Evaluation of Sequencing Library Preparation Protocols for Viral Metagenomic Analysis from Pristine Aquifer Groundwaters


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Library Construction and Sequencing
2.3. Sequencing Read Processing and Assembly
2.4. Viral Contig Recovery
2.5. Virome Diversity Measures and Comparison of Library Preparation Methods
2.6. Viral Taxonomic Assignment
2.7. Data Availability
3. Results
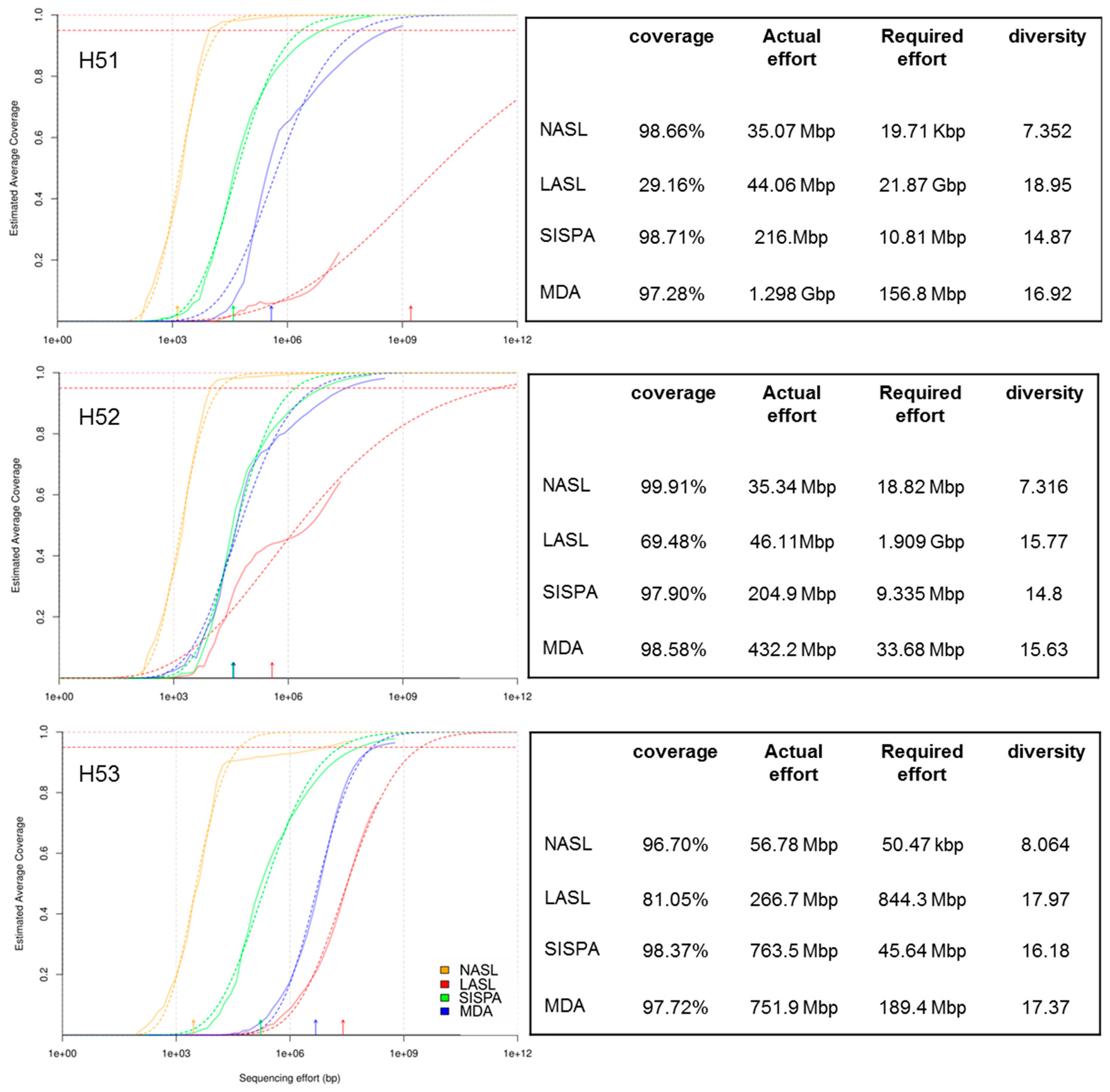
3.1. Raw Sequencing Output Statistics
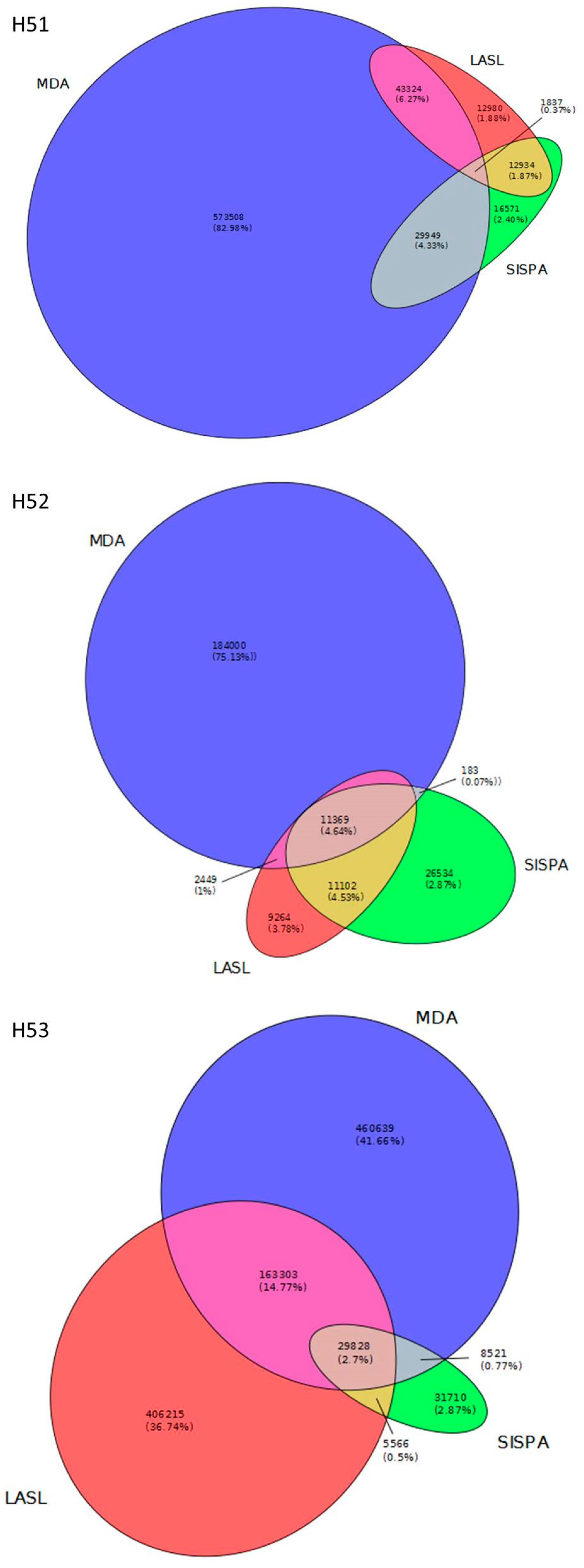
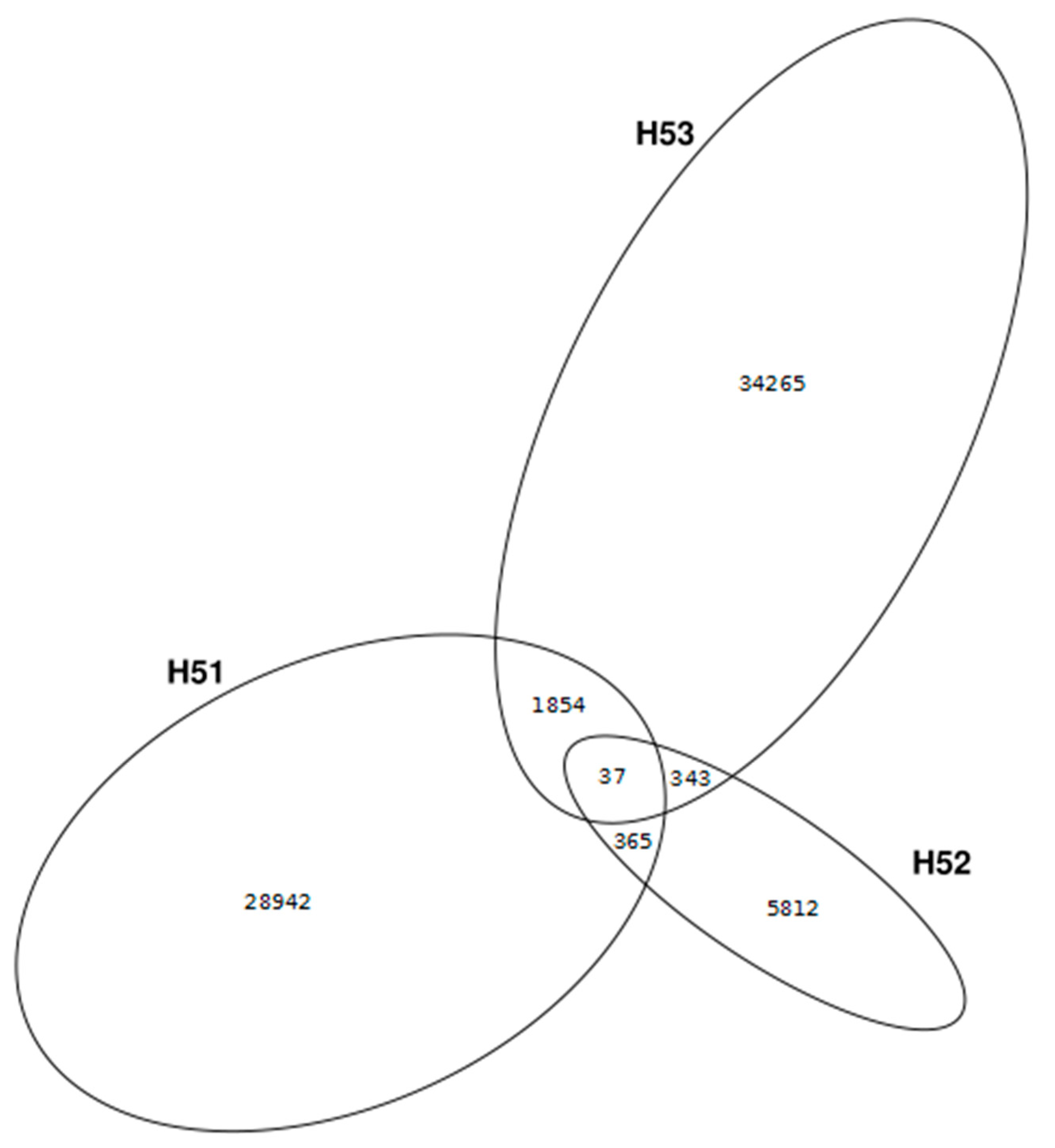
3.2. Data Set Comparison
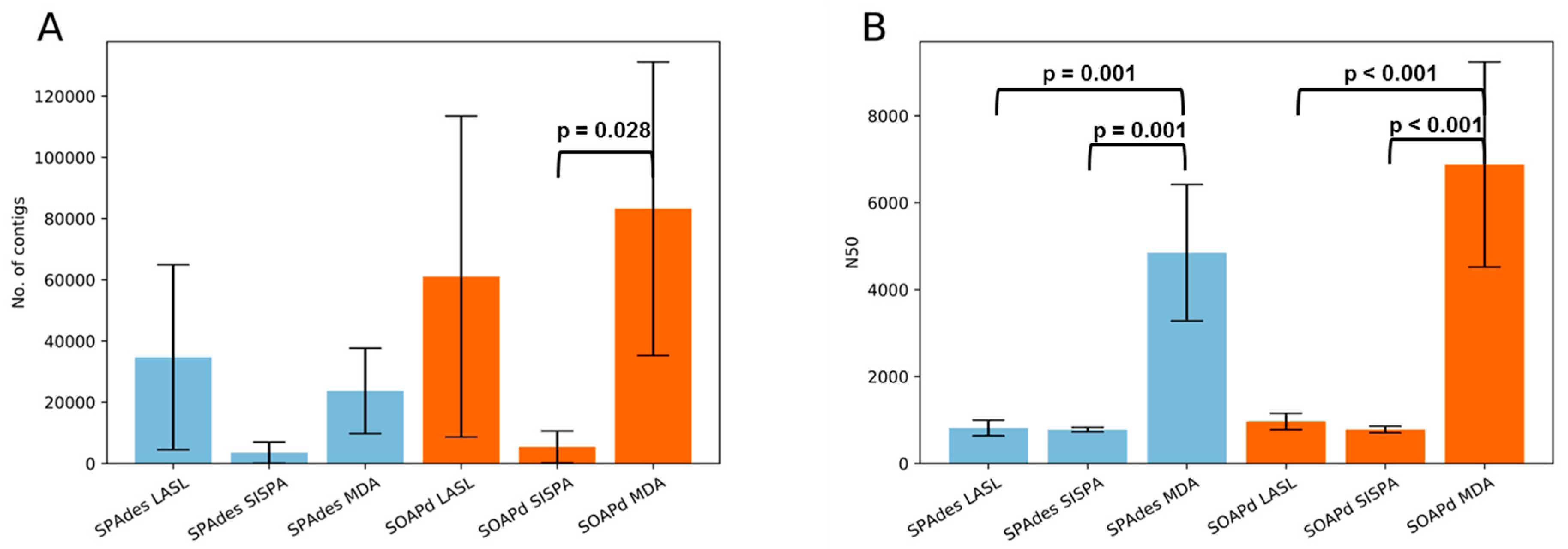
3.3. Assembly Statistics and Evaluation of Viral Identification Tools
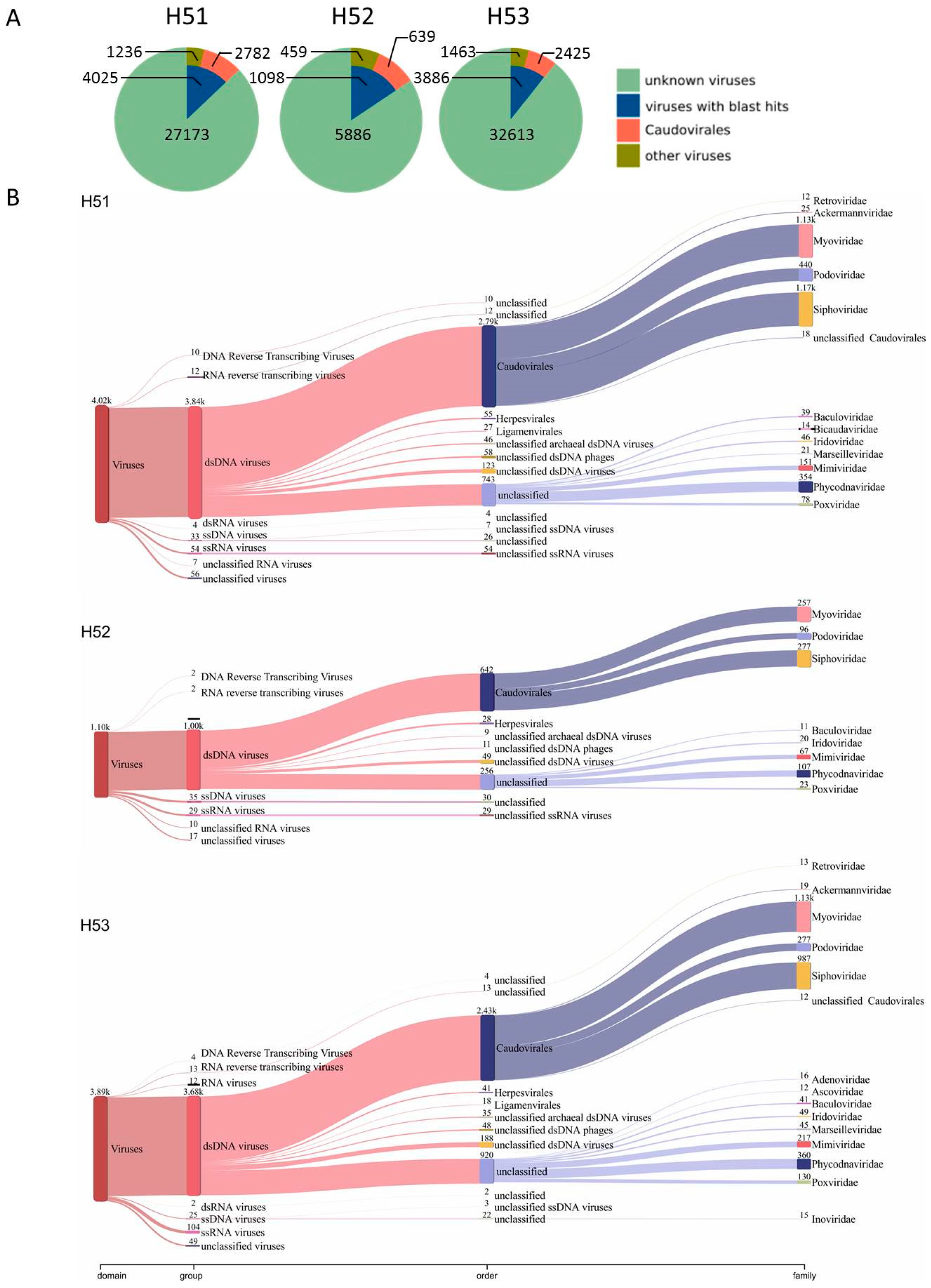
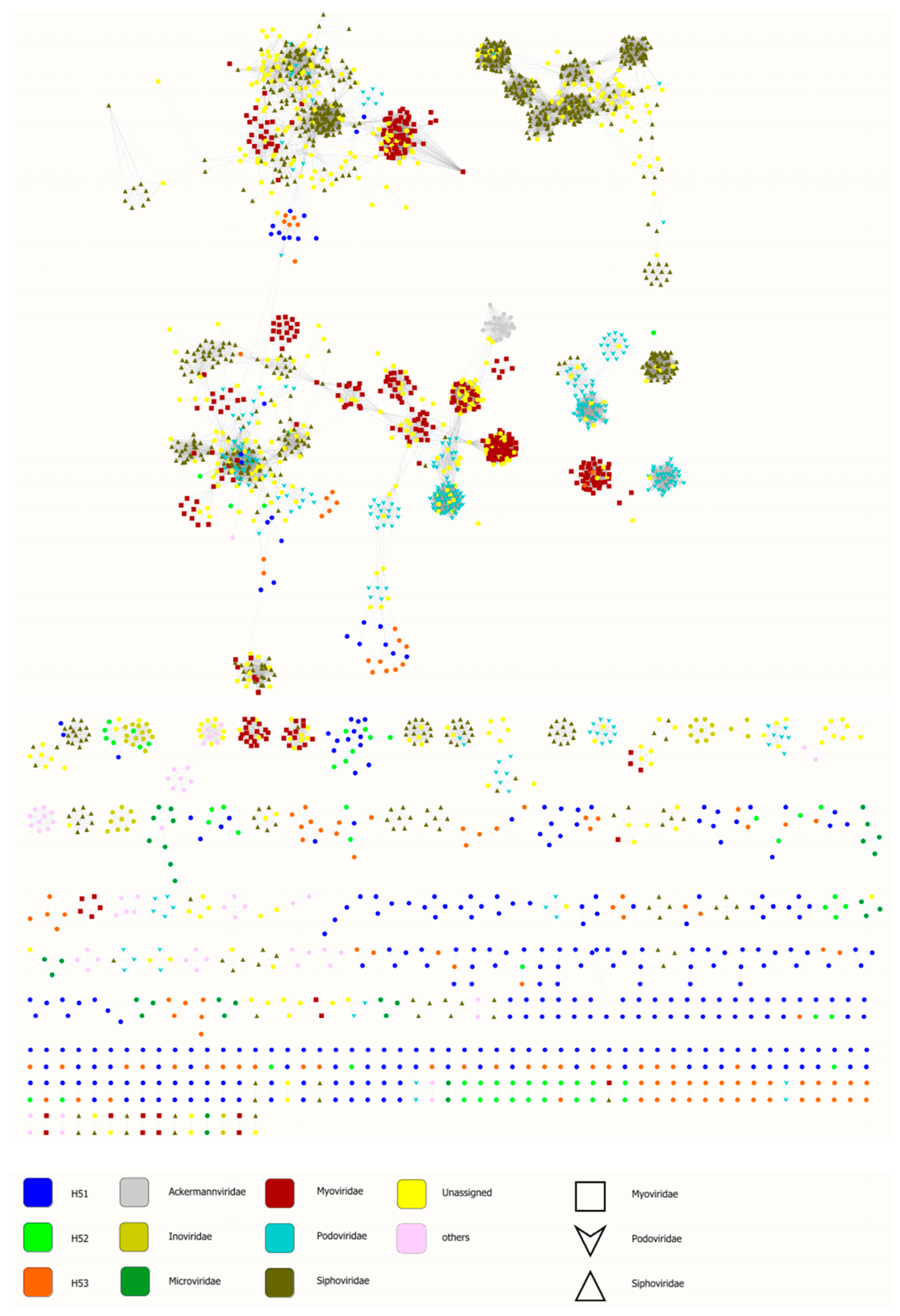
3.4. First Insights into Viral Taxonomic Composition of Hainich Groundwater
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Danielopol, D.L.; Pospisil, P.; Rouch, R. Biodiversity in groundwater: A large-scale view. Trends Ecol. Evol. 2000, 15, 223–224. [Google Scholar] [CrossRef]
- Griebler, C.; Avramov, M. Groundwater ecosystem services: A review. Freshw. Sci. 2015, 34, 355–367. [Google Scholar] [CrossRef]
- Griebler, C.; Lueders, T. Microbial biodiversity in groundwater ecosystems. Freshw. Biol. 2009, 54, 649–677. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses — major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M. Marine Viruses: Truth or Dare. Annu. Rev. Mar. Sci. 2011, 4, 425–448. [Google Scholar] [CrossRef]
- Daly, R.A.; Borton, M.A.; Wilkins, M.J.; Hoyt, D.W.; Kountz, D.J.; Wolfe, R.A.; Welch, S.A.; Marcus, D.N.; Trexler, R.V.; MacRae, J.D.; et al. Microbial metabolisms in a 2.5-km-deep ecosystem created by hydraulic fracturing in shales. Nat. Microbiol. 2016, 1, 16146. [Google Scholar] [CrossRef]
- Daly, R.A.; Roux, S.; Borton, M.A.; Morgan, D.M.; Johnston, M.D.; Booker, A.E.; Hoyt, D.W.; Meulia, T.; Wolfe, R.A.; Hanson, A.J.; et al. Viruses control dominant bacteria colonizing the terrestrial deep biosphere after hydraulic fracturing. Nat. Microbiol. 2019, 4, 352–361. [Google Scholar] [CrossRef]
- Kyle, J.E.; Eydal, H.S.C.; Ferris, F.G.; Pedersen, K. Viruses in granitic groundwater from 69 to 450 m depth of the Äspö hard rock laboratory, Sweden. ISME J. 2008, 2, 571. [Google Scholar] [CrossRef]
- Smith, R.J.; Jeffries, T.C.; Roudnew, B.; Seymour, J.R.; Fitch, A.J.; Simons, K.L.; Speck, P.G.; Newton, K.; Brown, M.H.; Mitchell, J.G. Confined aquifers as viral reservoirs. Environ. Microbiol. Rep. 2013, 5, 725–730. [Google Scholar] [CrossRef]
- Pan, D.; Watson, R.; Wang, D.; Tan, Z.H.; Snow, D.D.; Weber, K.A. Correlation between viral production and carbon mineralization under nitrate-reducing conditions in aquifer sediment. ISME J. 2014, 8, 1691–1703. [Google Scholar] [CrossRef]
- Wooley, J.C.; Ye, Y. Metagenomics: Facts and Artifacts, and Computational Challenges. J. Comput. Sci. Technol. 2009, 25, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Wilhartitz, I.C.; Kirschner, A.K.T.; Brussaard, C.P.D.; Fischer, U.R.; Wieltschnig, C.; Stadler, H.; Farnleitner, A.H. Dynamics of natural prokaryotes, viruses, and heterotrophic nanoflagellates in alpine karstic groundwater. Microbiol. Open 2013, 2, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Roudnew, B.; Lavery, T.J.; Seymour, J.R.; Smith, R.J.; Mitchell, J.G. Spatially varying complexity of bacterial and virus-like particle communities within an aquifer system. Aquat. Microb. Ecol. 2013, 68, 259–266. [Google Scholar] [CrossRef][Green Version]
- Ruby, J.G.; Bellare, P.; Derisi, J.L. PRICE: Software for the targeted assembly of components of (Meta) genomic sequence data. G3 Bethesda Md 2013, 3, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M. Challenges in the analysis of viral metagenomes. Virus Evol. 2016, 2, vew022. [Google Scholar] [CrossRef] [PubMed]
- Parras-Moltó, M.; Rodríguez-Galet, A.; Suárez-Rodríguez, P.; López-Bueno, A. Evaluation of bias induced by viral enrichment and random amplification protocols in metagenomic surveys of saliva DNA viruses. Microbiome 2018, 6, 119. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Bae, J.-W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef]
- Froussard, P. A random-PCR method (rPCR) to construct whole cDNA library from low amounts of RNA. Nucleic Acids Res. 1992, 20, 2900. [Google Scholar] [CrossRef] [PubMed]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; Depasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Dean, F.B.; Nelson, J.R.; Giesler, T.L.; Lasken, R.S. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001, 11, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, e368. [Google Scholar] [CrossRef] [PubMed]
- Henn, M.R.; Sullivan, M.B.; Stange-Thomann, N.; Osburne, M.S.; Berlin, A.M.; Kelly, L.; Yandava, C.; Kodira, C.; Zeng, Q.; Weiand, M.; et al. Analysis of high-throughput sequencing and annotation strategies for phage genomes. PloS ONE 2010, 5, e9083. [Google Scholar] [CrossRef] [PubMed]
- Solonenko, S.A.; Ignacio-Espinoza, J.C.; Alberti, A.; Cruaud, C.; Hallam, S.; Konstantinidis, K.; Tyson, G.; Wincker, P.; Sullivan, M.B. Sequencing platform and library preparation choices impact viral metagenomes. BMC Genom. 2013, 14, 320. [Google Scholar] [CrossRef] [PubMed]
- Duhaime, M.B.; Deng, L.; Poulos, B.T.; Sullivan, M.B. Towards quantitative metagenomics of wild viruses and other ultra-low concentration DNA samples: A rigorous assessment and optimization of the linker amplification method. Environ. Microbiol. 2012, 14, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Székely, A.J.; Breitbart, M. Single-stranded DNA phages: From early molecular biology tools to recent revolutions in environmental microbiology. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenck, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbart, M.; Sullivan, M.B. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016, 4, e2777. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef]
- Karlsson, O.E.; Belák, S.; Granberg, F. The Effect of Preprocessing by Sequence-Independent, Single-Primer Amplification (SISPA) on Metagenomic Detection of Viruses. Biosecurity Bioterrorism Biodefense Strategy Pract. Sci. 2013, 11, S227–S234. [Google Scholar] [CrossRef] [PubMed]
- Blanco, L.; Bernad, A.; Lázaro, J.M.; Martín, G.; Garmendia, C.; Salas, M. Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical mode of DNA replication. J. Biol. Chem. 1989, 264, 8935–8940. [Google Scholar] [PubMed]
- Lasken, R.S.; Stockwell, T.B. Mechanism of chimera formation during the Multiple Displacement Amplification reaction. BMC Biotechnol. 2007, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Martiny, A.C.; Reppas, N.B.; Barry, K.W.; Malek, J.; Chisholm, S.W.; Church, G.M. Sequencing genomes from single cells by polymerase cloning. Nat. Biotechnol. 2006, 24, 680. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Chang, H.-W.; Nam, Y.-D.; Roh, S.W.; Kim, M.-S.; Sung, Y.; Jeon, C.O.; Oh, H.-M.; Bae, J.-W. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl. Environ. Microbiol. 2008, 74, 5975–5985. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Low, S.; Woodcroft, B.J.; Raina, J.-B.; Skarshewski, A.; Le, X.H.; Butler, M.K.; Stocker, R.; Seymour, J.; Tyson, G.W.; et al. Validation of picogram- and femtogram-input DNA libraries for microscale metagenomics. PeerJ 2016, 4, e2486. [Google Scholar] [CrossRef]
- Bowers, R.M.; Clum, A.; Tice, H.; Lim, J.; Singh, K.; Ciobanu, D.; Ngan, C.Y.; Cheng, J.-F.; Tringe, S.G.; Woyke, T. Impact of library preparation protocols and template quantity on the metagenomic reconstruction of a mock microbial community. BMC Genom. 2015, 16, 856. [Google Scholar] [CrossRef]
- Küsel, K.; Totsche, K.U.; Trumbore, S.E.; Lehmann, R.; Steinhäuser, C.; Herrmann, M. How Deep Can Surface Signals Be Traced in the Critical Zone? Merging Biodiversity with Biogeochemistry Research in a Central German Muschelkalk Landscape. Front. Earth Sci. 2016, 4, 32. [Google Scholar] [CrossRef]
- Kohlhepp, B.; Lehmann, R.; Seeber, P.; Küsel, K.; Trumbore, S.E.; Totsche, K.U. Aquifer configuration and geostructural links control the groundwater quality in thin-bedded carbonate–siliciclastic alternations of the Hainich CZE, central Germany. Hydrol. Earth Syst. Sci. 2017, 21, 6091–6116. [Google Scholar] [CrossRef]
- Kumar, S.; Herrmann, M.; Thamdrup, B.; Schwab, V.F.; Geesink, P.; Trumbore, S.E.; Totsche, K.-U.; Küsel, K. Nitrogen Loss from Pristine Carbonate-Rock Aquifers of the Hainich Critical Zone Exploratory (Germany) Is Primarily Driven by Chemolithoautotrophic Anammox Processes. Front. Microbiol. 2017, 8, 1951. [Google Scholar] [CrossRef]
- Palacios, G.; Quan, P.-L.; Jabado, O.J.; Conlan, S.; Hirschberg, D.L.; Liu, Y.; Zhai, J.; Renwick, N.; Hui, J.; Hegyi, H. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg. Infect. Dis. 2007, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef]
- Hölzer, M.; Marz, M. De novo transcriptome assembly: A comprehensive cross-species comparison of short-read RNA-Seq assemblers. GigaScience 2019, 8, giz039. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef]
- Song, L.; Florea, L.; Langmead, B. Lighter: Fast and memory-efficient sequencing error correction without counting. Genome Biol. 2014, 15, 509. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-r, L.M.; Konstantinidis, K.T. Nonpareil: A redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 2013, 30, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.M.; Konstantinidis, K.T. Estimating coverage in metagenomic data sets and why it matters. ISME J. 2014, 8, 2349. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-R, L.M.; Gunturu, S.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. Nonpareil 3: Fast Estimation of Metagenomic Coverage and Sequence Diversity. MSystems 2018, 3, e00039-18. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R. Available online: http://www.rstudio.com (accessed on 27 May 2019).
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 119. [Google Scholar] [CrossRef]
- Boratyn, G.M.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 12. [Google Scholar] [CrossRef]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.-Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Youens-Clark, K.; Roux, S.; Hurwitz, B.L.; Sullivan, M.B. iVirus: Facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. Isme J. 2016, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.S.; Baker, B.J.; Thomas, B.C.; Singer, S.W.; Banfield, J.F. EMIRGE: Reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011, 12, R44. [Google Scholar] [CrossRef]
- Emerson, J.B.; Roux, S.; Brum, J.R.; Bolduc, B.; Woodcroft, B.J.; Jang, H.B.; Singleton, C.M.; Solden, L.M.; Naas, A.E.; Boyd, J.A.; et al. Host-linked soil viral ecology along a permafrost thaw gradient. Nat. Microbiol. 2018, 3, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689. [Google Scholar] [CrossRef] [PubMed]
- Reisser, W. The Hidden Life of Algae Underground. In Algae and Cyanobacteria in Extreme Environments; Seckbach, J., Ed.; Springer Netherlands: Dordrecht, The Netherlands, 2007; pp. 47–58. ISBN 978-1-4020-6112-7. [Google Scholar]
- Rosario, K.; Duffy, S.; Breitbart, M. Diverse circovirus-like genome architectures revealed by environmental metagenomics. J. Gen. Virol. 2009, 90, 2418–2424. [Google Scholar] [CrossRef]
- Tucker, K.P.; Parsons, R.; Symonds, E.M.; Breitbart, M. Diversity and distribution of single-stranded DNA phages in the North Atlantic Ocean. Isme J. 2010, 5, 822. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Salzberg, S.L. Pavian: Interactive analysis of metagenomics data for microbiomics and pathogen identification. bioRxiv 2016. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef] [PubMed]
- Wegner, C.-E.; Gaspar, M.; Geesink, P.; Herrmann, M.; Marz, M.; Küsel, K. Biogeochemical Regimes in Shallow Aquifers Reflect the Metabolic Coupling of the Elements Nitrogen, Sulfur, and Carbon. Appl. Environ. Microbiol. 2019, 85, e02346-18. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.E.; Brazelton, W.J.; Baross, J.A. Is the genetic landscape of the deep subsurface biosphere affected by viruses? Front. Microbiol. 2011, 2, 219. [Google Scholar] [CrossRef] [PubMed]
- Hölzer, M.; Marz, M. Chapter Nine—Software Dedicated to Virus Sequence Analysis “Bioinformatics Goes Viral.”. In Advances in Virus Research; Beer, M., Höper, D., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 99, pp. 233–257. ISBN 0065-3527. [Google Scholar]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Ponsero, A.; Thornton, J.; U’Ren, J.M. Phage hunters: Computational strategies for finding phages in large-scale ‘omics datasets. Virus Res. 2018, 244, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Bzhalava, Z.; Tampuu, A.; Bała, P.; Vicente, R.; Dillner, J. Machine Learning for detection of viral sequences in human metagenomic datasets. BMC Bioinform. 2018, 19, 336. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in Aquatic Ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Sullivan, M.B. The Pacific Ocean Virome (POV): A Marine Viral Metagenomic Dataset and Associated Protein Clusters for Quantitative Viral Ecology. PLoS ONE 2013, 8, e57355. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef]
- Earl, P.L.; Jones, E.V.; Moss, B. Homology between DNA polymerases of poxviruses, herpesviruses, and adenoviruses: Nucleotide sequence of the vaccinia virus DNA polymerase gene. Proc. Natl. Acad. Sci. USA 1986, 83, 3659–3663. [Google Scholar] [CrossRef]
- Villarreal, L.P.; DeFilippis, V.R. A Hypothesis for DNA Viruses as the Origin of Eukaryotic Replication Proteins. J. Virol. 2000, 74, 7079. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Raw Reads | ||||||||
| Library Preparation | NASL | LASL | SISPA | MDA | ||||
| NASL | n/a | >0.05 | 0.008 | 0.002 | ||||
| LASL | n/a | >0.05 | 0.023 | |||||
| SISPA | n/a | >0.05 | ||||||
| MDA | n/a | |||||||
| Clusters at 90% Read Identity | ||||||||
| Relative proportion | Number of clusters | |||||||
| Library Preparation | NASL | LASL | SISPA | MDA | NASL | LASL | SISPA | MDA |
| NASL | n/a | <0.001 | >0.05 | >0.05 | n/a | 0.018 | >0.05 | 0.008 |
| LASL | n/a | <0.001 | <0.001 | n/a | >0.05 | >0.05 | ||
| SISPA | n/a | >0.05 | n/a | >0.05 | ||||
| MDA | n/a | n/a | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kallies, R.; Hölzer, M.; Brizola Toscan, R.; Nunes da Rocha, U.; Anders, J.; Marz, M.; Chatzinotas, A. Evaluation of Sequencing Library Preparation Protocols for Viral Metagenomic Analysis from Pristine Aquifer Groundwaters. Viruses 2019, 11, 484. https://doi.org/10.3390/v11060484
Kallies R, Hölzer M, Brizola Toscan R, Nunes da Rocha U, Anders J, Marz M, Chatzinotas A. Evaluation of Sequencing Library Preparation Protocols for Viral Metagenomic Analysis from Pristine Aquifer Groundwaters. Viruses. 2019; 11(6):484. https://doi.org/10.3390/v11060484
Chicago/Turabian StyleKallies, René, Martin Hölzer, Rodolfo Brizola Toscan, Ulisses Nunes da Rocha, John Anders, Manja Marz, and Antonis Chatzinotas. 2019. "Evaluation of Sequencing Library Preparation Protocols for Viral Metagenomic Analysis from Pristine Aquifer Groundwaters" Viruses 11, no. 6: 484. https://doi.org/10.3390/v11060484
APA StyleKallies, R., Hölzer, M., Brizola Toscan, R., Nunes da Rocha, U., Anders, J., Marz, M., & Chatzinotas, A. (2019). Evaluation of Sequencing Library Preparation Protocols for Viral Metagenomic Analysis from Pristine Aquifer Groundwaters. Viruses, 11(6), 484. https://doi.org/10.3390/v11060484