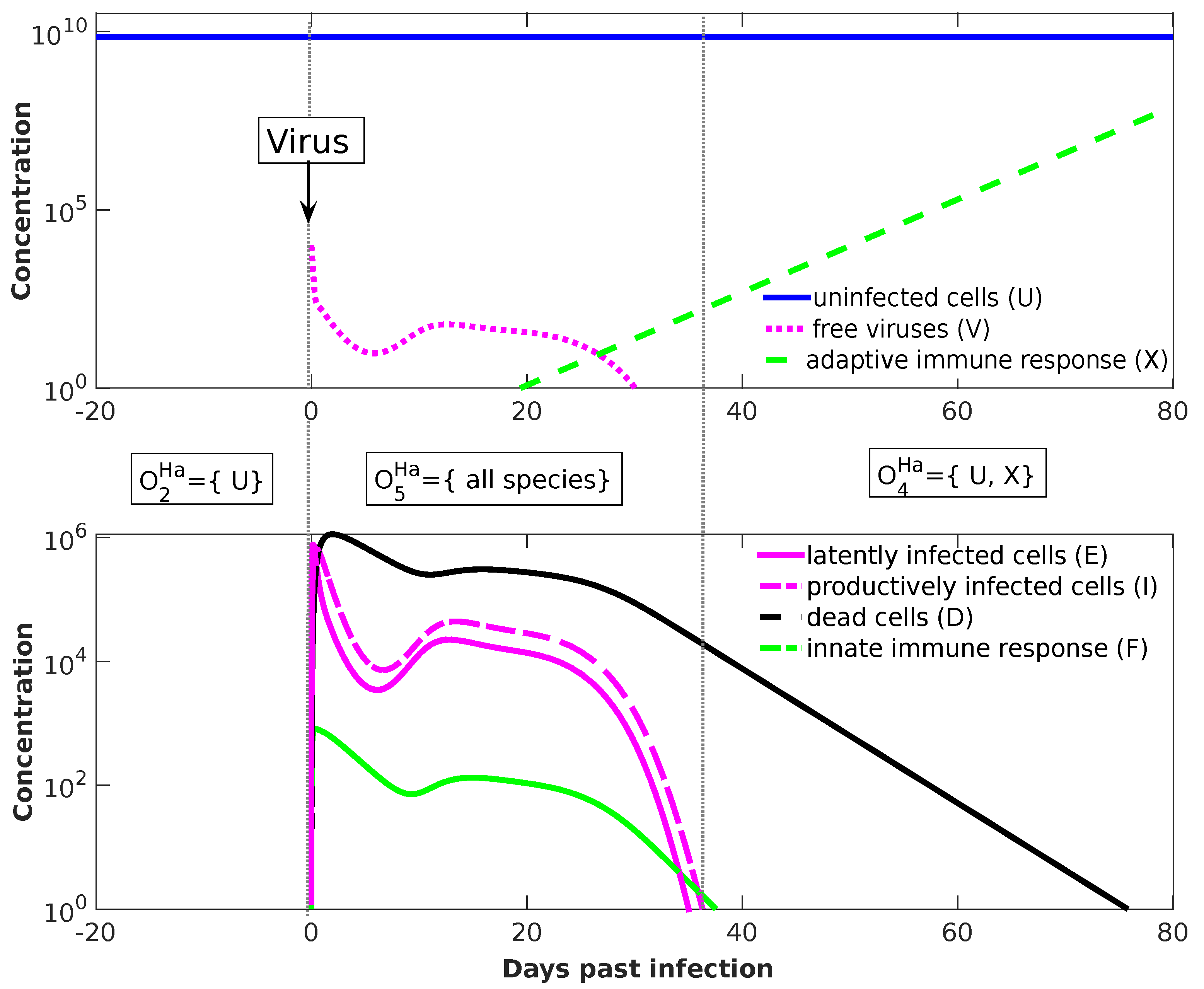
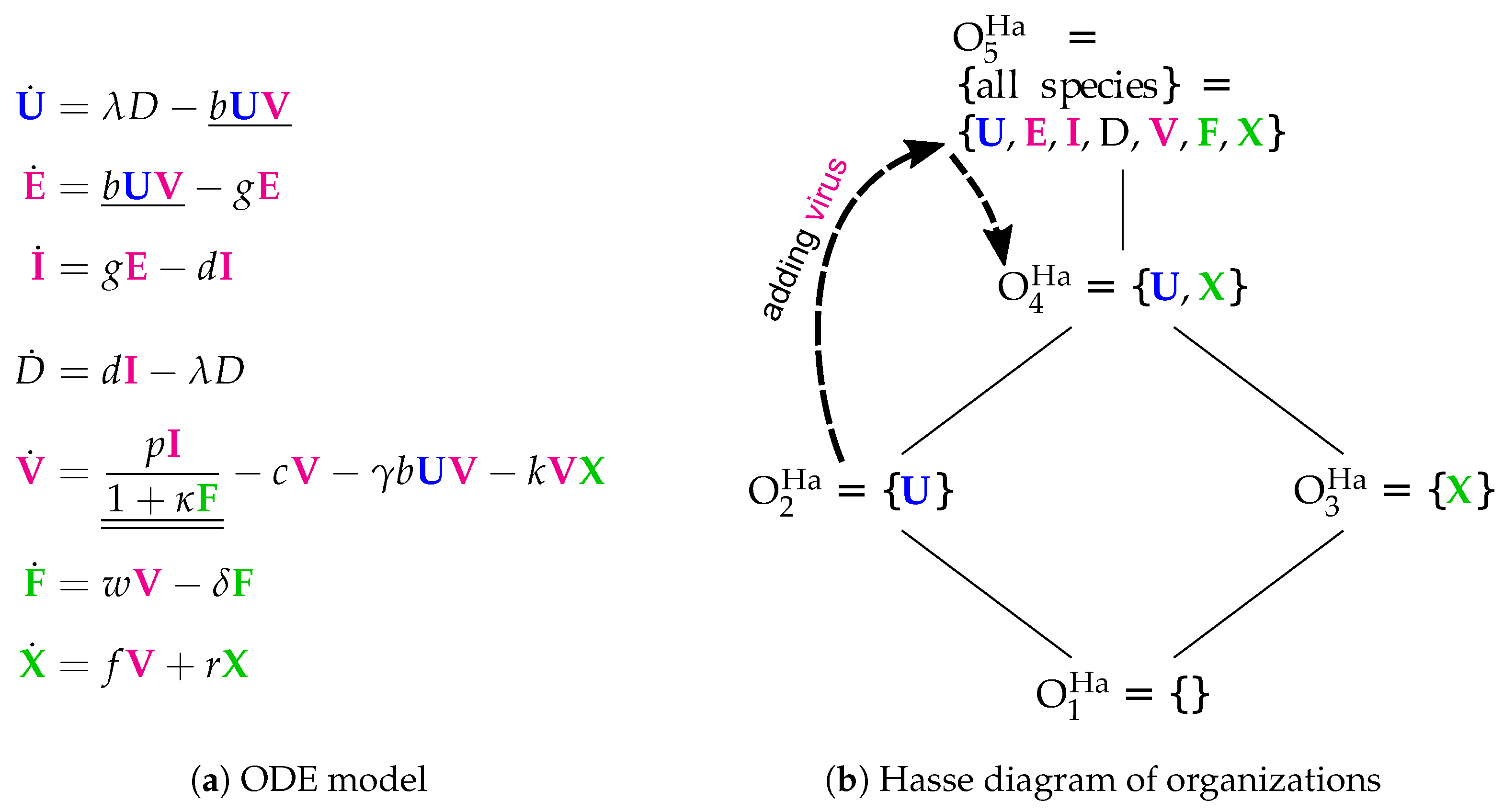
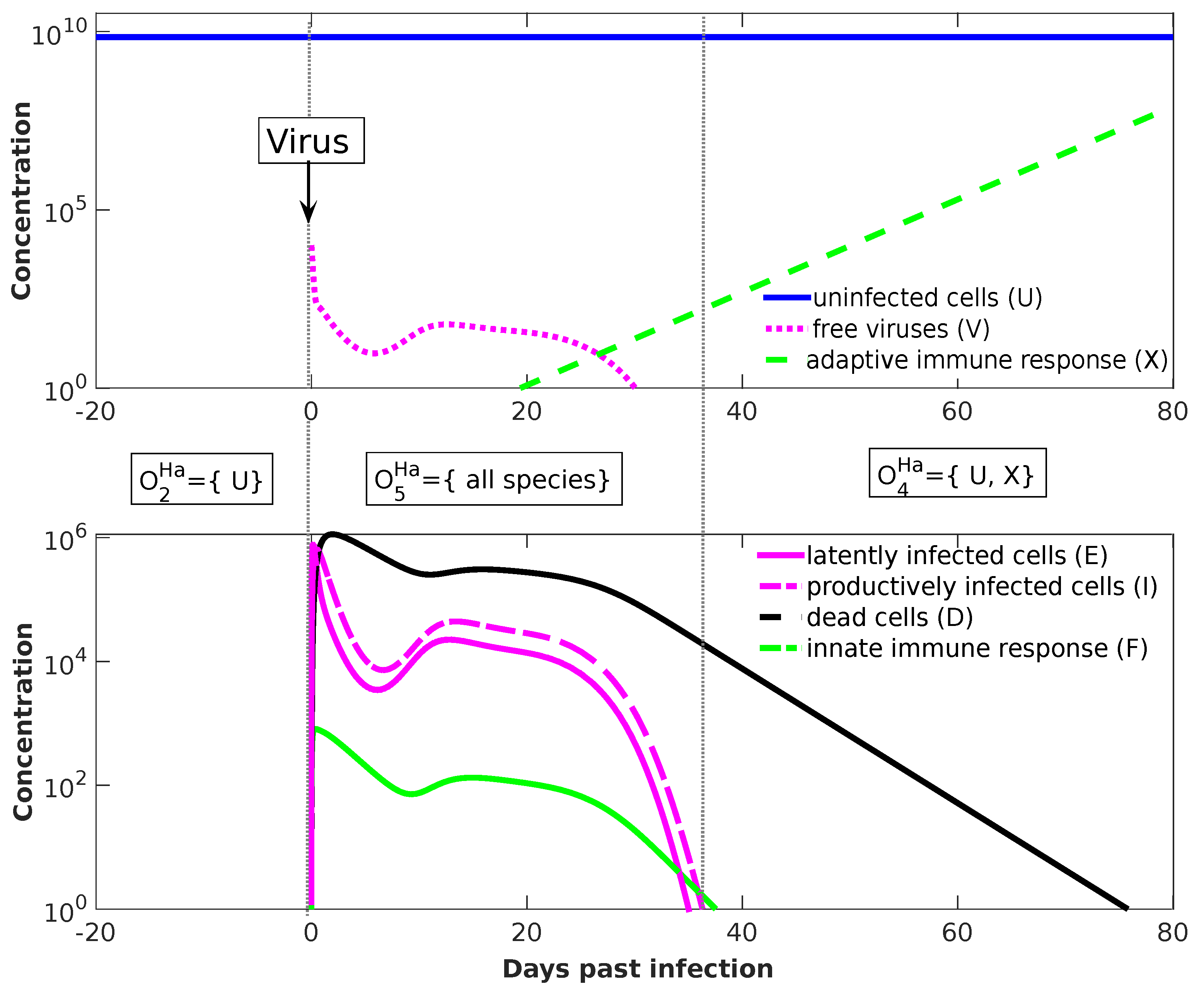
3.5. Innate and Adaptive Immune Response (Handel Model, Ha, 2009)
- Infection is catalyzed by viruses V and transforms uninfected cells U to latently infected cells E and viruses V are consumed thereby. Latently infected cells E transform into infected cells I autonomously, which in turn transform into dead cells D autonomously too. Finally, the transformation of dead cells D into non-infected cells U closes the circle.
- The remaining three species V, F and X form an almost totally separate subsystem since the only interaction with the four species from the "circle" mentioned above is the catalysis of the infection by viruses V.
- The interactions within the subsystem consisting of viruses V and immune responses F and X are as follows:
- –
- Viruses V catalyze the proliferation of F and X. In the Hernandez model, proliferation of interferon F is catalyzed by infected cells instead of viruses.
- –
- There is no direct interaction between innate immune response F and adaptive immune response X.
- –
- The adaptive immune response X deletes viruses directly. Innate immune response F inhibits the self-replication of the viruses which is represented by the denominator of the fraction . We ignore the inhibition because whether the rate is zero or not is independent of F.
Temporal Dynamics
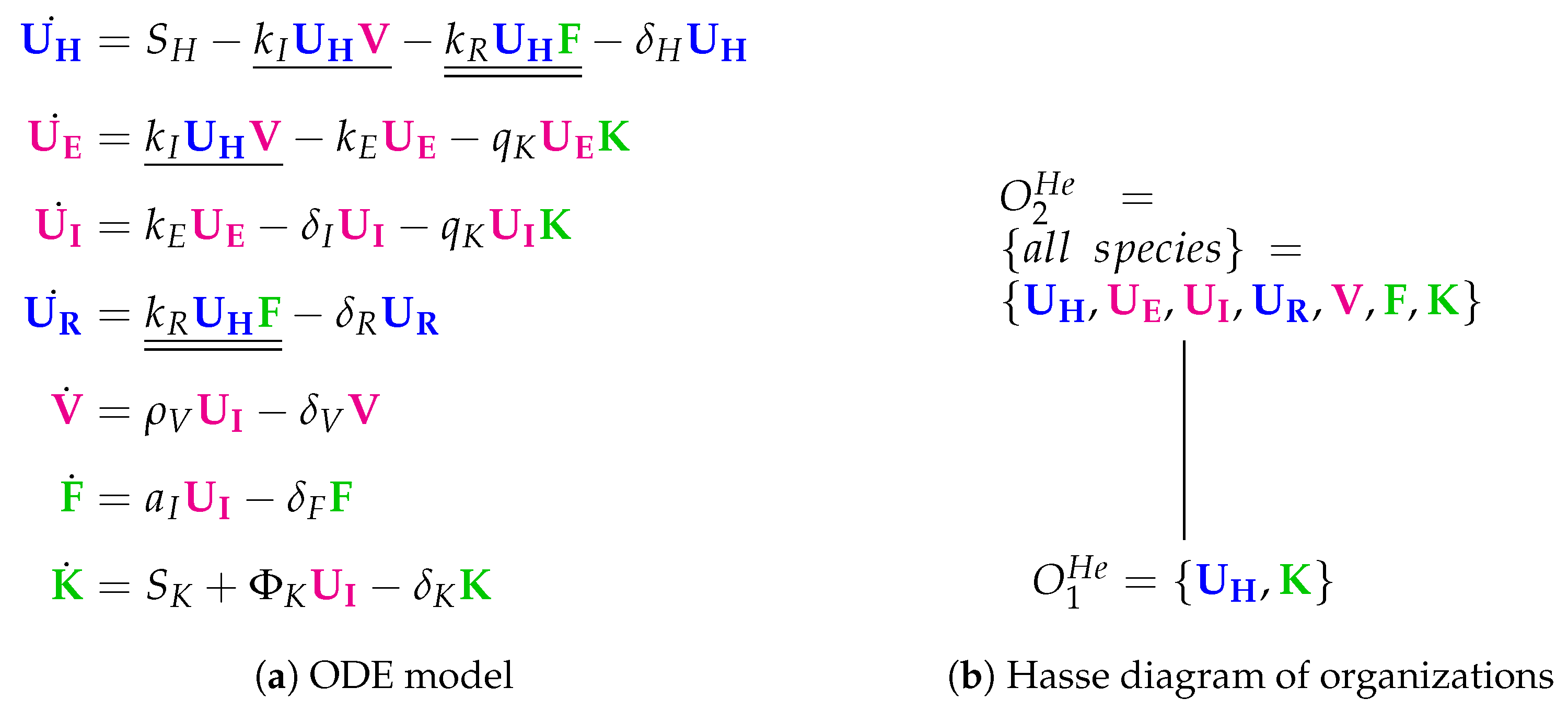
3.6. Innate Immune Response and Resistance to Infection (Hernandez Model, He, 2012)
- There is an infection reaction catalyzed by viruses like in all previous models but with one difference: during infection, healthy cells first transform to partially infected cells and only after that they transform spontaneously to infected cells at a rate .
- Interferon catalyzes the transformation of healthy cells to resistant cells , like in the Pawelek Model. However, in the Pawelek Model, interferon removes infected cells. Here, interferon’s production is catalyzed by infected cells at a rate . There is no further influence of interferon on any other species.
- Infected cells are removed by natural killers K, which also delete partially infected cells in this model. The production of killers K is catalyzed by infected cells at a rate .
- Note that here we have an constant inflow of healthy cells at a rate (first differential equation). Thus, healthy cells cannot converge to zero.
3.7. A Model with More Detailed Immune Response (Cao Model, C, 2015)
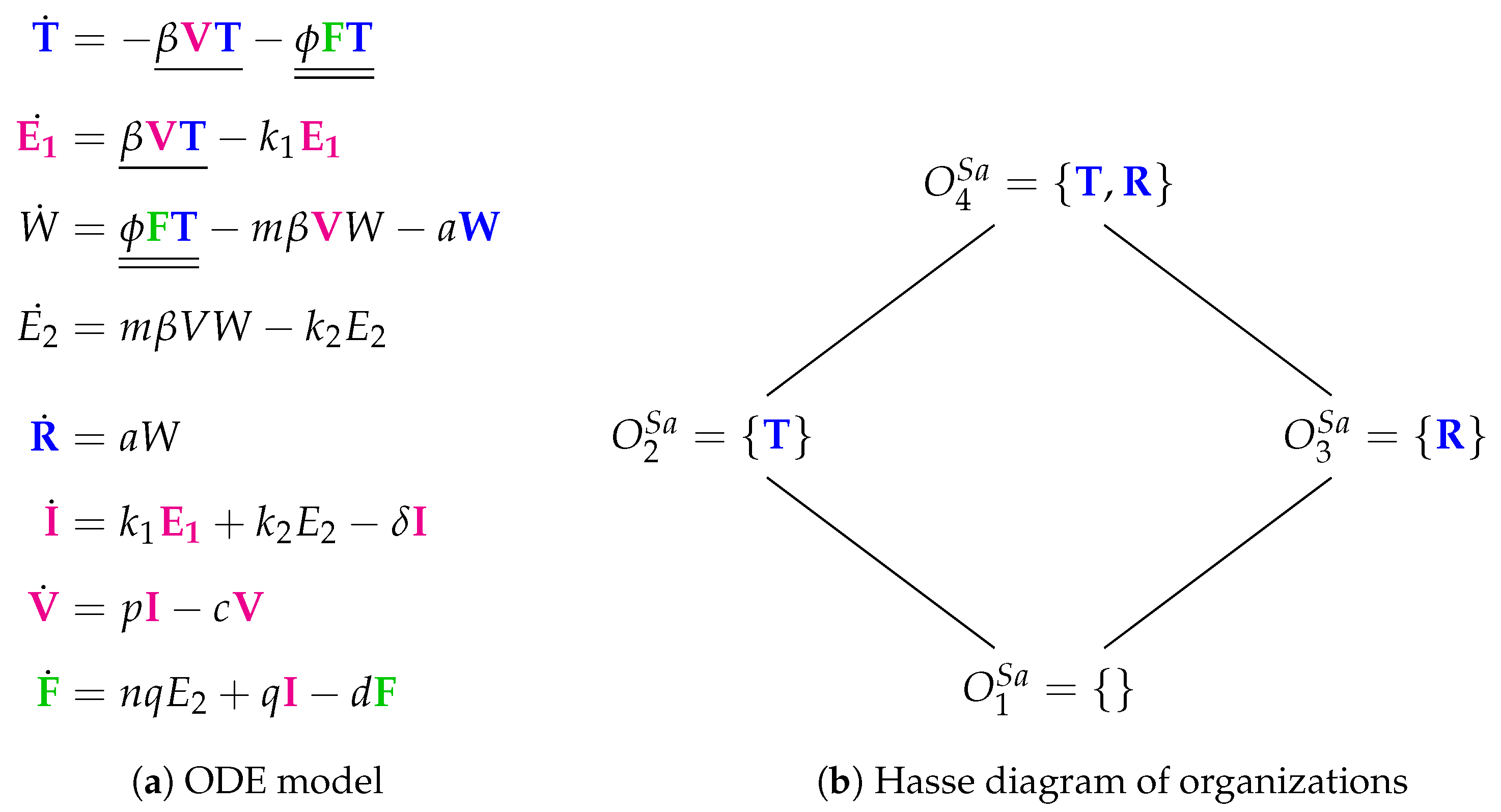
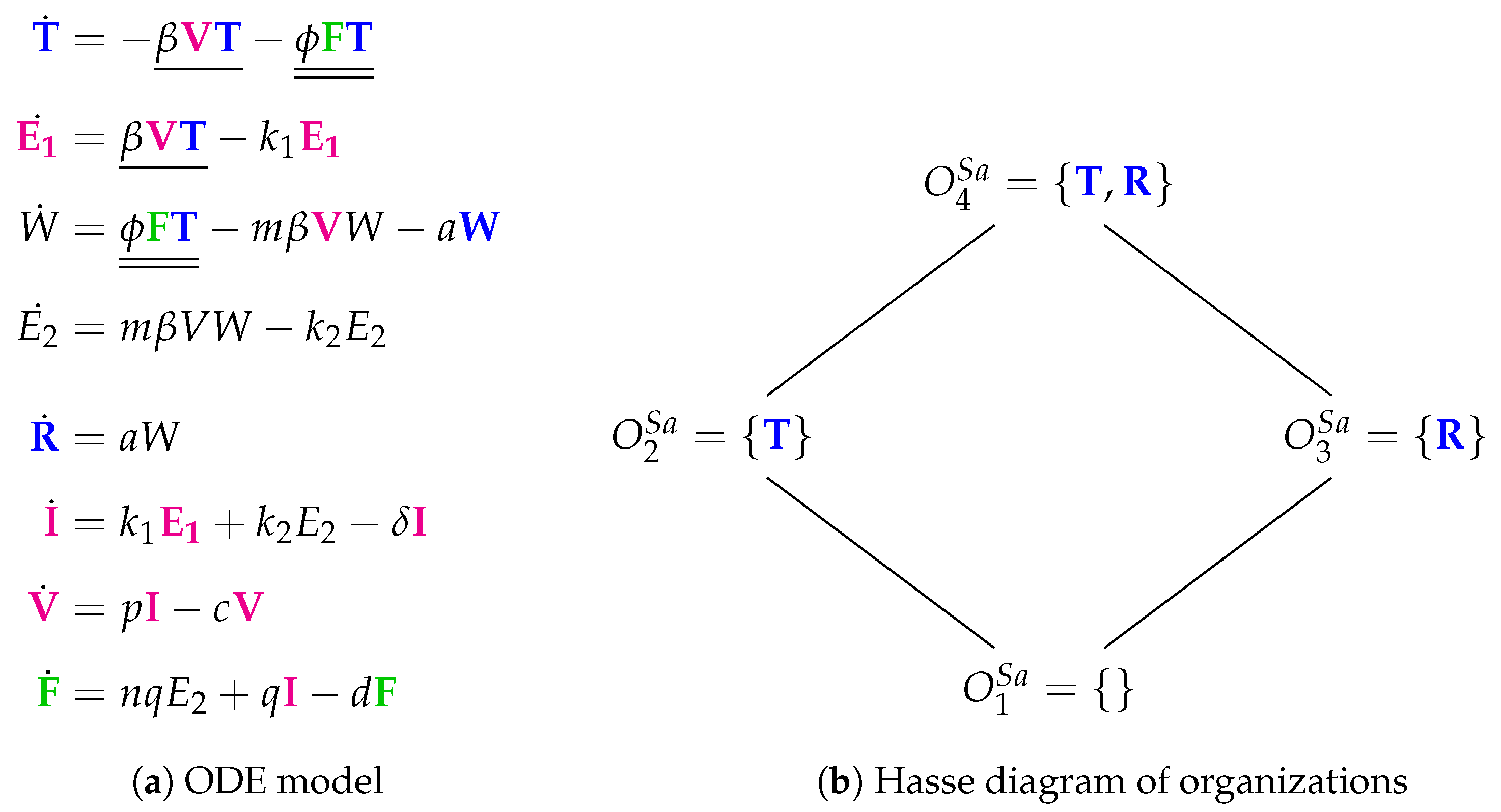
3.8. Innate Immune Response and Eclipse Phase (Saenz Model, Sa, 2010)
- The “full” organization is missing here. For sure, one of the reasons is that there is no reaction producing susceptible cells T. Thus, when viruses V or interferon F are present susceptible cells T can not survive and the “full” organization neither.
- Adaptive immune response is replaced by refractory cells in the organizations here.
3.9. Focusing on Innate and Adaptive Immunity (Hancioglu Model, Hcg, 2007)
- The smallest one is , which contains all the species responsible for the immune response.
- is a subset of , which additionally contains species H and R, representing the healthy organism without infection but with the immune response turned on.
- is the “full” organization containing all the species of the models and thus representing the organism with infection and immune response.
3.10. Model with Delay Differential Equations (Bocharov Model, Bo, 1994)
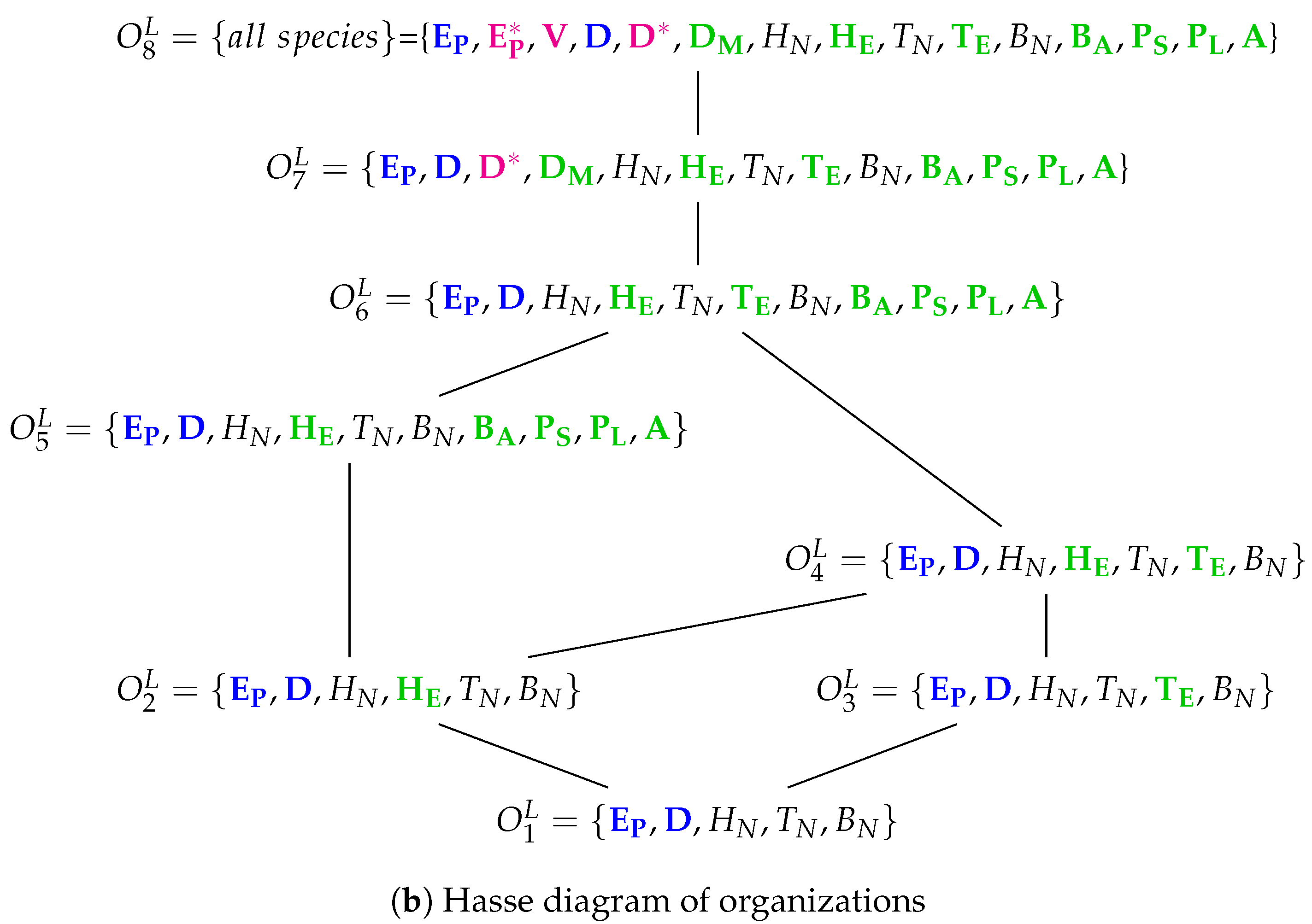
3.11. Complex Dual-Compartment Model (Lee Model, L, 2009)
3.12. Hierarchy of Influenza A Virus Models
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

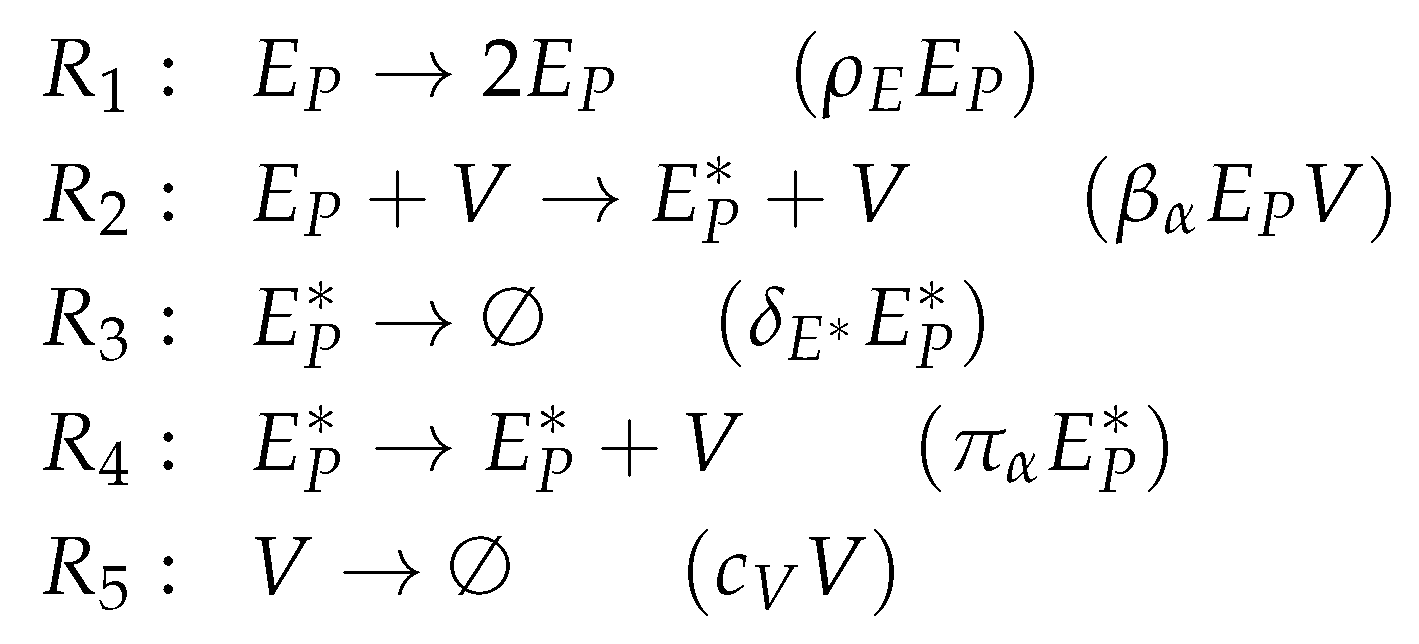

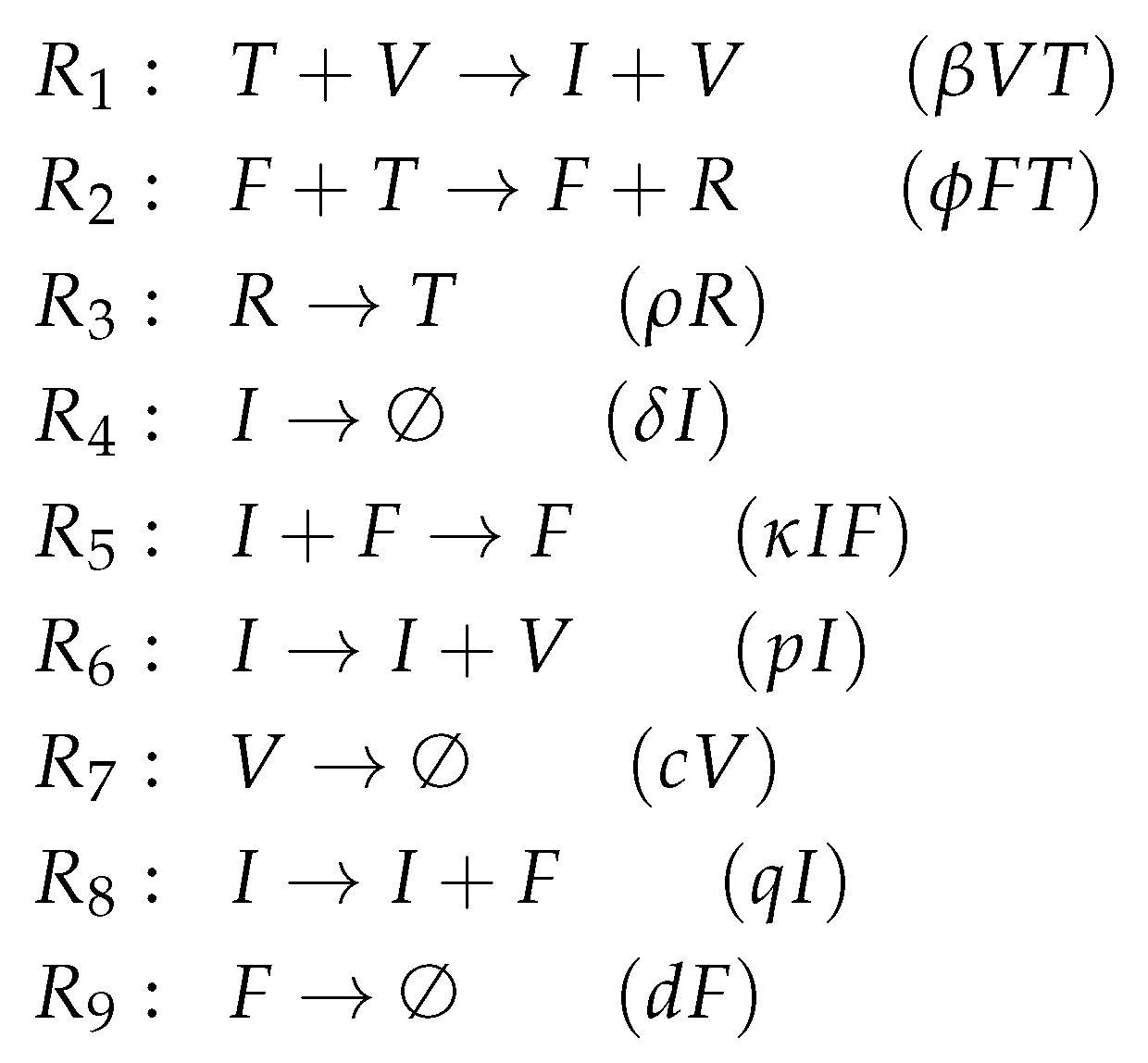
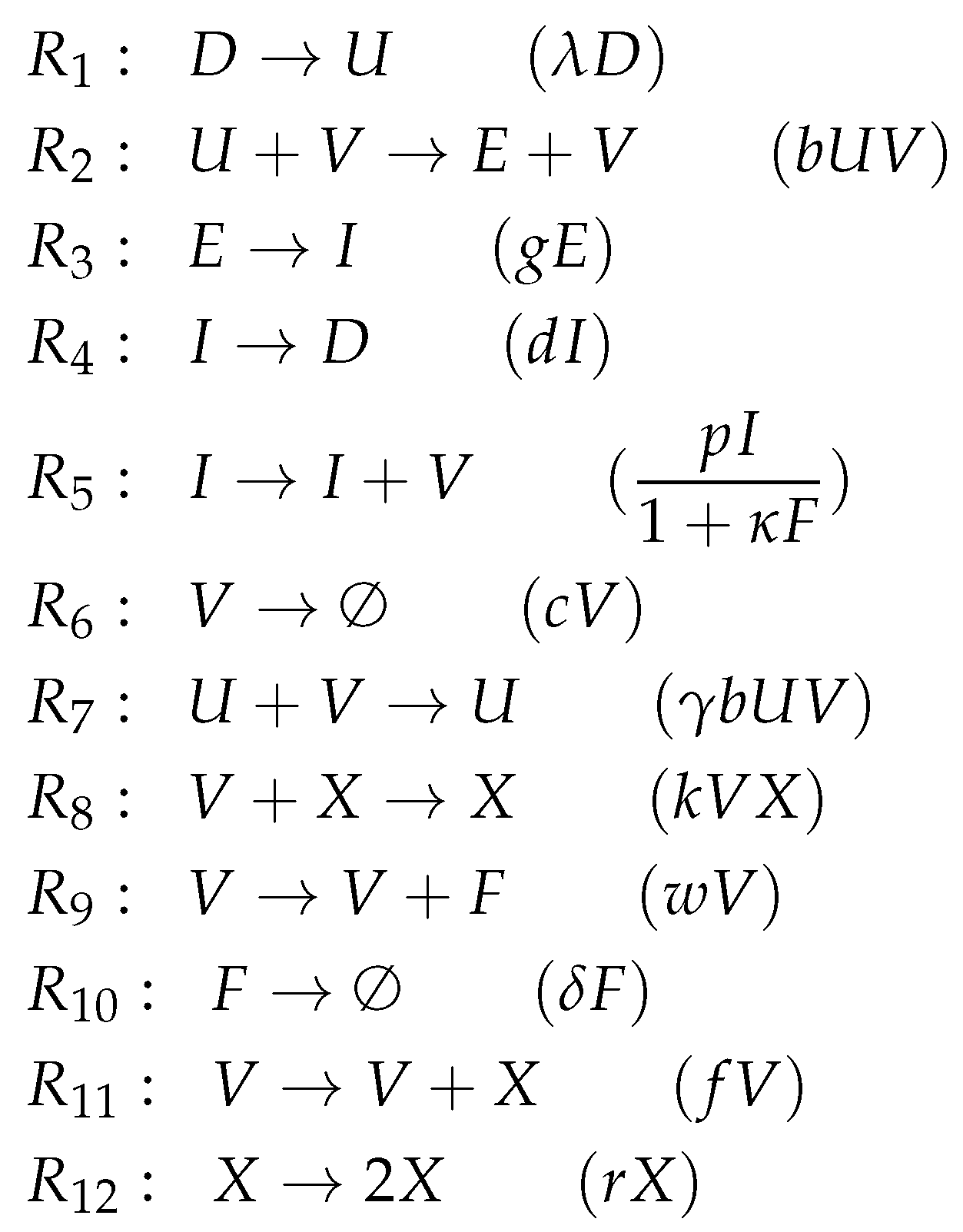
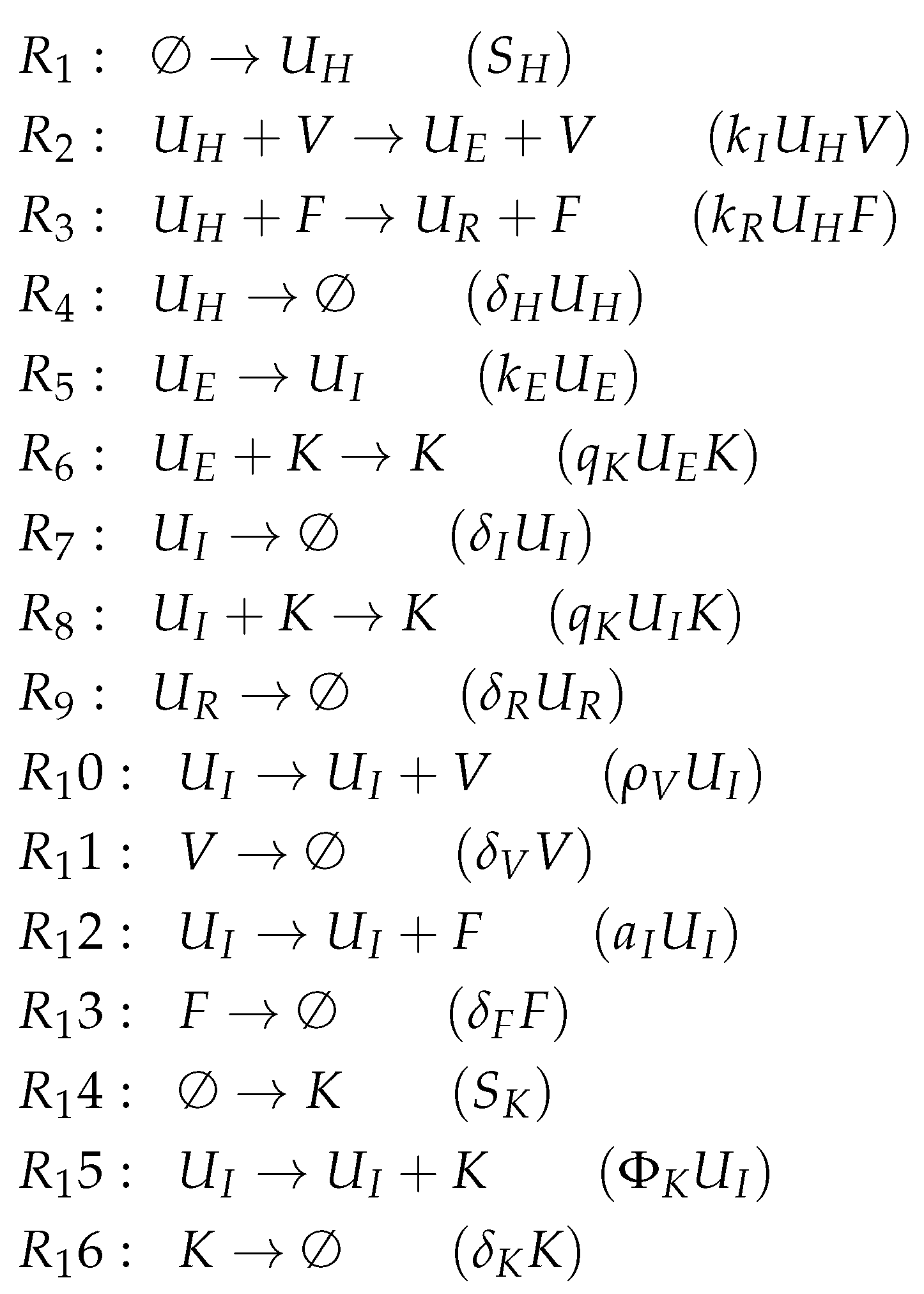

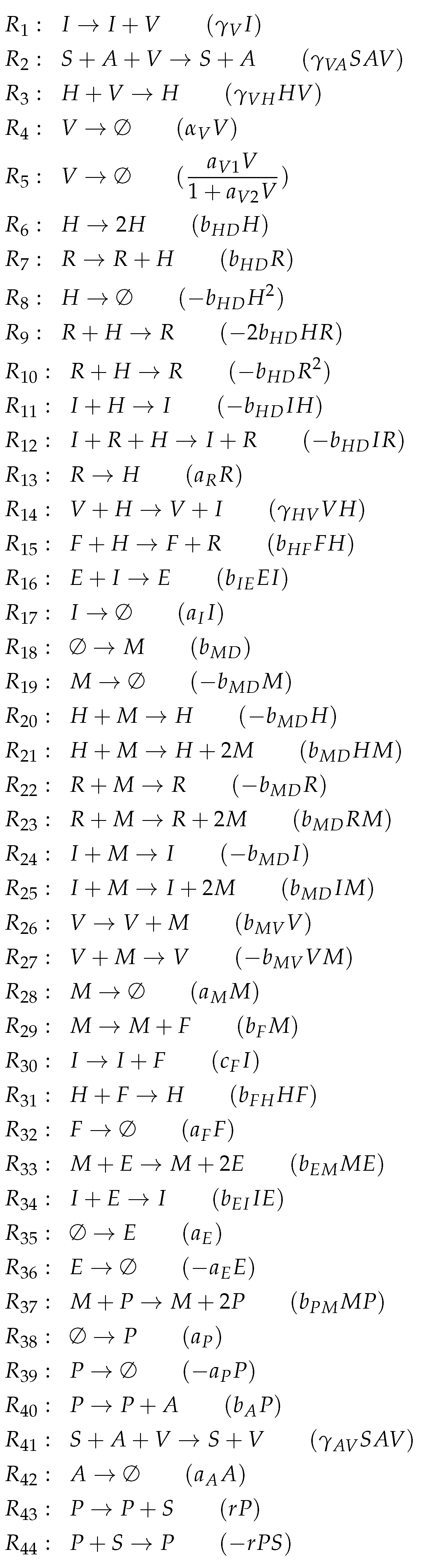
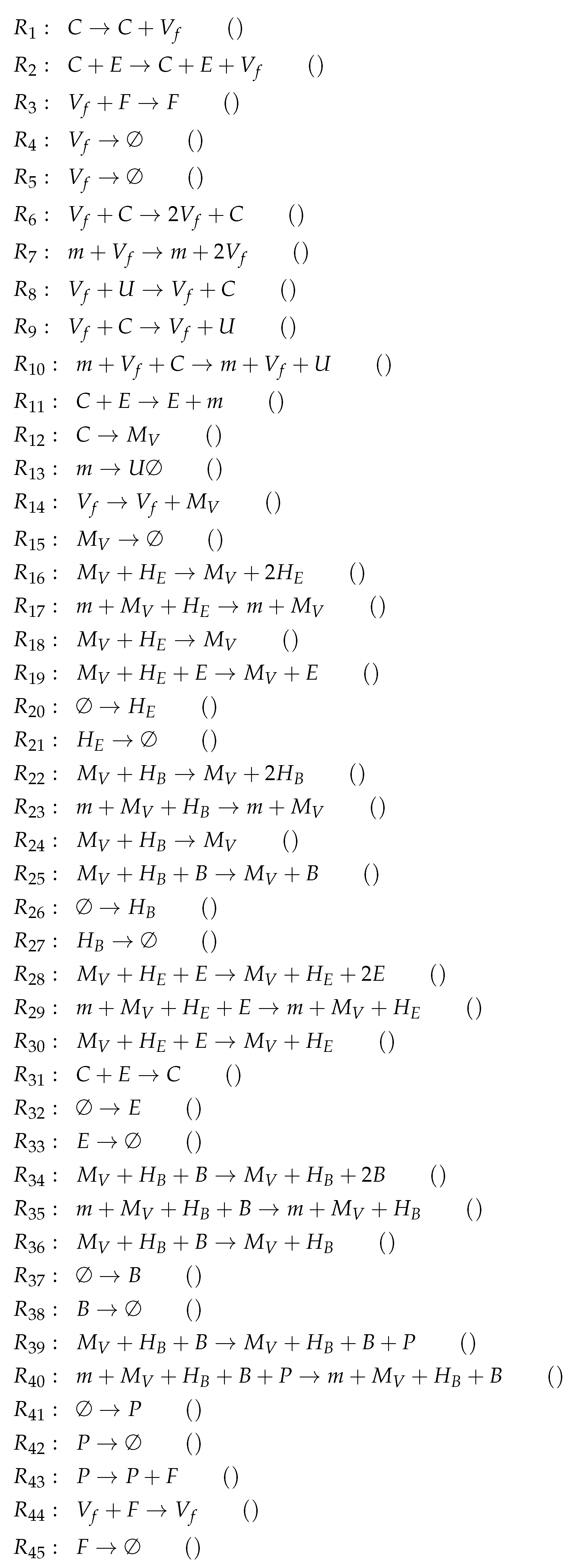
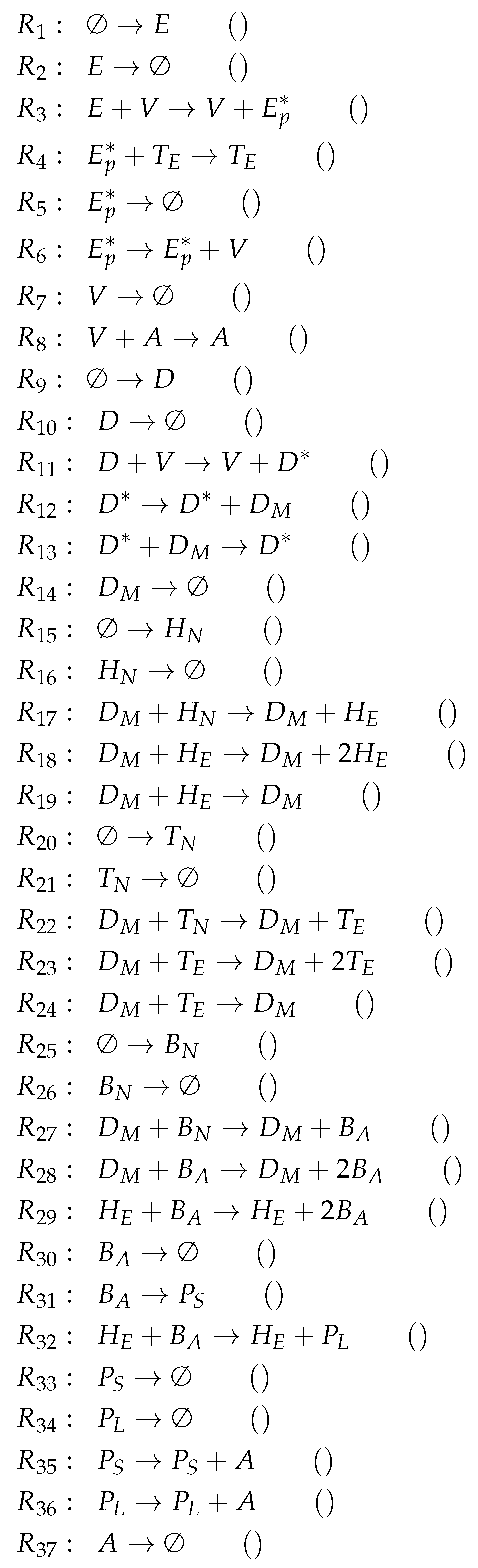
References
- Stöhr, K. Influenza—WHO cares. Lancet Infect. Dis. 2002, 2, 517. [Google Scholar] [CrossRef]
- Petrova, V.N.; Russell, C.A. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F.; Smith, G.J.; Fouchier, R.A.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Prim. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Perelson, A.S. Influenza A virus infection kinetics: Quantitative data and models. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011, 3, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Beauchemin, C.A.; Handel, A. A review of mathematical models of influenza A infections within a host or cell culture: Lessons learned and challenges ahead. BMC Public Health 2011, 11 (Suppl. 1), S7. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, H.M.; Reddy, M.B.; Kamal, M.A.; Rayner, C.R.; Beauchemin, C.A. Assessing mathematical models of influenza infections using features of the immune response. PLoS ONE 2013, 8, e57088. [Google Scholar] [CrossRef] [PubMed]
- Boianelli, A.; Nguyen, V.K.; Ebensen, T.; Schulze, K.; Wilk, E.; Sharma, N.; Stegemann-Koniszewski, S.; Bruder, D.; Toapanta, F.R.; Guzman, C.A.; et al. Modeling Influenza Virus Infection: A Roadmap for Influenza Research. Viruses 2015, 7, 5274–5304. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.; Liao, L.E.; Beauchemin, C.A. Progress and trends in mathematical modelling of influenza A virus infections. Curr. Opin. Syst. Biol. 2018, 12, 30–36. [Google Scholar] [CrossRef]
- Lee, H.Y.; Topham, D.J.; Park, S.Y.; Hollenbaugh, J.; Treanor, J.; Mosmann, T.R.; Jin, X.; Ward, B.M.; Miao, H.; Holden-Wiltse, J.; et al. Simulation and prediction of the adaptive immune response to influenza A virus infection. J. Virol. 2009, 83, 7151–7165. [Google Scholar] [CrossRef]
- Dittrich, P.; Speroni di Fenizio, P. Chemical Organization Theory. Bull. Math. Biol. 2007, 69, 1199–1231. [Google Scholar] [CrossRef]
- Matsumaru, N.; Centler, F.; di Fenizio, P.S.; Dittrich, P. Chemical organization theory applied to virus dynamics. IT-Inf. Technol. 2006, 48, 154–160. [Google Scholar]
- Peter, S.; Dittrich, P. On the Relation between Organizations and Limit Sets in Chemical Reaction Systems. Adv. Complex Syst. 2011, 14, 77–96. [Google Scholar] [CrossRef]
- Baccam, P.; Beauchemin, C.; Macken, C.A.; Hayden, F.G.; Perelson, A.S. Kinetics of influenza A virus infection in humans. J. Virol. 2006, 80, 7590–7599. [Google Scholar] [CrossRef]
- Miao, H.; Hollenbaugh, J.A.; Zand, M.S.; Holden-Wiltse, J.; Mosmann, T.R.; Perelson, A.S.; Wu, H.; Topham, D.J. Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus. J. Virol. 2010, 84, 6687–6698. [Google Scholar] [CrossRef]
- Smith, A.M.; Smith, A.P. A Critical, Nonlinear Threshold Dictates Bacterial Invasion and Initial Kinetics During Influenza. Sci. Rep. 2016, 6, 38703. [Google Scholar] [CrossRef]
- Soliman, S.; Heiner, M. A unique transformation from ordinary differential equations to reaction networks. PLoS ONE 2010, 5, e14284. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, R.; Schuster, S. The Regulation of Cellular Systems; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Fontana, W.; Buss, L.W. “The arrival of the fittest”: Toward a theory of biological organization. Bull. Math. Biol. 1994, 56, 1–64. [Google Scholar]
- Kreyssig, P.; Wozar, C.; Peter, S.; Veloz, T.; Ibrahim, B.; Dittrich, P. Effects of small particle numbers on long-term behaviour in discrete biochemical systems. Bioinformatics 2014, 30, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, B. Toward a systems-level view of mitotic checkpoints. Prog. Biophys. Mol. Biol. 2015, 117, 217–224. [Google Scholar] [CrossRef]
- Kreyssig, P.; Escuela, G.; Reynaert, B.; Veloz, T.; Ibrahim, B.; Dittrich, P. Cycles and the qualitative evolution of chemical systems. PLoS ONE 2012, 7, e45772. [Google Scholar] [CrossRef]
- Smith, A.P.; Moquin, D.J.; Bernhauerova, V.; Smith, A.M. Influenza virus infection model with density dependence supports biphasic viral decay. Front. Microbiol. 2018, 9, 1554. [Google Scholar] [CrossRef]
- Pawelek, K.A.; Huynh, G.T.; Quinlivan, M.; Cullinane, A.; Rong, L.; Perelson, A.S. Modeling within-host dynamics of influenza virus infection including immune responses. PLoS Comput. Biol. 2012, 8, e1002588. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.; Longini, I.M., Jr.; Antia, R. Towards a quantitative understanding of the within-host dynamics of influenza A infections. J. R. Soc. Interface 2009, 7, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Handel, A.; Longini, I.M., Jr.; Antia, R. Neuraminidase inhibitor resistance in influenza: Assessing the danger of its generation and spread. PLoS Comput. Biol. 2007, 3, e240. [Google Scholar] [CrossRef]
- Hernandez-Vargas, A.E.; Meyer-Hermann, M. Innate immune system dynamics to influenza virus. IFAC Proc. Vol. 2012, 45, 260–265. [Google Scholar] [CrossRef]
- Cao, P.; Yan, A.W.; Heffernan, J.M.; Petrie, S.; Moss, R.G.; Carolan, L.A.; Guarnaccia, T.A.; Kelso, A.; Barr, I.G.; McVernon, J.; et al. Innate Immunity and the Inter-exposure Interval Determine the Dynamics of Secondary Influenza Virus Infection and Explain Observed Viral Hierarchies. PLoS Comput. Biol. 2015, 11, e1004334. [Google Scholar] [CrossRef] [PubMed]
- Saenz, R.A.; Quinlivan, M.; Elton, D.; Macrae, S.; Blunden, A.S.; Mumford, J.A.; Daly, J.M.; Digard, P.; Cullinane, A.; Grenfell, B.T.; et al. Dynamics of influenza virus infection and pathology. J. Virol. 2010, 84, 3974–3983. [Google Scholar] [CrossRef] [PubMed]
- Hancioglu, B.; Swigon, D.; Clermont, G. A dynamical model of human immune response to influenza A virus infection. J. Theor. Biol. 2007, 246, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, G.A.; Romanyukha, A.A. Mathematical model of antiviral immune response. III. Influenza A virus infection. J. Theor. Biol. 1994, 167, 323–360. [Google Scholar] [CrossRef]
- Cao, P.; McCaw, J. The mechanisms for within-host influenza virus control affect model-based assessment and prediction of antiviral treatment. Viruses 2017, 9, 197. [Google Scholar] [CrossRef]
- Zitzmann, C.; Kaderali, L. Mathematical Analysis of Viral Replication Dynamics and Antiviral Treatment Strategies: From Basic Models to Age-Based Multi-Scale Modeling. Front. Microbiol. 2018, 9, 1546. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Dittrich, P.; Parker, D.; Rowe, J.E. Organisation-Oriented Coarse Graining and Refinement of Stochastic Reaction Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Henze, R.; Mu, C.; Puljiz, M.; Kamaleson, N.; Huwald, J.; Haslegrave, J.; di Fenizio, P.S.; Parker, D.; Good, C.; Rowe, J.E.; et al. Multi-scale stochastic organization-oriented coarse-graining exemplified on the human mitotic checkpoint. Sci. Rep. 2019, 9, 3902. [Google Scholar] [CrossRef] [PubMed]
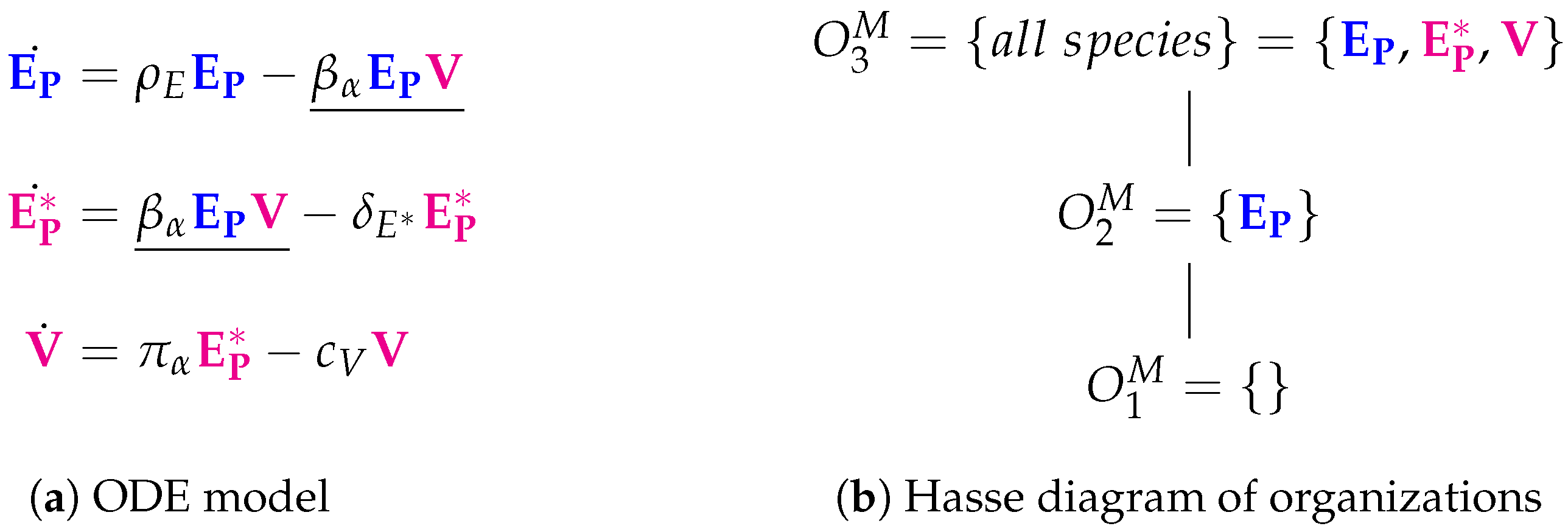
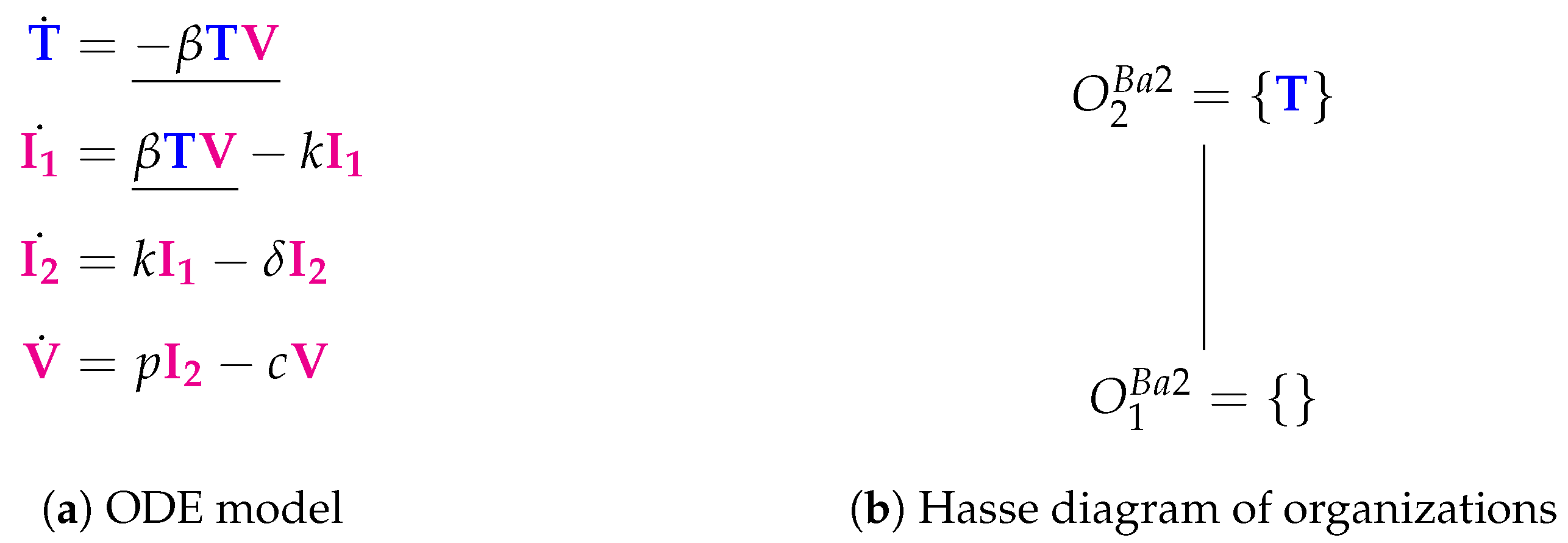


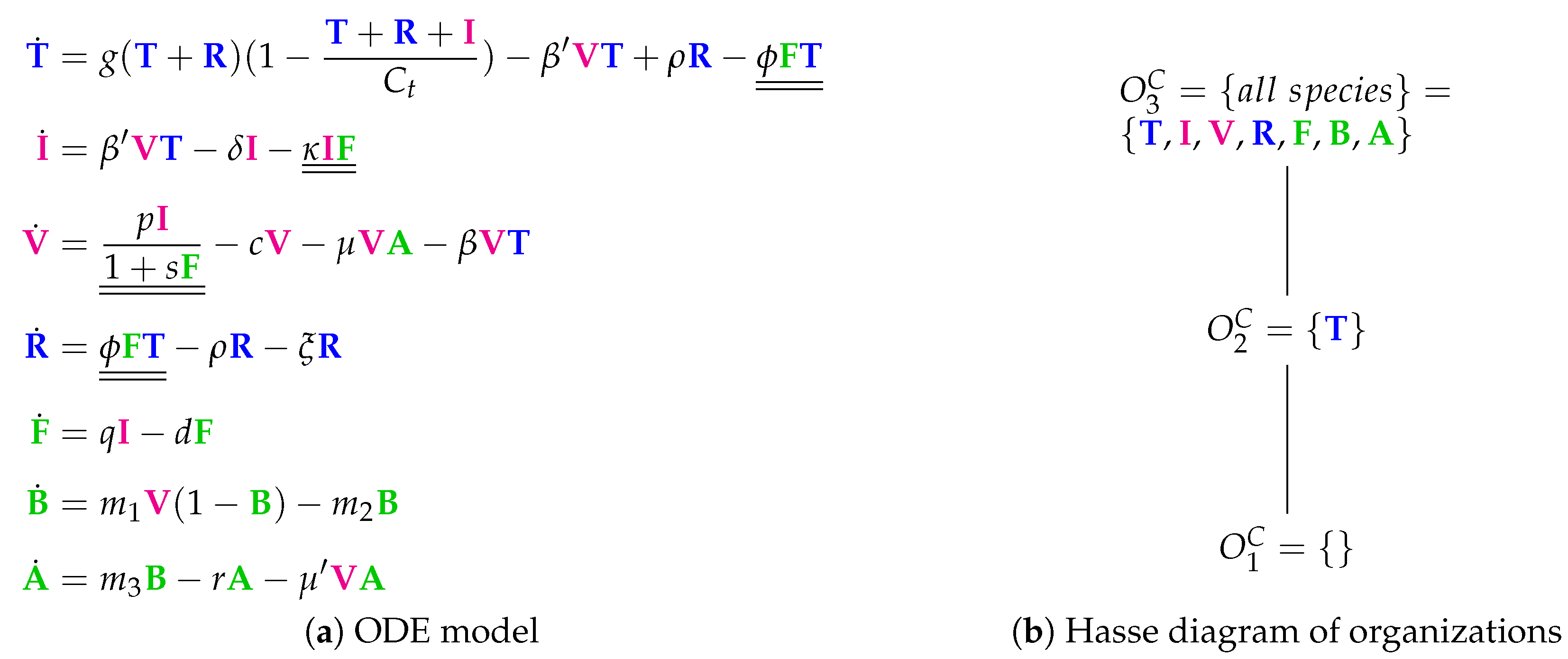
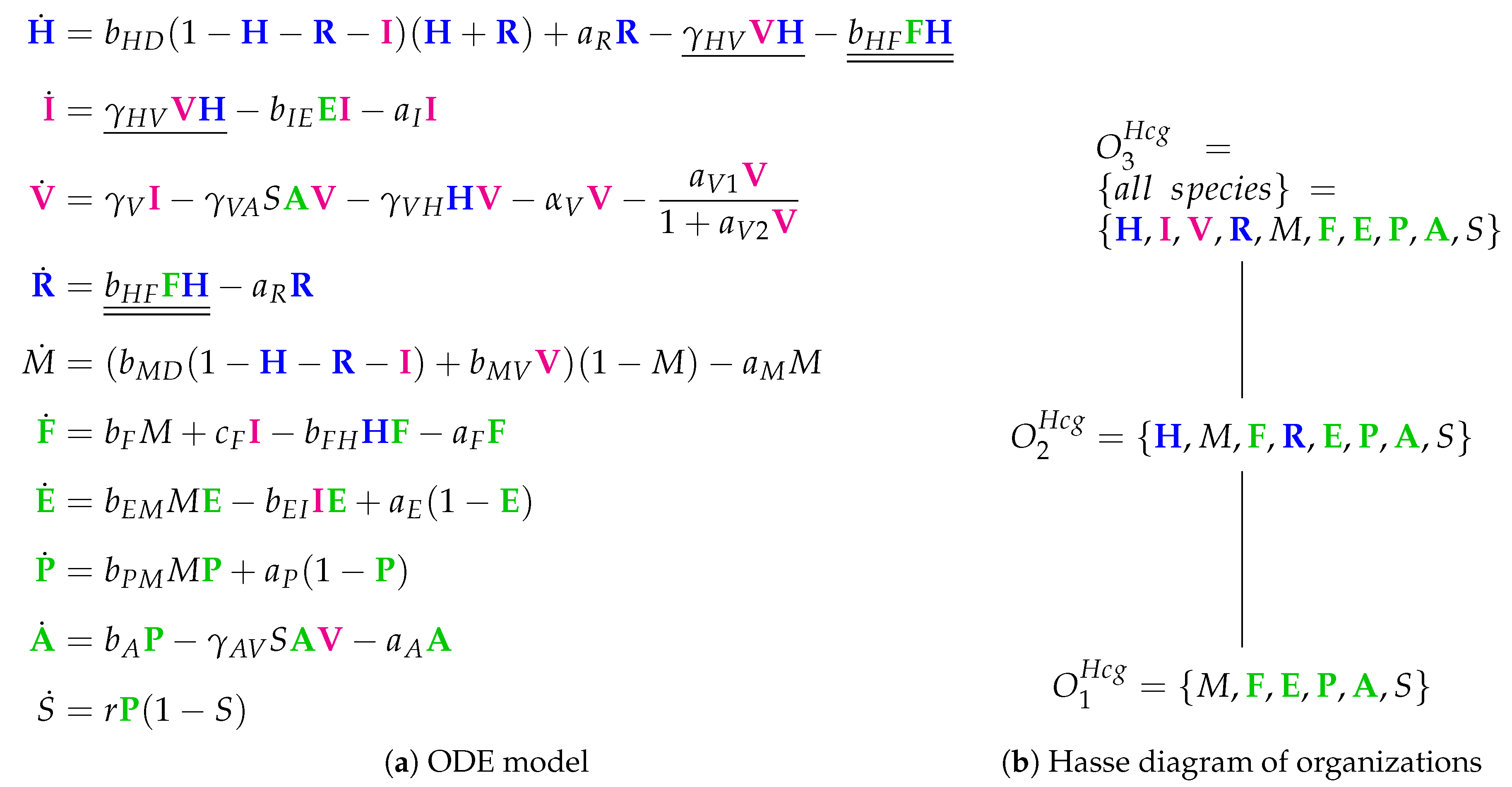
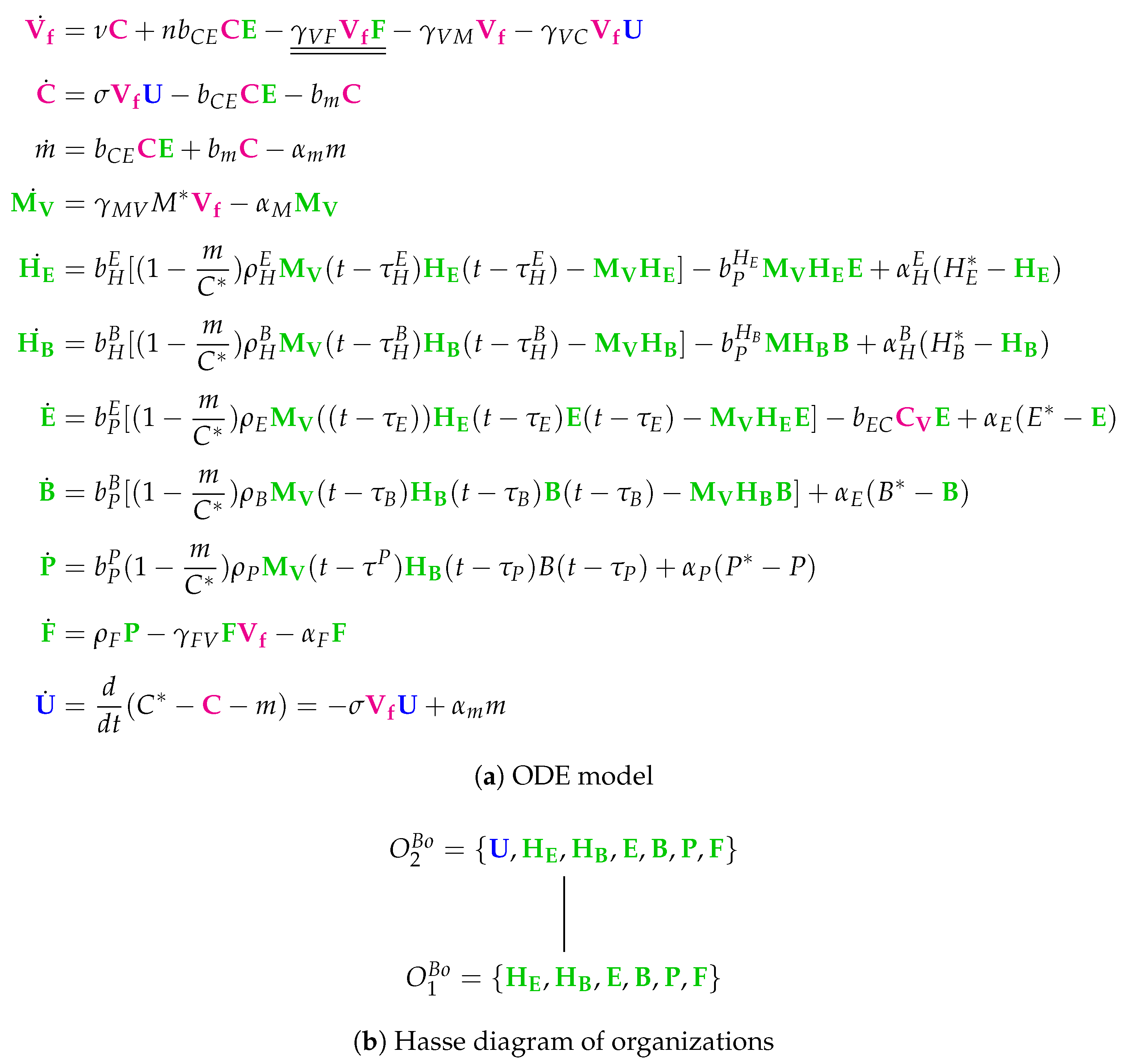
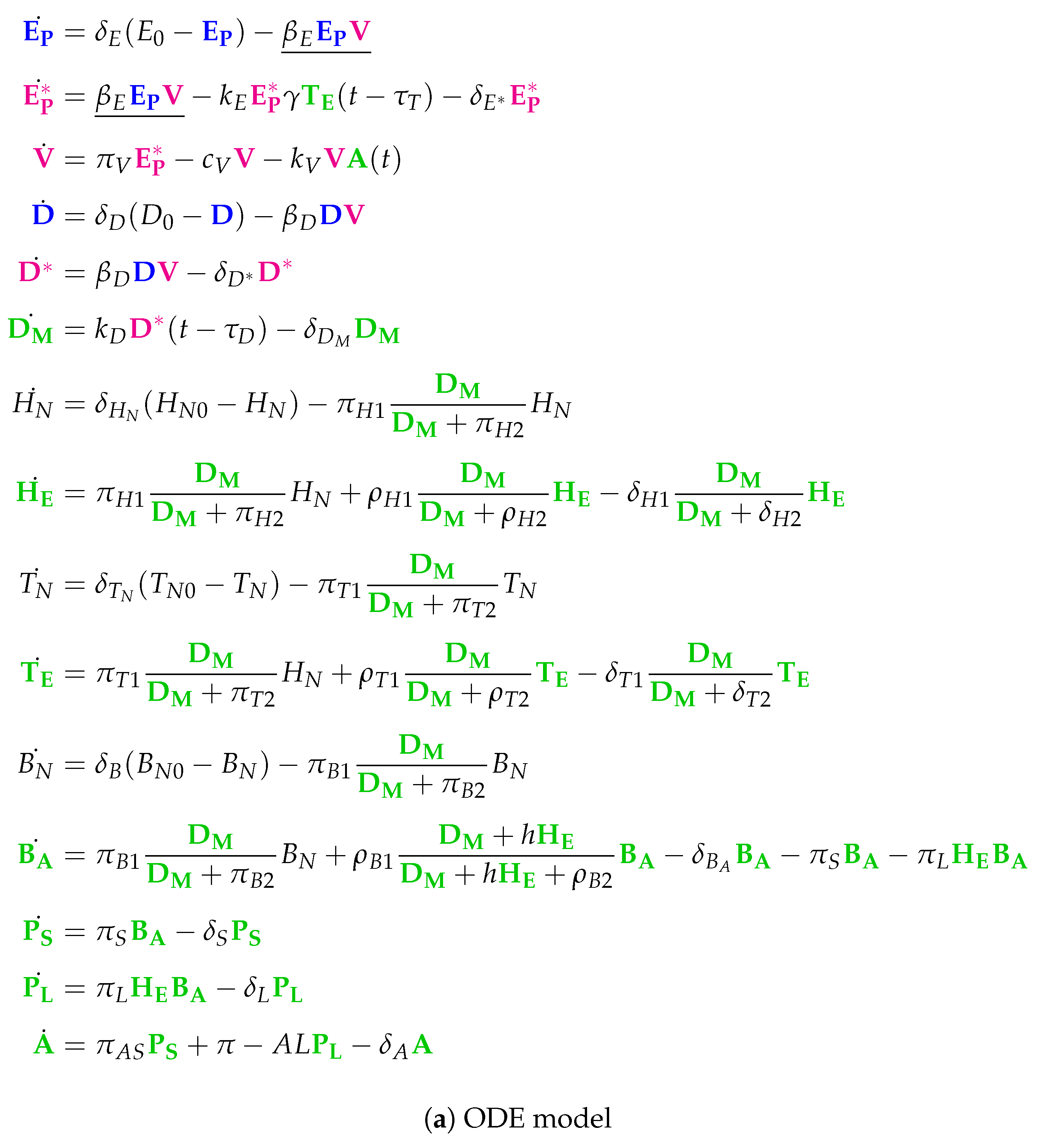

{kind=link}
{kind=link}
{kind=link}
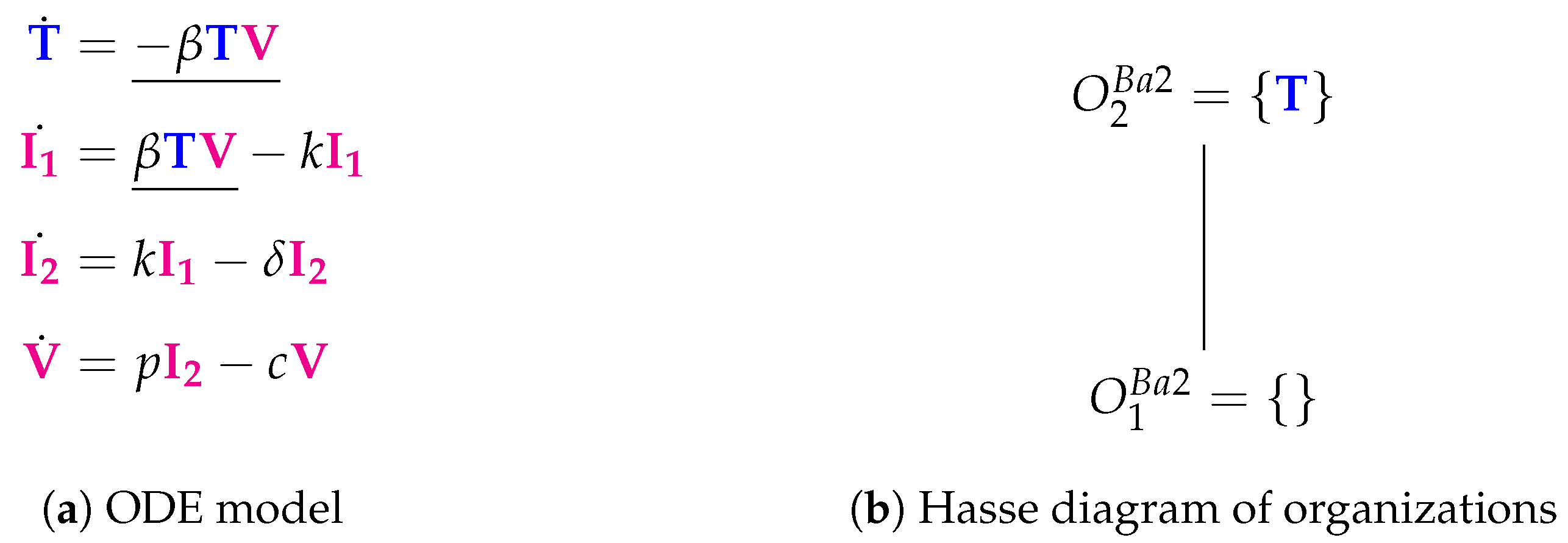{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
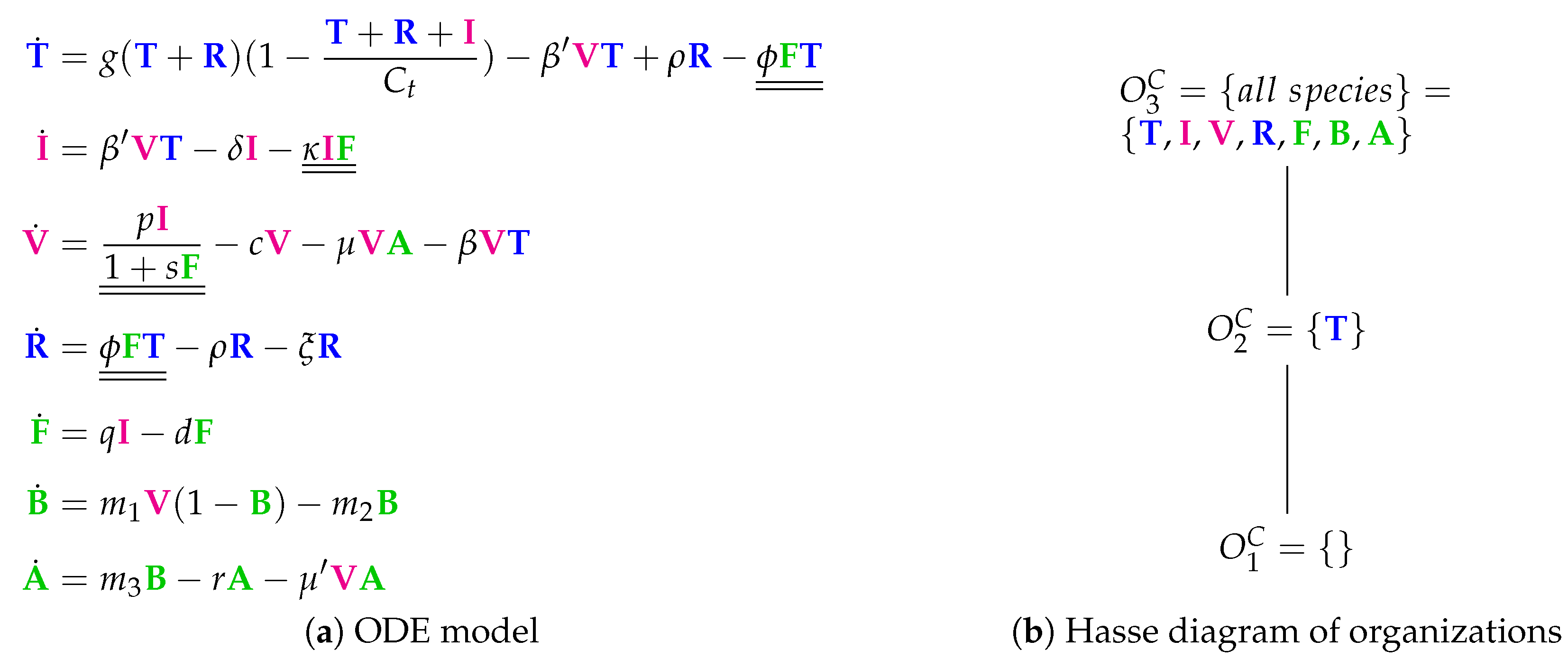{kind=link}
{kind=link}
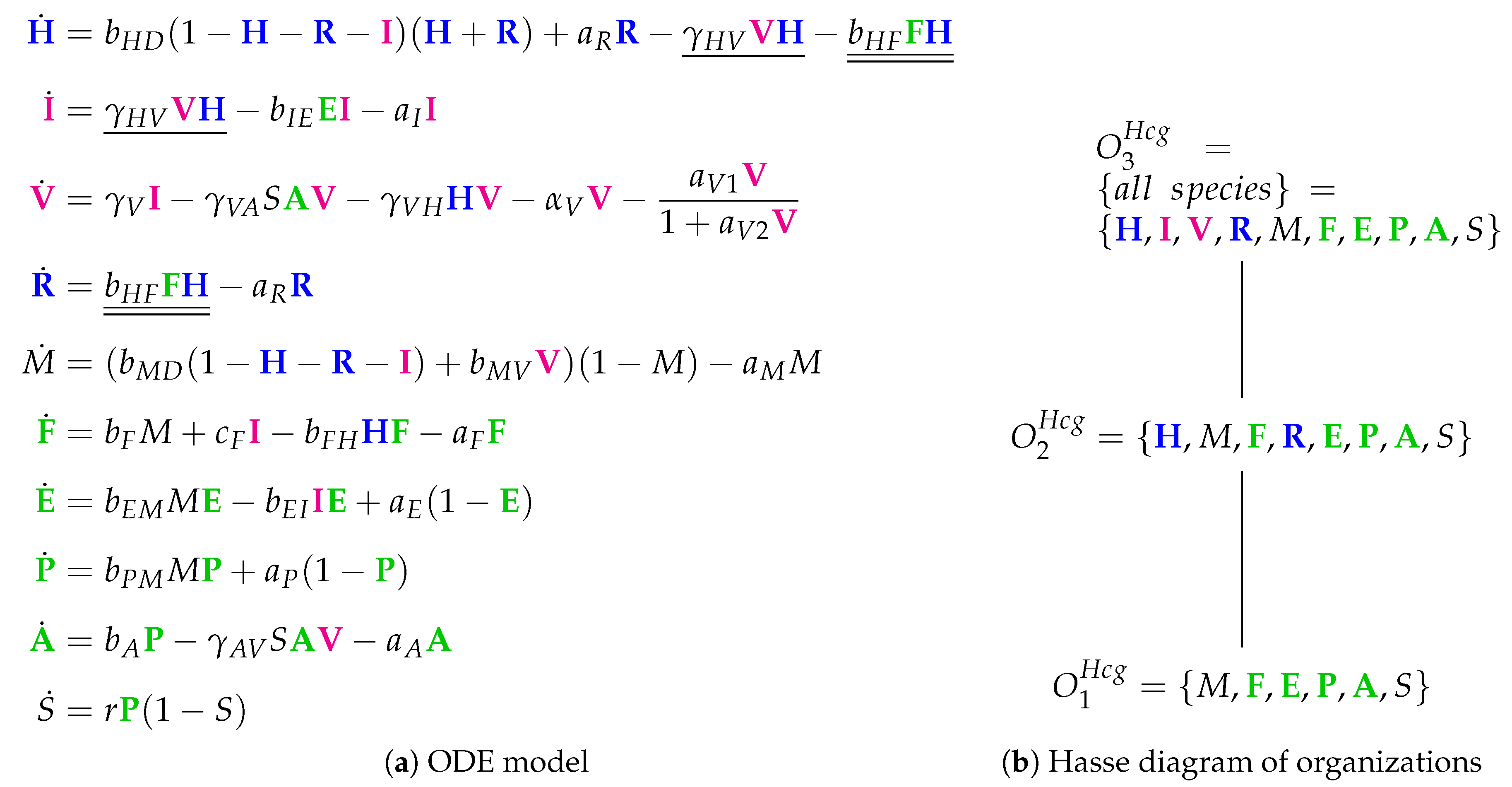{kind=link}
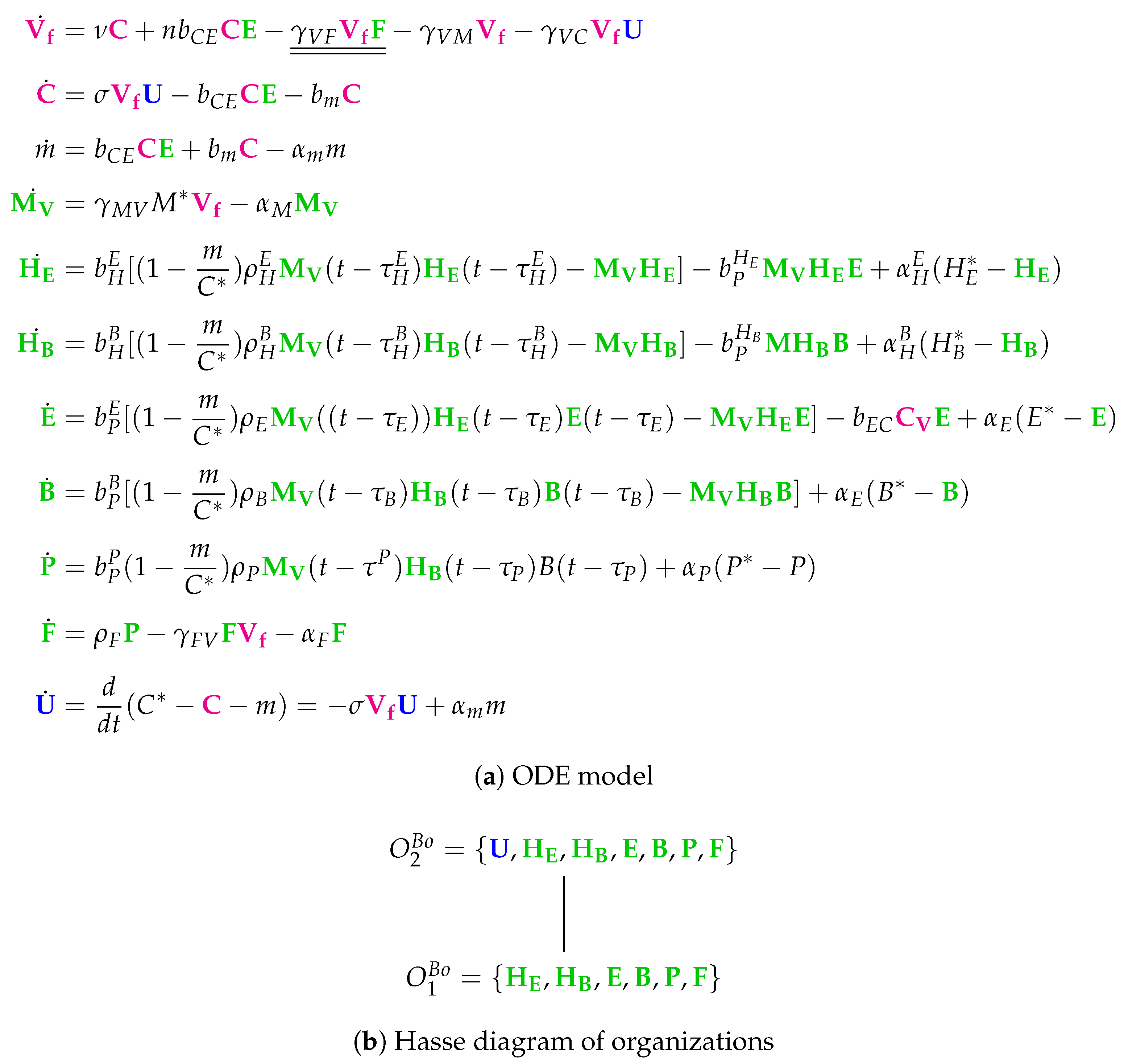{kind=link}
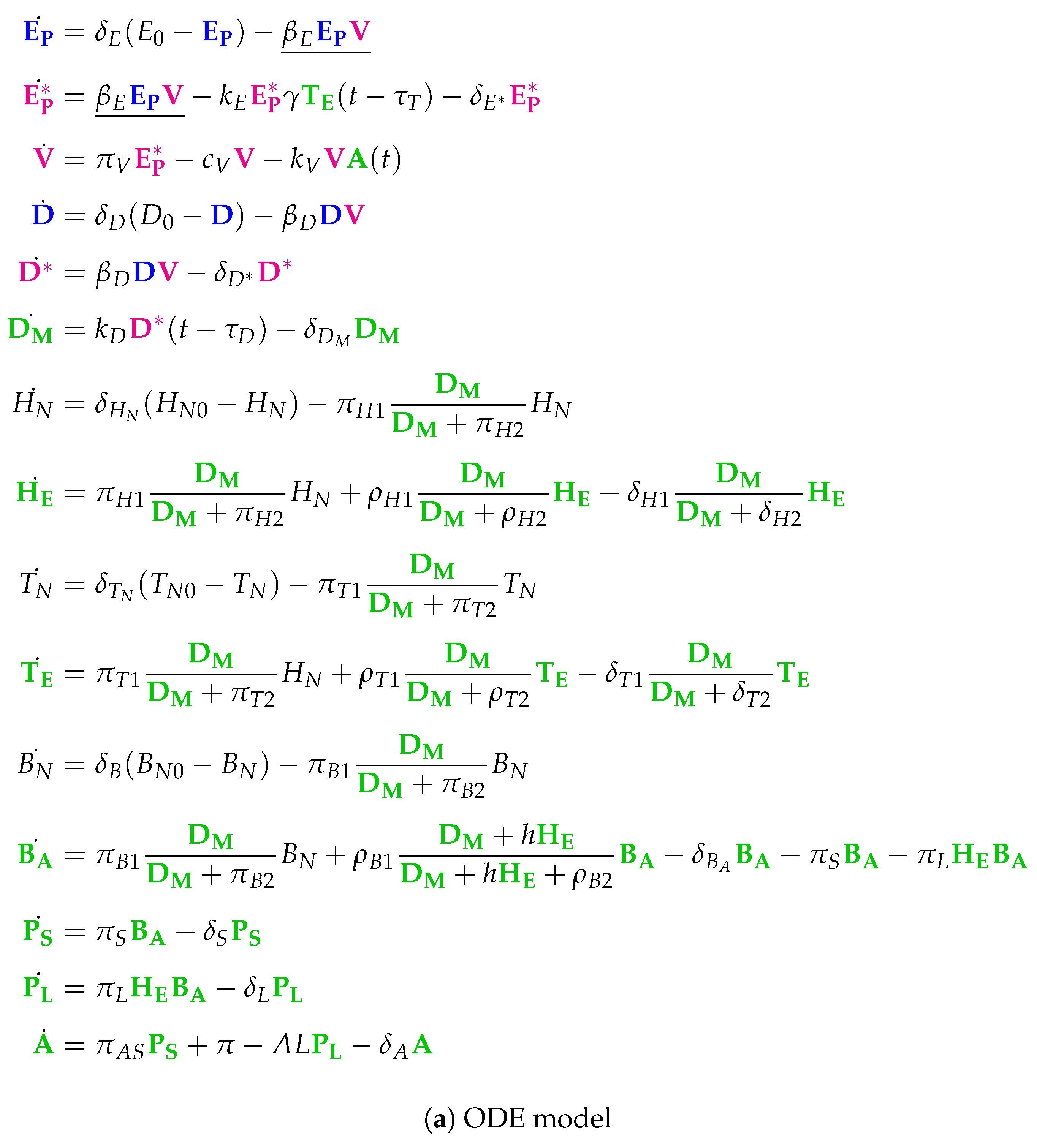{kind=link}
{kind=link}
{kind=link}
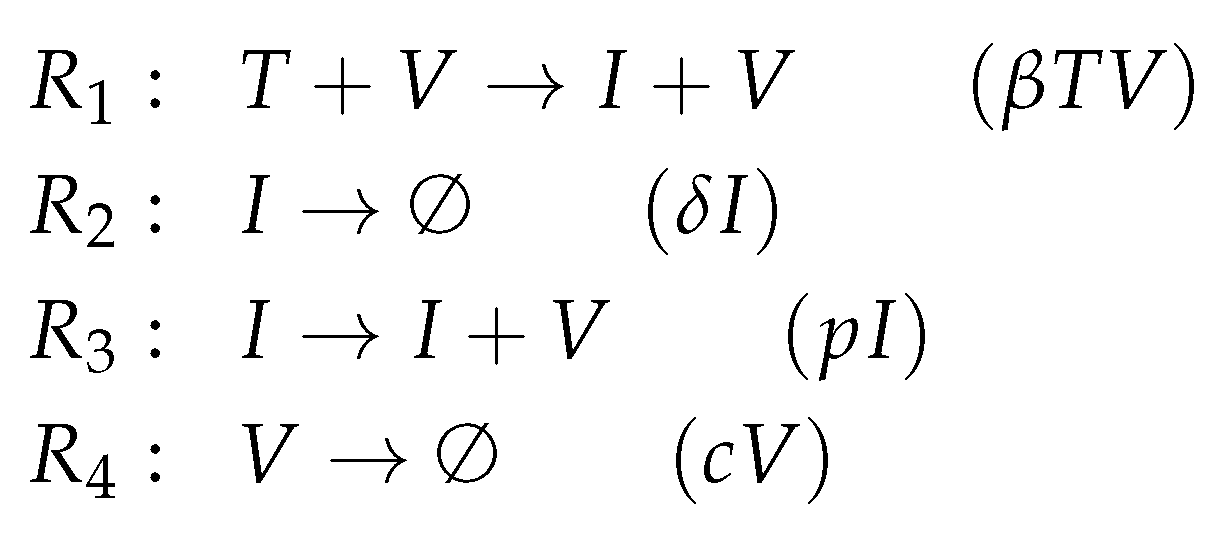{kind=link}
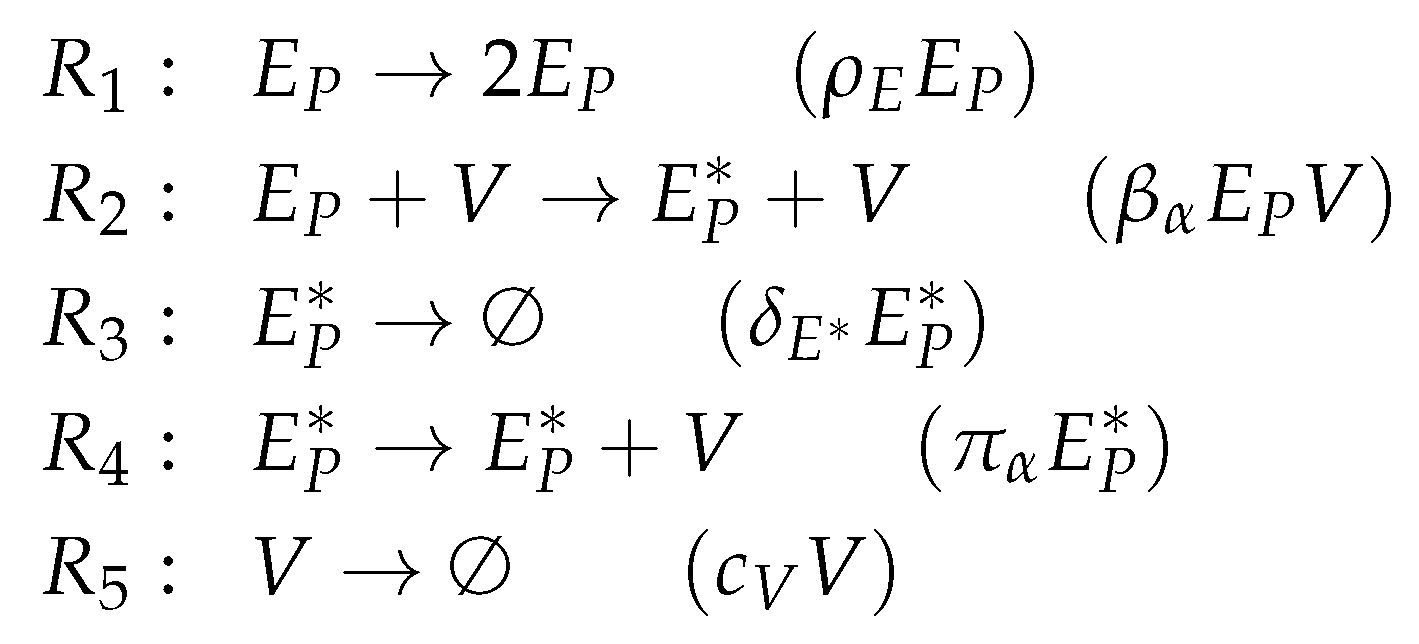{kind=link}
{kind=link}
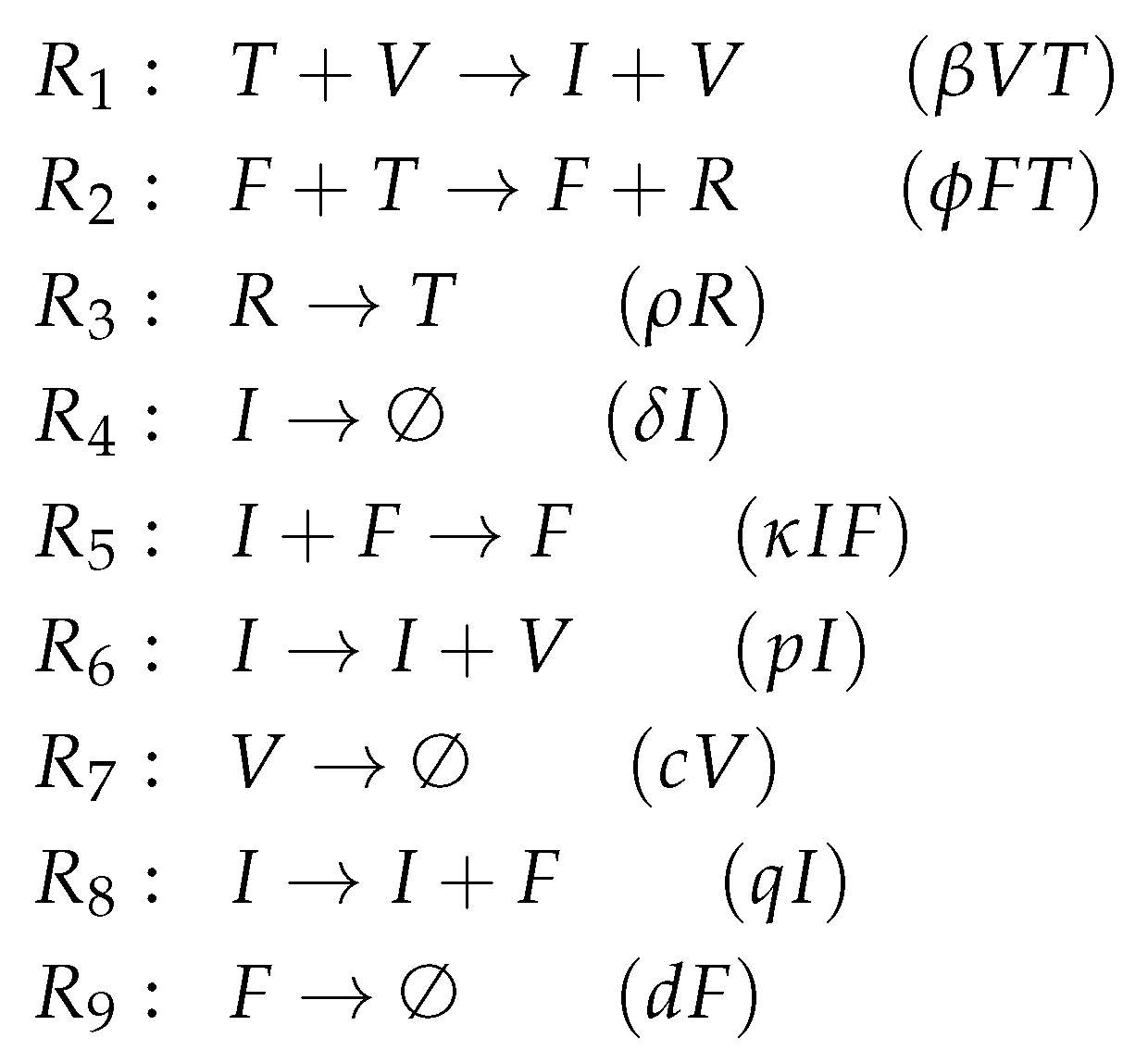{kind=link}
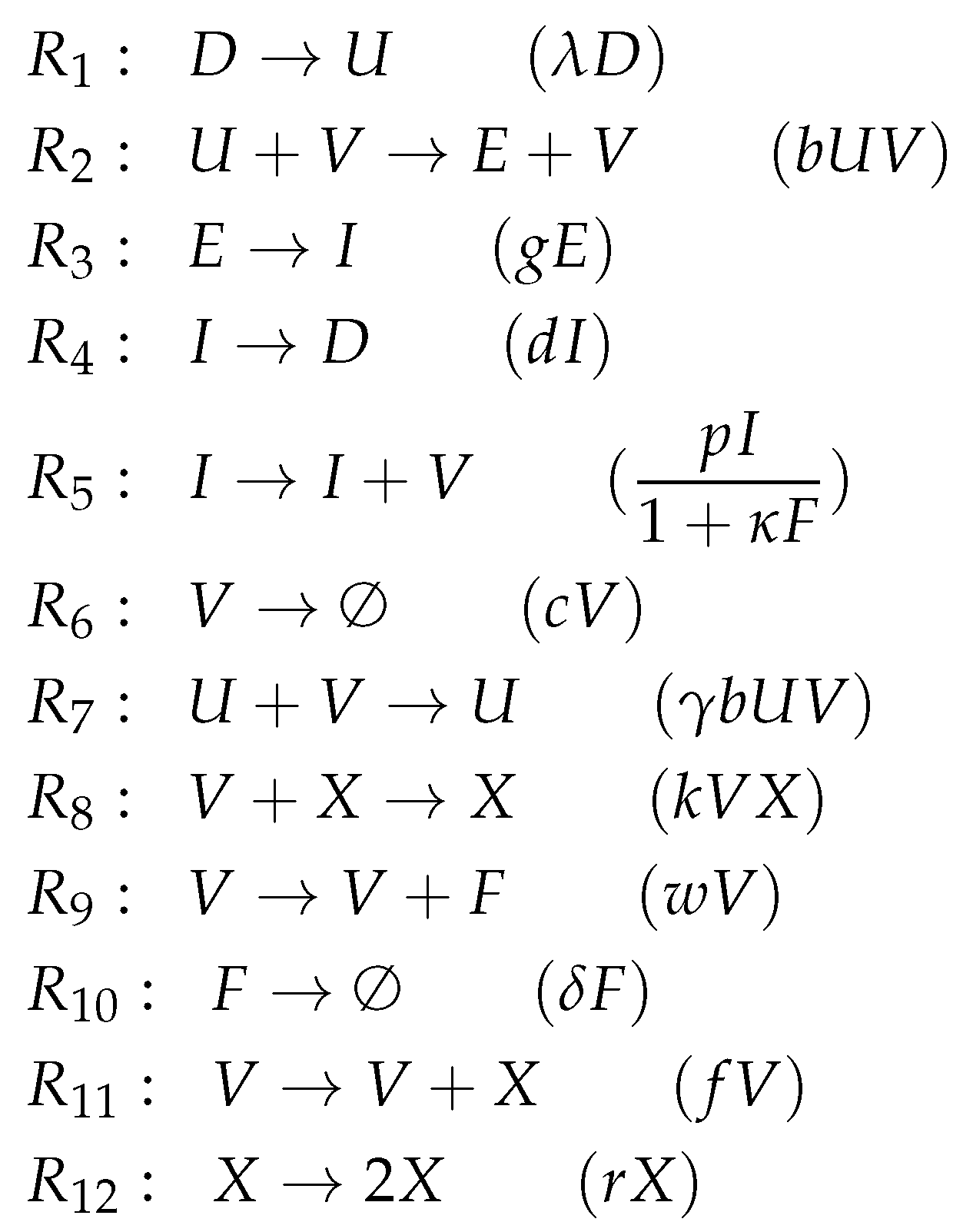{kind=link}
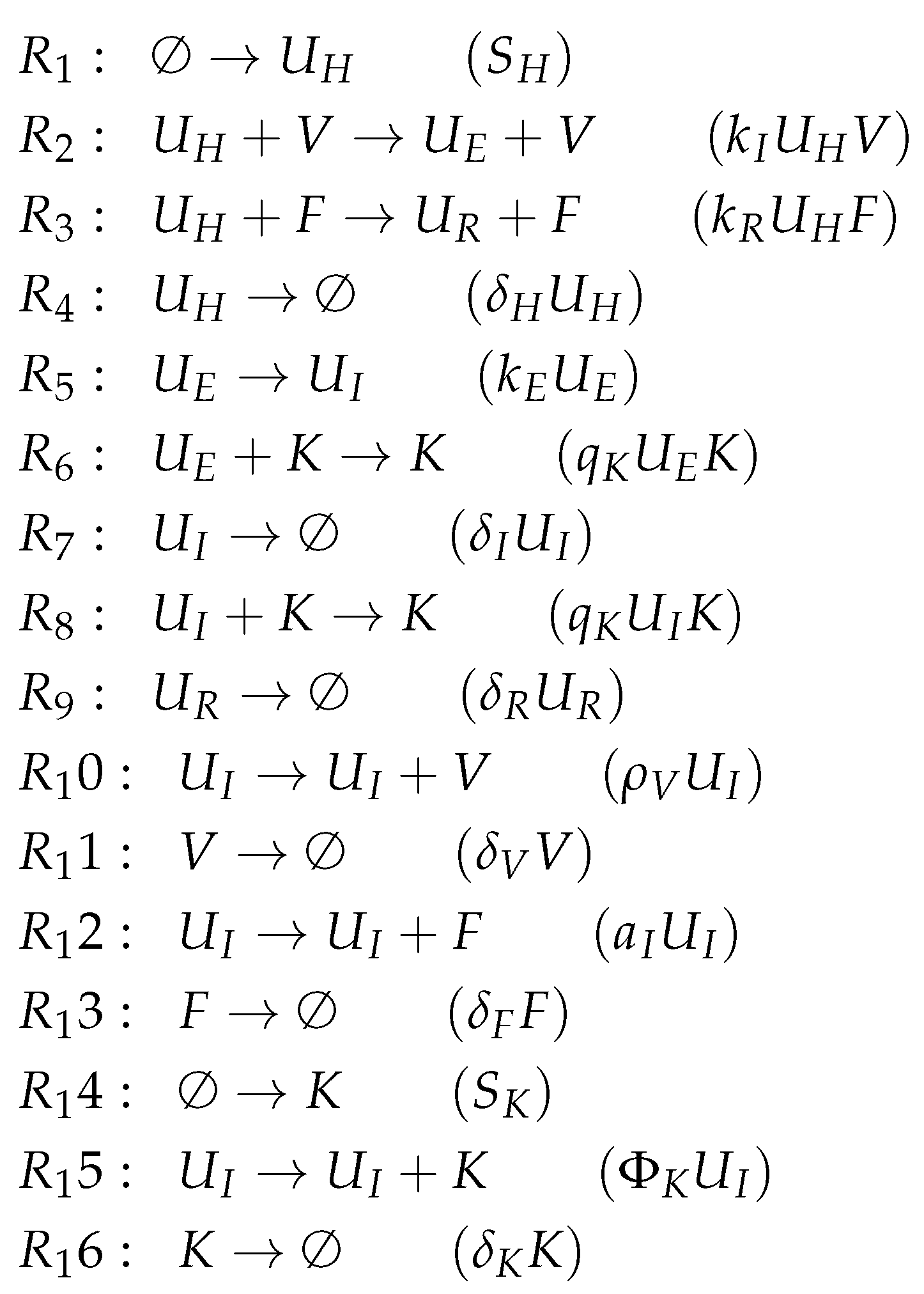{kind=link}
{kind=link}
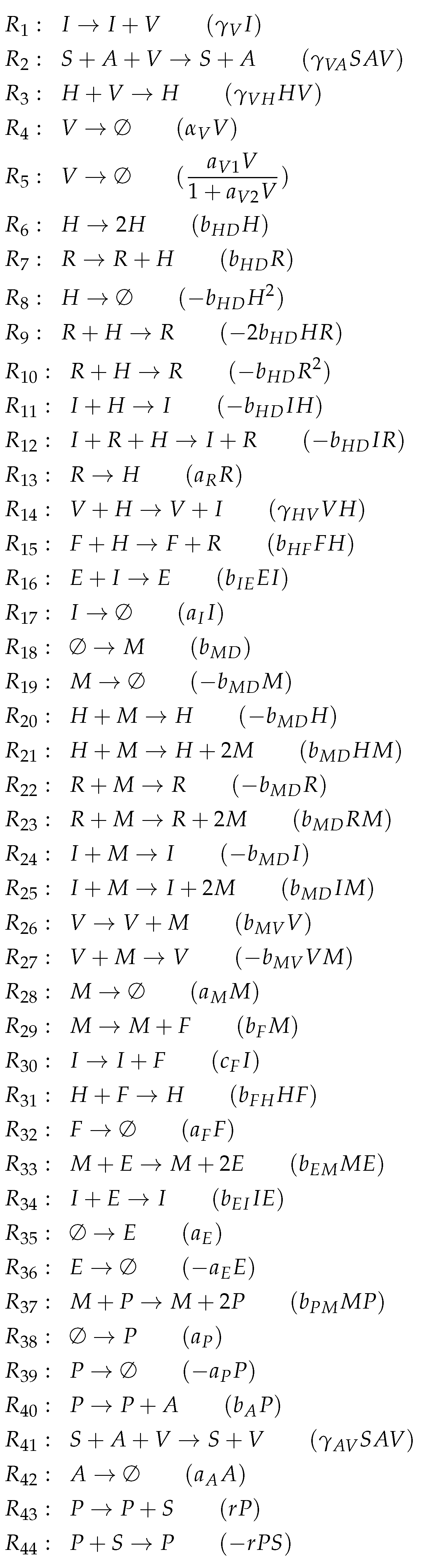{kind=link}
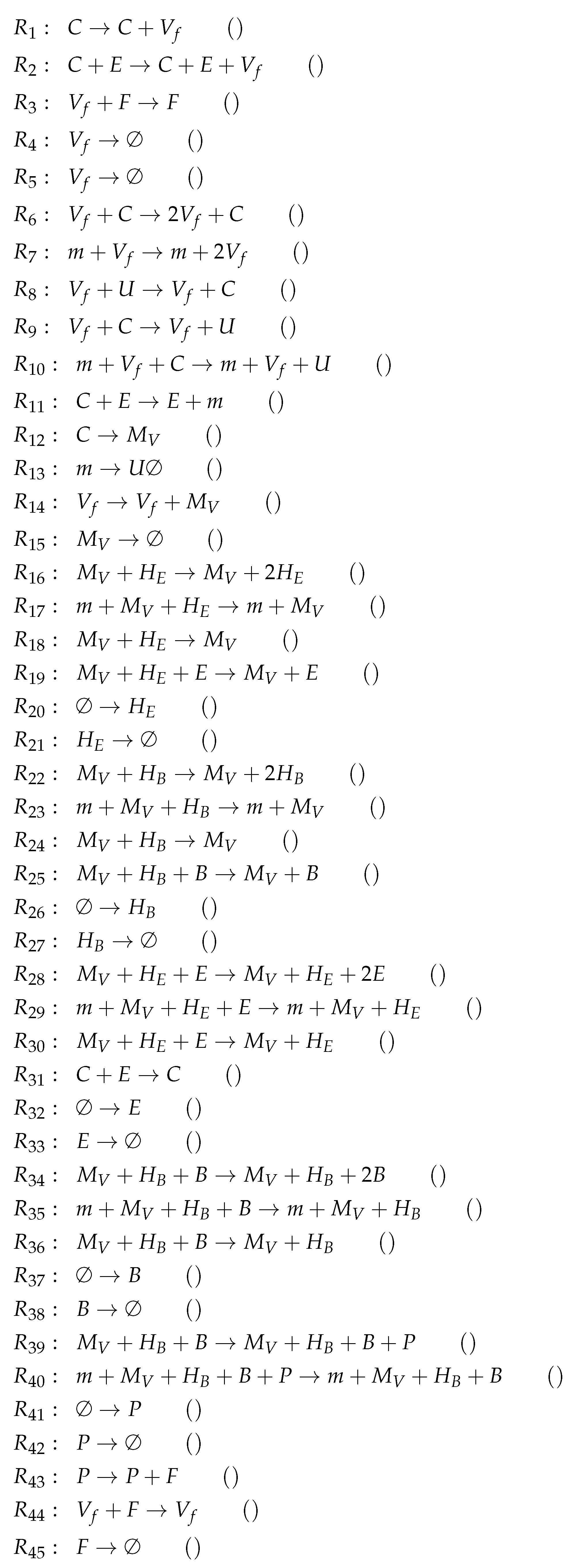{kind=link}
{kind=link}
| Model | Number of Variables | Number of Reactions | Number of Organizations | Organizations & Signature | |||
|---|---|---|---|---|---|---|---|
| Baccam [13] 2006 | 3 | 4 | 2 | ||||
| X | |||||||
| Miao [14] 2010 | 3 | 5 | 3 | ||||
| X | |||||||
| X | X | ||||||
| Baccam II [13] 2006 | 4 | 5 | 2 | ||||
| X | |||||||
| Pawelek [23] 2012 | 5 | 9 | 2 | ||||
| X | |||||||
| Smith [15] 2016 | 5 | 12 | 4 | ||||
| X | |||||||
| X | |||||||
| X | X | ||||||
| Handel [24] 2010 | 7 | 12 | 5 | ||||
| X | |||||||
| X | |||||||
| X | X | ||||||
| X | X | X | |||||
| Hernandez [26] 2012 | 7 | 16 | 2 | X | X | ||
| X | X | X | |||||
| Cao [27] 2015 | 7 | 26 | 3 | ||||
| X | |||||||
| X | X | X | |||||
| Saenz [28] 2010 | 8 | 12 | 4 | ||||
| , , | X | ||||||
| Hancioglu [29] 2007 | 10 | 44 | 3 | X | |||
| X | X | ||||||
| X | X | X | |||||
| Bocharov [30] 1994 | 10 | 45 | 2 | X | |||
| X | X | ||||||
| Lee [9] 2009 | 15 | 37 | 8 | X | |||
| , , , , | X | X | |||||
| , | X | X | X | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peter, S.; Hölzer, M.; Lamkiewicz, K.; di Fenizio, P.S.; Al Hwaeer, H.; Marz, M.; Schuster, S.; Dittrich, P.; Ibrahim, B. Structure and Hierarchy of Influenza Virus Models Revealed by Reaction Network Analysis. Viruses 2019, 11, 449. https://doi.org/10.3390/v11050449
Peter S, Hölzer M, Lamkiewicz K, di Fenizio PS, Al Hwaeer H, Marz M, Schuster S, Dittrich P, Ibrahim B. Structure and Hierarchy of Influenza Virus Models Revealed by Reaction Network Analysis. Viruses. 2019; 11(5):449. https://doi.org/10.3390/v11050449
Chicago/Turabian StylePeter, Stephan, Martin Hölzer, Kevin Lamkiewicz, Pietro Speroni di Fenizio, Hassan Al Hwaeer, Manja Marz, Stefan Schuster, Peter Dittrich, and Bashar Ibrahim. 2019. "Structure and Hierarchy of Influenza Virus Models Revealed by Reaction Network Analysis" Viruses 11, no. 5: 449. https://doi.org/10.3390/v11050449
APA StylePeter, S., Hölzer, M., Lamkiewicz, K., di Fenizio, P. S., Al Hwaeer, H., Marz, M., Schuster, S., Dittrich, P., & Ibrahim, B. (2019). Structure and Hierarchy of Influenza Virus Models Revealed by Reaction Network Analysis. Viruses, 11(5), 449. https://doi.org/10.3390/v11050449