The Utility of Data Transformation for Alignment, De Novo Assembly and Classification of Short Read Virus Sequences

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
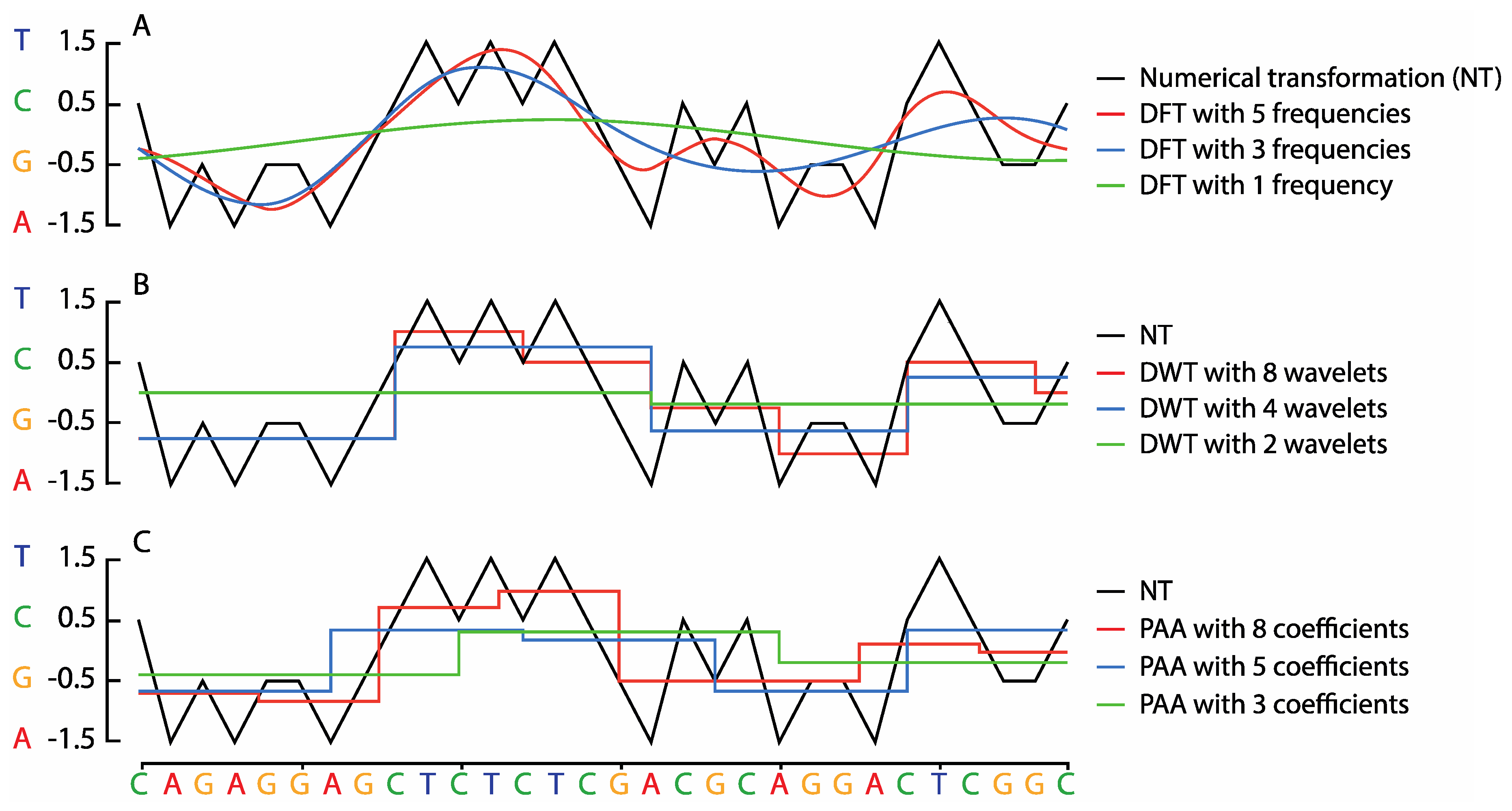
2.1. Symbolic to Numeric Sequence Representations
2.2. Sequence Transformation
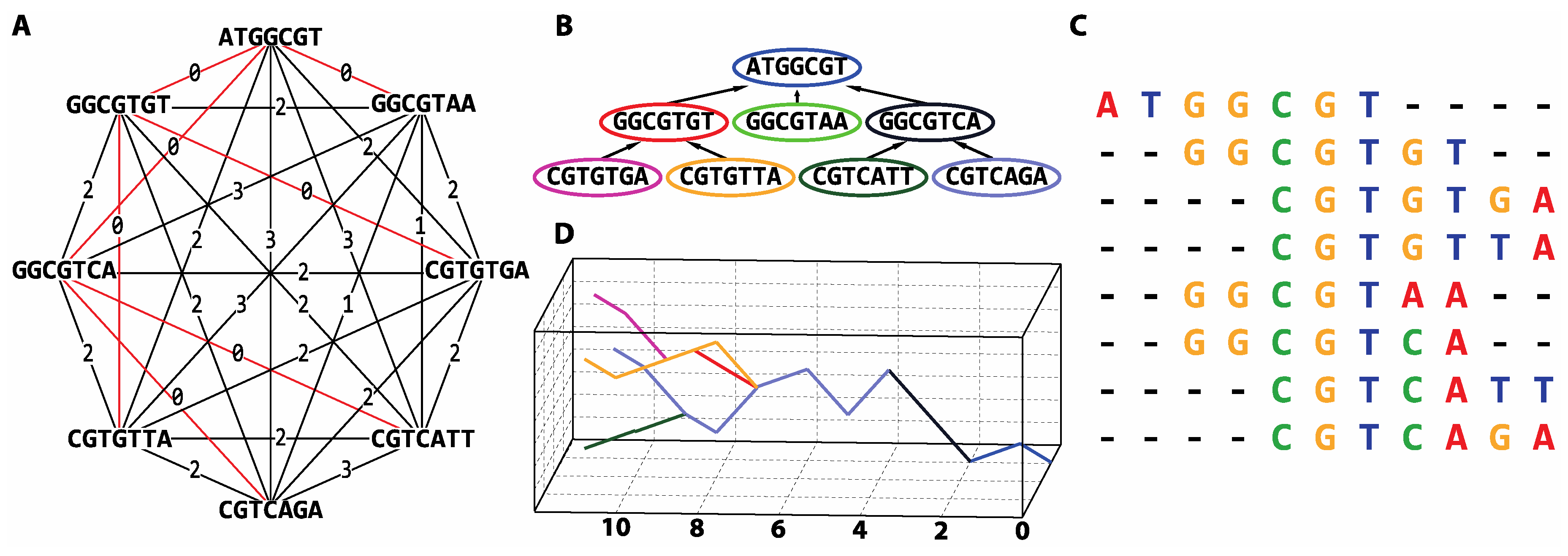
2.3. Similarity Search Approaches for Sequential Data
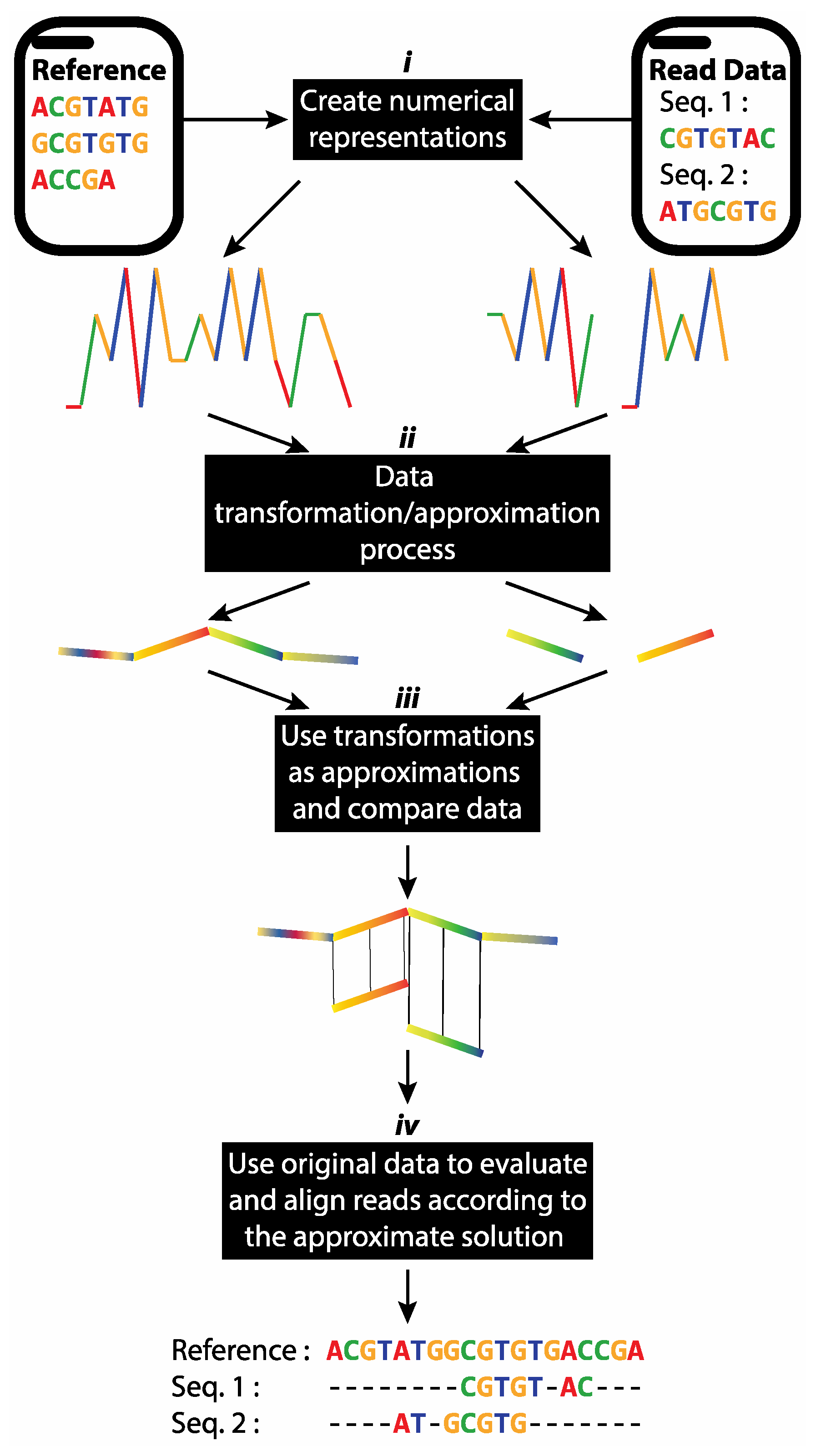
2.4. Proposed Short Reads Processing Methodology
2.5. Data
2.6. Classification and Alignment Evaluation
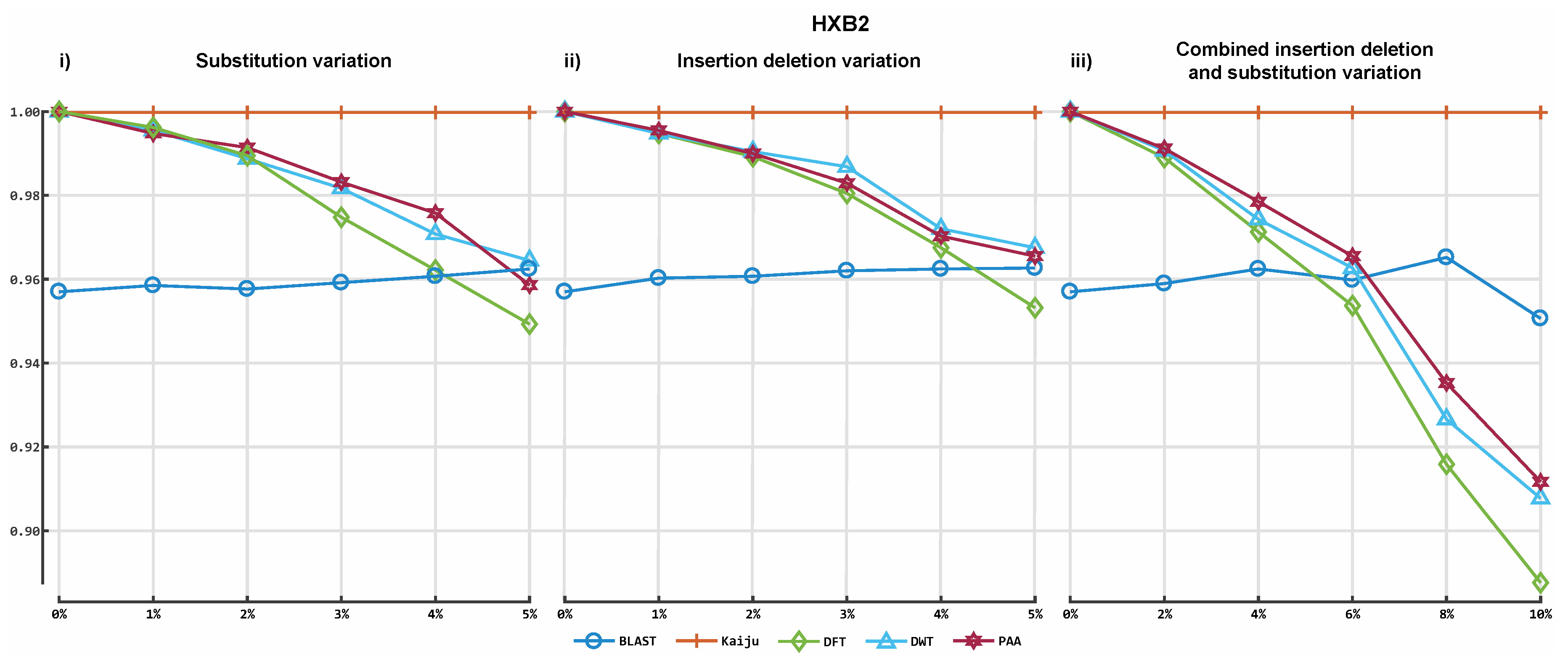
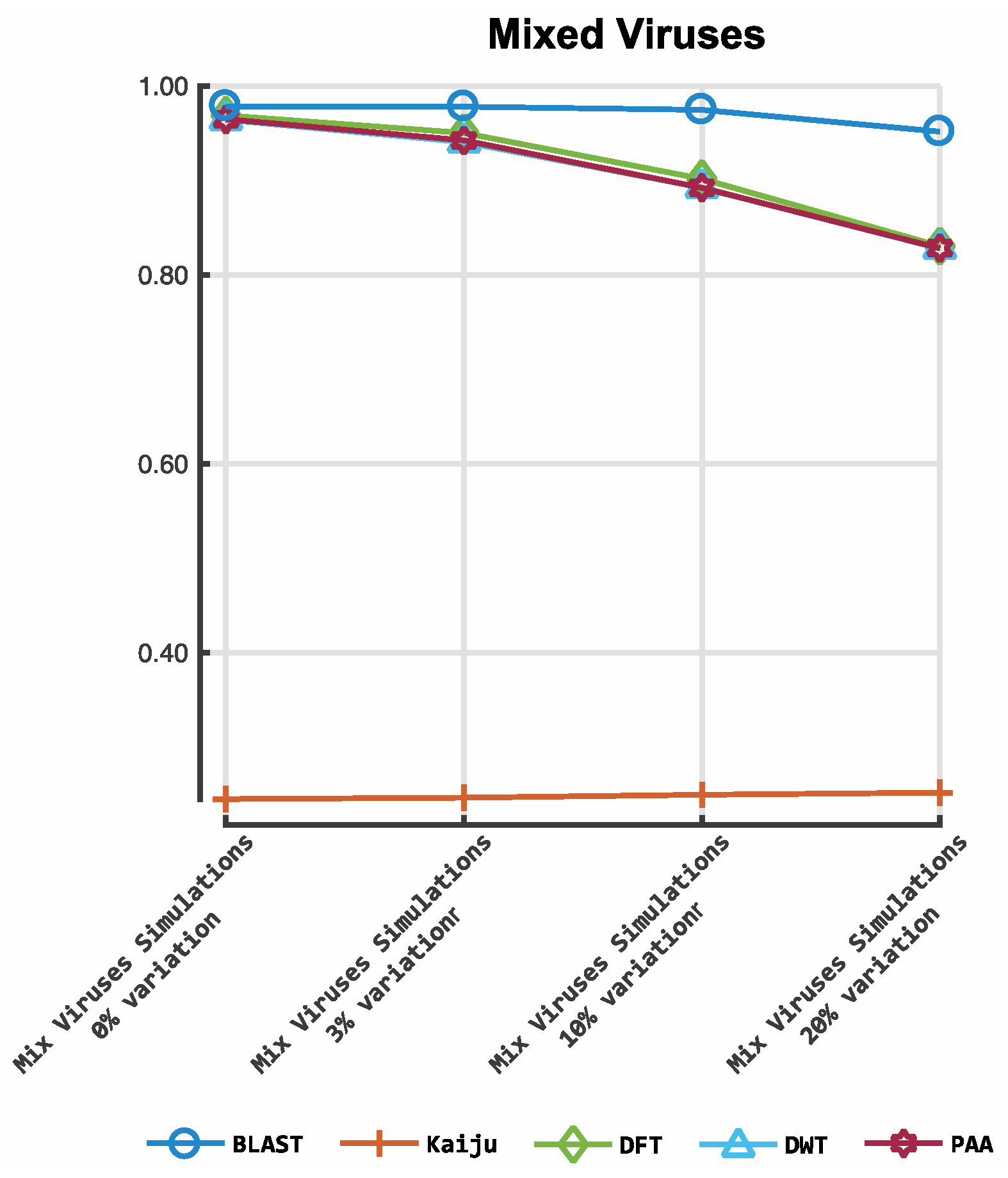
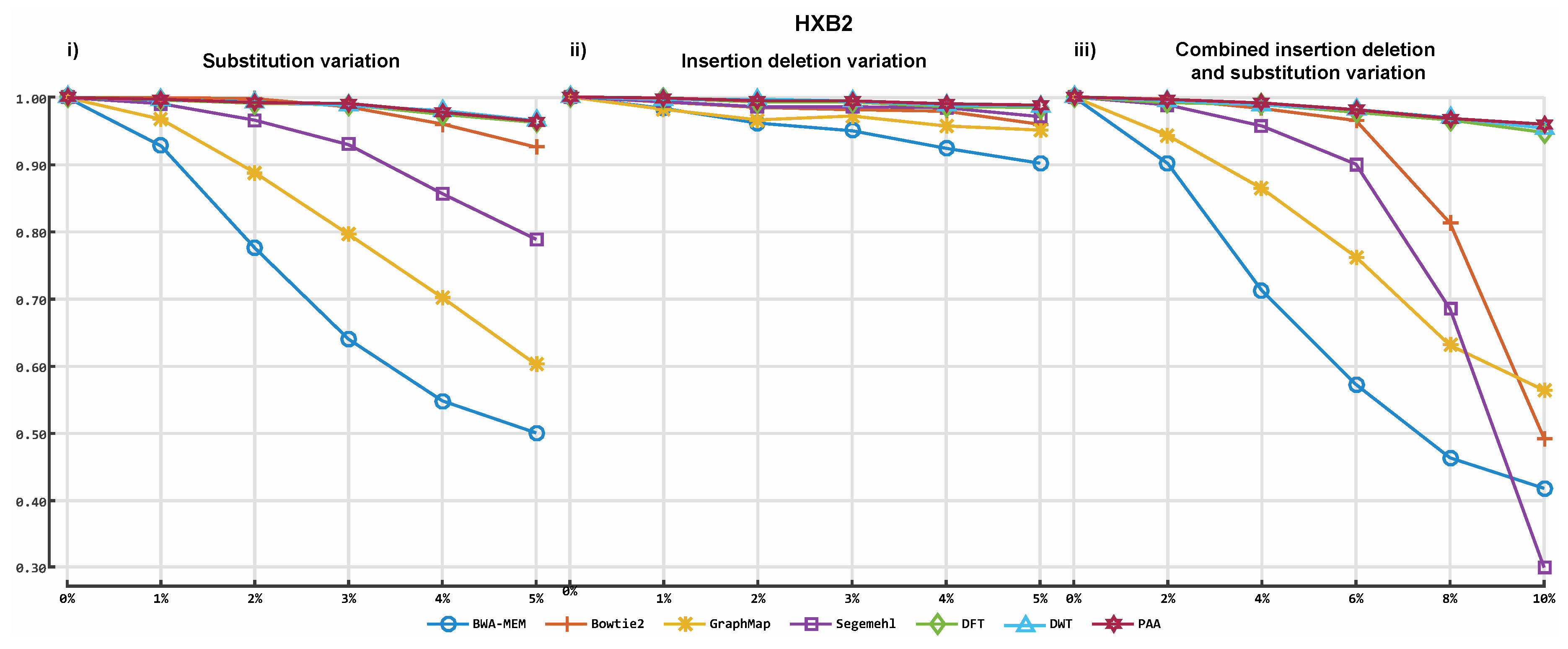
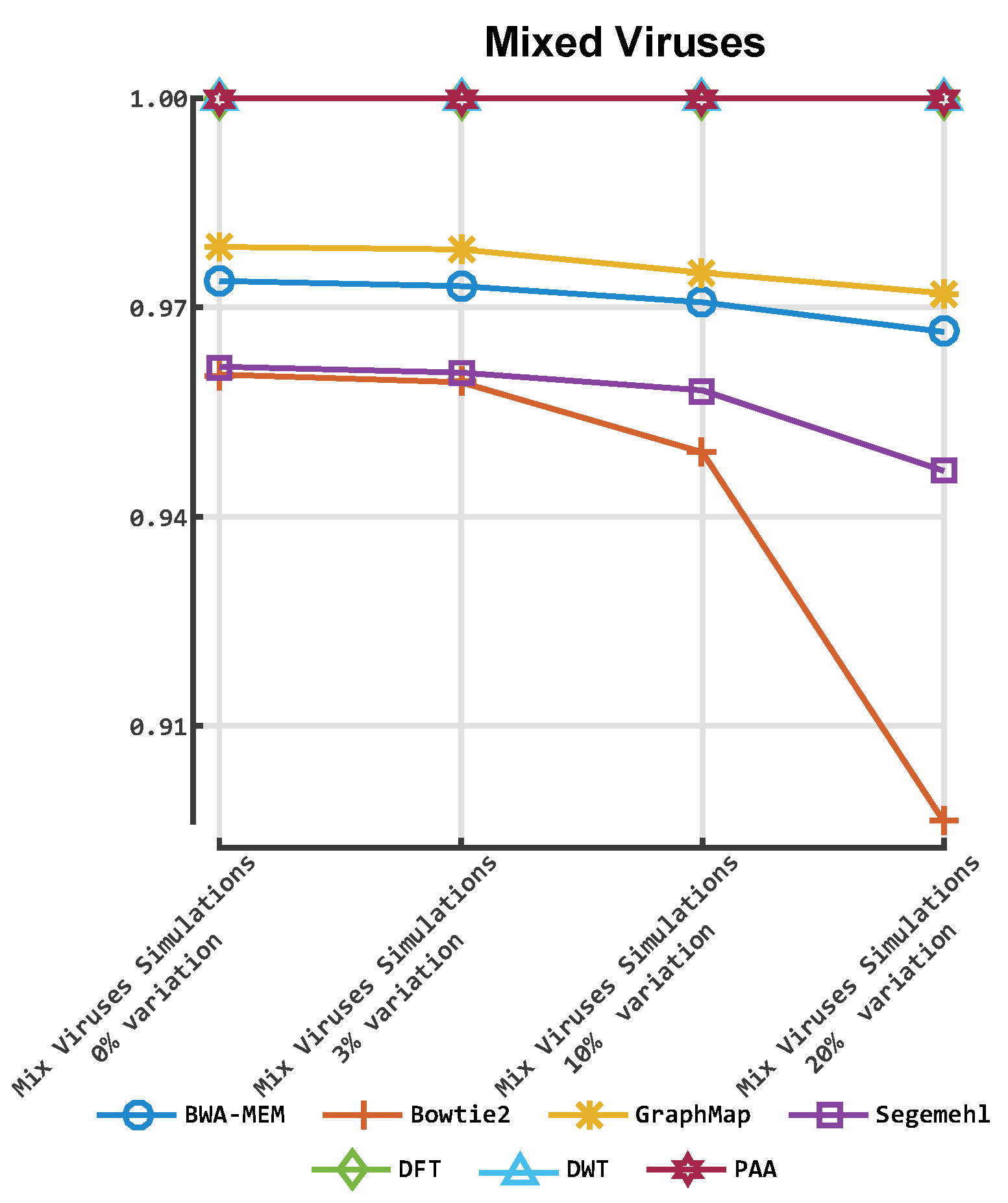
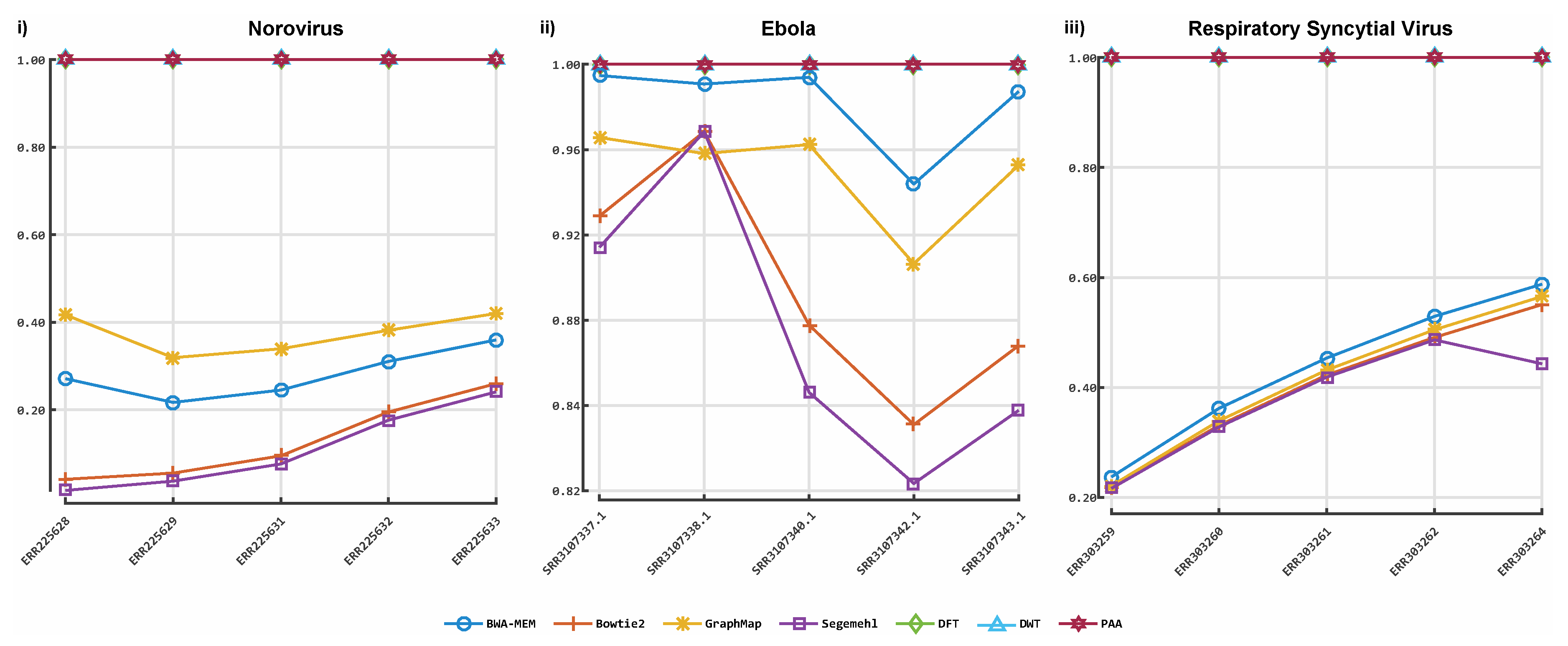
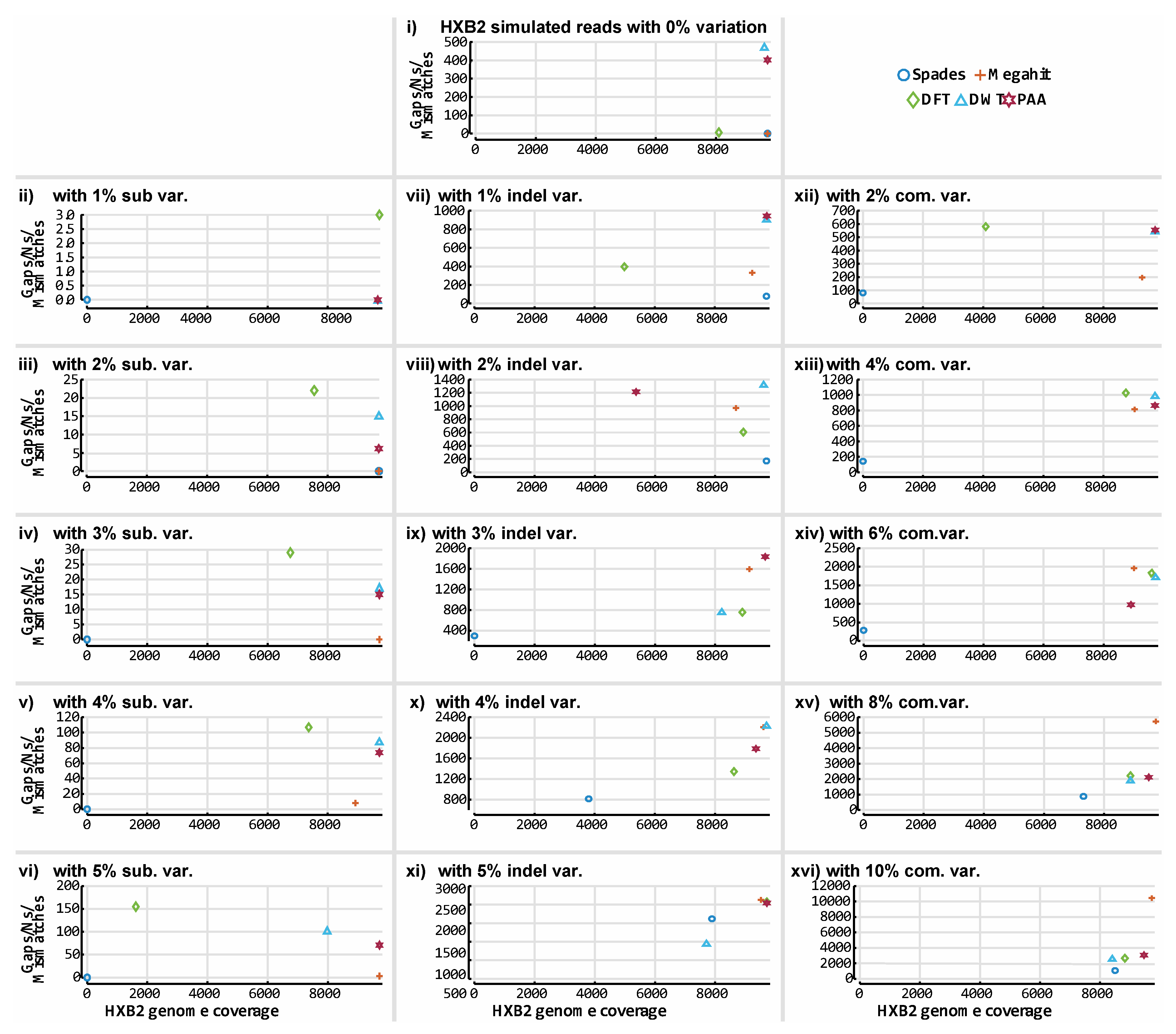
3. Results
3.1. Classification by Numbers (CBN)
3.2. Alignment by Numbers (ALBN)
3.3. De novo Assembly by Numbers
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Salipante, S.J.; Roach, D.J.; Kitzman, J.O.; Snyder, M.W.; Stackhouse, B.; Butler-Wu, S.M.; Lee, C.; Cookson, B.T.; Shendure, J. Large-scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains. Genome Res. 2015, 25, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.; Constantinides, B.; Tapinos, A.; Robertson, D.L.; Prosperi, M. Challenges in the analysis of viral metagenomes. Virus Evol. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.M.S.; Frith, M.C.; Horton, P. A bioinformatician’s guide to the forefront of suffix array construction algorithms. Brief. Bioinform. 2014, 15, 138–154. [Google Scholar] [CrossRef]
- Myers, E.W. Toward simplifying and accurately formulating fragment assembly. J. Comput. Biol. 1995, 2, 275–290. [Google Scholar] [CrossRef]
- Kececioglu, J.D.; Myers, E.W. Combinatorial algorithms for DNA sequence assembly. Algorithmica 1995, 13, 7–51. [Google Scholar] [CrossRef]
- Earl, D.; Bradnam, K.; John, J.S.; Darling, A.; Lin, D.; Fass, J.; Yu, H.O.K.; Buffalo, V.; Zerbino, D.R.; Diekhans, M. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res. 2011, 21, 2224–2241. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Caccamo, M.; Turner, I.; Flicek, P.; McVean, G. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet. 2012, 44, 226–232. [Google Scholar] [CrossRef]
- Pevzner, P.A.; Tang, H.; Waterman, M.S. An Eulerian path approach to DNA fragment assembly. Proc. Natl. Acad. Sci. USA 2001, 98, 9748–9753. [Google Scholar] [CrossRef]
- Bradnam, K.R.; Fass, J.N.; Alexandrov, A.; Baranay, P.; Bechner, M.; Birol, I.; Boisvert, S.; Chapman, J.A.; Chapuis, G.; Chikhi, R. Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. Gigascience 2013, 2, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Archer, J.; Rambaut, A.; Taillon, B.E.; Harrigan, P.R.; Lewis, M.; Robertson, D.L. The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time—An ultra-deep approach. PLoS Comput. Biol. 2010, 6, e1001022. [Google Scholar] [CrossRef] [PubMed]
- Clement, N.L.; Thompson, L.P.; Miranker, D.P. ADaM: Augmenting existing approximate fast matching algorithms with efficient and exact range queries. BMC Bioinform. 2014, 15, S1. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Faloutsos, C.; Swami, A. Efficient similarity search in sequence databases. In Proceedings of the 4th International Conference on Foundations of Data Organization and Algorithms, Chicago, IL, USA, 13–15 October 1993. [Google Scholar]
- Chan, K.-P.; Fu, A.-C. Efficient time series matching by wavelets. In Proceedings of the 15th International Conference on Data Engineering, Sydney, Australia, 23–26 March 1999; pp. 126–133. [Google Scholar]
- Woodward, A.M.; Rowland, J.J.; Kell, D.B. Fast automatic registration of images using the phase of a complex wavelet transform: Application to proteome gels. Analyst 2004, 129, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Geurts, P. Pattern extraction for time series classification. In Proceedings of the 5th European Conference on Principles of Data Mining and Knowledge Discovery, Freiburg, Germany, 3–7 September 2001; pp. 115–127. [Google Scholar]
- Keogh, E.; Chakrabarti, K.; Pazzani, M.; Mehrotra, S. Locally adaptive dimensionality reduction for indexing large time series databases. ACM SIGMOD Record 2001, 30, 151–162. [Google Scholar] [CrossRef]
- Shumway, R.H.; Stoffer, D.S.; Stoffer, D.S. Time Series Analysis and Its Applications with R examples, 2nd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Silverman, B.; Linsker, R. A measure of DNA periodicity. J. Theor. Biol. 1986, 118, 295–300. [Google Scholar] [CrossRef]
- Cheever, E.; Searls, D.; Karunaratne, W.; Overton, G. Using signal processing techniques for DNA sequence comparison. In Proceedings of the Fifteenth Annual Northeast Bioengineering Conference, Boston, MA, USA, 27–28 March 1989; pp. 173–174. [Google Scholar]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.K.; Arniker, S.B. Numerical representation of DNA sequences. In Proceedings of the 2009 IEEE International Conference on Electro/Information Technology, Windsor, ON, Canada, 7–9 June 2009; pp. 307–310. [Google Scholar]
- Yi, B.-K.; Faloutsos, C. Fast time sequence indexing for arbitrary Lp norms. In Proceedings of the 26th roceedings of 26th International Conference on Very Large Data Bases, Cairo, Egypt, 10–14 September 2000; pp. 385–394. [Google Scholar]
- Keogh, E.; Ratanamahatana, C.A. Exact indexing of dynamic time warping. Knowl. Inf. Syst. 2005, 7, 358–386. [Google Scholar] [CrossRef]
- Vlachos, M.; Kollios, G.; Gunopulos, D. Discovering similar multidimensional trajectories. In Proceedings of the 18th International Conference on Data Engineering, San Jose, CA, USA, 26 February–1 March 2002; pp. 673–684. [Google Scholar]
- Kotsakos, D.; Trajcevski, G.; Gunopulos, D.; Aggarwal, C.C. In Data Clustering: Algorithms and Applications; Aggarwal, C.C., Reddy, C., Eds.; CRC Press: Boca Raton, FL, USA, 2013; Chapter 15; pp. 357–379. [Google Scholar]
- Chávez, E.; Navarro, G.; Baeza-Yates, R.; Marroquín, J.L. Searching in metric spaces. ACM Comput. Surv. (CSUR) 2001, 33, 273–321. [Google Scholar] [CrossRef]
- Beckmann, N.; Kriegel, H.-P.; Schneider, R.; Seeger, B. The R*-tree: An efficient and robust access method for points and rectangles. SIGMOD Rec. 1990, 19, 322–331. [Google Scholar] [CrossRef]
- Agrawal, R.; Lin, K.; Sawhney, H.S.; Shim, K. Fast similarity search in the presence of noise, scaling, and translation in time-series databases. In Proceedings of the 21th International Conference on Very Large Data Bases, Zurich, Switzerland, 11–15 September 1995; pp. 490–501. [Google Scholar]
- Bingham, S.; Kot, M. Multidimensional trees, range searching, and a correlation dimension algorithm of reduced complexity. Phys. Lett. A 1989, 140, 327–330. [Google Scholar] [CrossRef]
- Bellman, R. Adaptive Control Processes: A Guided Tour; Princeton University Press: London, UK, 1961; Volume 4. [Google Scholar]
- Verleysen, M.; François, D. The Curse of Dimensionality in Data Mining and Time Series Prediction. In Proceedings of the 8th International Work-Conference on Artificial Neural Networks, Barcelona, Spain, 8–10 June 2005; pp. 758–770. [Google Scholar]
- Yianilos, P.N. Data structures and algorithms for nearest neighbor search in general metric spaces. In Proceedings of the 4th annual ACM-SIAM Symposium on Discrete Algorithms, Austin, TX, USA; 1993; pp. 311–321. [Google Scholar]
- Bozkaya, T.; Ozsoyoglu, M. Indexing large metric spaces for similarity search queries. ACM Trans. Database Syst. (TODS) 1999, 24, 361–404. [Google Scholar] [CrossRef]
- Uhlmann, J.K. Satisfying general proximity/similarity queries with metric trees. Inf. Process. Lett. 1991, 40, 175–179. [Google Scholar] [CrossRef]
- Nair, A.S.; Sreenadhan, S.P. A coding measure scheme employing electron-ion interaction pseudopotential (EIIP). Bioinformation 2006, 1, 197. [Google Scholar] [PubMed]
- Holden, T.; Subramaniam, R.; Sullivan, R.; Cheung, E.; Schneider, C.; Tremberger, G.; Flamholz, A.; Lieberman, D.H.; Cheung, T.D. ATCG nucleotide fluctuation of Deinococcus radiodurans radiation genes. In Proceedings of the Instruments, Methods, and Missions for Astrobiology X, San Diego, CA, USA, 1 October 2007. [Google Scholar]
- Voss, R.F. Evolution of long-range fractal correlations and 1/f noise in DNA base sequences. Phys. Rev. Lett. 1992, 68, 3805. [Google Scholar] [CrossRef] [PubMed]
- Faloutsos, C.; Ranganathan, M.; Manolopoulos, Y. Fast subsequence matching in time-series databases. In Proceedings of the 1994 ACM SIGMOD International Conference on Management of data, Minneapolis, MN, USA, 24–27 May 1994. [Google Scholar]
- Mitsa, T. Temporal Data Mining; CRC Press: New York, NY, USA, 2010. [Google Scholar]
- Mörchen, F. Time Series Feature Extraction for Data Mining Using DWT and DFT; Technical Report 3; Departement of Mathematics and Computer Science Philipps-University Marburg: Marburg, Germany, 2003; pp. 735–739. [Google Scholar]
- Jensen, A.; la Cour-Harbo, A. Ripples in Mathematics: The Discrete Wavelet Transform; Springer: Berlin, Germany, 2001. [Google Scholar]
- Wu, Y.-L.; Agrawal, D.; El Abbadi, A. A comparison of DFT and DWT based similarity search in time-series databases. In Proceedings of the 9th International Conference on Information and Knowledge Management, Washington, DC, USA, 6–11 November 2000; pp. 488–495. [Google Scholar]
- Caboche, S.; Audebert, C.; Lemoine, Y.; Hot, D. Comparison of mapping algorithms used in high-throughput sequencing: Application to Ion Torrent data. BMC Genom. 2014, 15, 264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cotten, M.; Petrova, V.; Phan, M.V.; Rabaa, M.A.; Watson, S.J.; Ong, S.H.; Kellam, P.; Baker, S. Deep sequencing of norovirus genomes defines evolutionary patterns in an urban tropical setting. J. Virol. 2014, 88, 11056–11069. [Google Scholar] [CrossRef] [PubMed]
- Phan, M.V.; Anh, P.H.; Cuong, N.V.; Munnink, B.B.O.; van der Hoek, L.; My, P.T.; Tri, T.N.; Bryant, J.E.; Baker, S.; Thwaites, G. Unbiased whole-genome deep sequencing of human and porcine stool samples reveals circulation of multiple groups of rotaviruses and a putative zoonotic infection. Virus Evol. 2016, 2. [Google Scholar] [CrossRef]
- Kiyuka, P.K.; Agoti, C.N.; Munywoki, P.K.; Njeru, R.; Bett, A.; Otieno, J.R.; Otieno, G.P.; Kamau, E.; Clark, T.G.; van der Hoek, L. Human Coronavirus NL63 Molecular Epidemiology and Evolutionary Patterns in Rural Coastal Kenya. J. Infect. Dis. 2018, 217, 1728–1739. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; Watson, S.J.; Asogun, D.; Tobin, E.A.; Lu, J.; Phan, M.V.; Jah, U.; Wadoum, R.E.G.; Meredith, L.; Thorne, L. Rapid outbreak sequencing of Ebola virus in Sierra Leone identifies transmission chains linked to sporadic cases. Virus Evol. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Agoti, C.N.; Otieno, J.R.; Munywoki, P.K.; Mwihuri, A.G.; Cane, P.A.; Nokes, D.J.; Kellam, P.; Cotten, M. Local evolutionary patterns of human respiratory syncytial virus derived from whole-genome sequencing. J. Virol. 2015, 89, 3444–3454. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Sović, I.; Šikić, M.; Wilm, A.; Fenlon, S.N.; Chen, S.; Nagarajan, N. Fast and sensitive mapping of nanopore sequencing reads with GraphMap. Nat. Commun. 2016, 7, 11307. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Stadler, P.F.; Hoffmann, S. Lacking alignments? The next-generation sequencing mapper segemehl revisited. Bioinform. 2014, 30, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Tapinos, A.; Robertson, D.L. De novo assembly of nucleotide sequences in a compressed feature space. In Proceedings of the 2017 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Manchester, UK, 23–25 August 2017; pp. 1–7. [Google Scholar]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Anton, B.; Sergey, N.; Dmitry, A.; Alexey, A.; Mikhail, D.; Alexander, S.; Valery, M.; Sergey, I.; Son, P.; Andrey, D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455. [Google Scholar]
- Tapinos, A.; Mendes, P. A method for comparing multivariate time series with different dimensions. PloS ONE 2013, 8, e54201. [Google Scholar] [CrossRef]
- Sheybani, E.O. An Algorithm for Real-Time Blind Image Quality Comparison and Assessment. Int. J. Electr. Comput. Eng. (IJECE) 2011, 2, 120–129. [Google Scholar] [CrossRef]
- Hendriks, R.C.; Gerkmann, T.; Jensen, J. DFT-domain based single-microphone noise reduction for speech enhancement: A survey of the state of the art. In Synthesis Lectures on Speech and Audio Processing; Morgan & Claypool: San Rafael, CA, USA, 2013; pp. 1–80. [Google Scholar]
- Kouchaki, S.; Tapinos, A.; Robertson, D.L. A signal processing method for alignment-free metagenomic binning: Multi-resolution genomic binary patterns. Sci. Rep. 2019, 9, 2159. [Google Scholar] [CrossRef]
- Shi, H.; Schmidt, B.; Liu, W.; Müller-Wittig, W. A Parallel Algorithm for Error Correction in High-Throughput Short-Read Data on CUDA-Enabled Graphics Hardware. J. Comput. Biol. 2010, 17, 603–615. [Google Scholar] [CrossRef]
- Zhang, Q.; Pell, J.; Canino-Koning, R.; Howe, A.C.; Brown, C.T. These Are Not the K-mers You Are Looking For: Efficient Online K-mer Counting Using a Probabilistic Data Structure. PLoS ONE 2014, 9, e101271. [Google Scholar] [CrossRef]
- Salikhov, K.; Sacomoto, G.; Kucherov, G. Using cascading Bloom filters to improve the memory usage for de Brujin graphs. Algorithms Mol. Biol. 2014, 9, 364–376. [Google Scholar] [CrossRef]
- Berlin, K.; Koren, S.; Chin, C.-S.; Drake, J.P.; Landolin, J.M.; Phillippy, A.M. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 2015, 33, 623–630. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’neill, P.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the oxford nanopore technologies minion. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Fu, S.; Wang, A.; Au, K.F. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 2019, 20, 26. [Google Scholar] [CrossRef]
- Watson, M.; Warr, A. Errors in long-read assemblies can critically affect protein prediction. Nature Biotechnol. 2019, 37, 124. [Google Scholar] [CrossRef]
- Radovanović, M.; Nanopoulos, A.; Ivanović, M. Time-series classification in many intrinsic dimensions. In Proceedings of the 2010 SIAM International Conference on Data Mining, Columbus, OH, USA, 29 April–1 May 2010; pp. 677–688. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Numerical Representation |
|---|---|
| Integer number | |
| Real number | |
| EIIP | |
| Atomic | |
| Pair | |
| Complex number | |
| DNA Walk | |
| Tetrahedron | |
| Voss indicator |
| Family | Virus | GenBank Genome ID | Variation Type (%) | Reads | Simulator | ||
|---|---|---|---|---|---|---|---|
| Ins | Del | Sub | |||||
| HIV | HXB2 | K03455 | 0.0 | 0.0 | 0.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 0.0 | 0.0 | 1.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 0.0 | 0.0 | 2.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 0.0 | 0.0 | 3.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 0.0 | 0.0 | 4.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 0.0 | 0.0 | 5.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 0.5 | 0.5 | 0.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 1.0 | 1.0 | 0.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 1.5 | 1.5 | 0.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 2.0 | 2.0 | 0.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 2.5 | 2.5 | 0.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 0.5 | 0.5 | 1.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 1.0 | 1.0 | 2.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 1.5 | 1.5 | 3.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 2.0 | 2.0 | 4.0 | 2133 | CuReSim |
| HIV | HXB2 | K03455 | 2.5 | 2.5 | 5.0 | 2133 | CuReSim |
| Mixed Viruses: Caliciviridae, Filoviridae, Pneumoviridae | Norovirus, Ebola virus, RSV | KM198529, KM198528, KM198511, KM198500, KM198486, KU296608, KU296553, KU296549, KU296528, KU296416, KP317952, KP317946, KP317934, KP317923, KP317922 | 0.0 | 0.0 | 0.0 | 200,000 | WGSIM |
| Mixed Viruses: Caliciviridae, Filoviridae, Pneumoviridae | Norovirus, Ebola virus, RSV | KM198529, KM198528, KM198511, KM198500, KM198486, KU296608, KU296553, KU296549, KU296528, KU296416, KP317952, KP317946, KP317934, KP317923, KP317922 | 1.0 | 1.0 | 1.0 | 200,000 | WGSIM |
| Mixed Viruses, Caliciviridae, Filoviridae, Pneumoviridae | Norovirus, Ebola virus, RSV | KM198529, KM198528, KM198511, KM198500, KM198486, KU296608, KU296553, KU296549, KU296528, KU296416, KP317952, KP317946, KP317934, KP317923, KP317922 | 3.33 | 3.33 | 3.33 | 100,000 | WGSIM |
| Mixed Viruses, Caliciviridae, Filoviridae, Pneumoviridae | Norovirus, Ebola virus, RSV | KM198529, KM198528, KM198511, KM198500, KM198486, KU296608, KU296553, KU296549, KU296528, KU296416, KP317952, KP317946, KP317934, KP317923, KP317922 | 6.66 | 6.66 | 6.66 | 200,000 | WGSIM |
| Family | Virus | Amplicon/Random Primer | GenBank Genome ID | ENA/SRA_ID | Reads | Sequencing Technology |
|---|---|---|---|---|---|---|
| Caliciviridae | Norovirus | Amplicon | KM198486 | ERR225628 | 2126502 | Illumina MiSeq |
| Caliciviridae | Norovirus | Amplicon | KM198500 | ERR225629 | 3037674 | Illumina MiSeq |
| Caliciviridae | Norovirus | Amplicon | KM198511 | ERR225631 | 3285078 | Illumina MiSeq |
| Caliciviridae | Norovirus | Amplicon | KM198528 | ERR225632 | 4361884 | Illumina MiSeq |
| Caliciviridae | Norovirus | Amplicon | KM198529 | ERR225633 | 5187234 | Illumina MiSeq |
| Filoviridae | Ebola virus | Amplicon | KU296608 | SRR3107337 | 522968 | Ion Torrent PGM |
| Filoviridae | Ebola virus | Amplicon | KU296549 | SRR3107338 | 771031 | Ion Torrent PGM |
| Filoviridae | Ebola virus | Amplicon | KU296416 | SRR3107340 | 186657 | Ion Torrent PGM |
| Filoviridae | Ebola virus | Amplicon | KU296553 | SRR3107342 | 478346 | Ion Torrent PGM |
| Filoviridae | Ebola virus | Amplicon | KU296528 | SRR3107343 | 42410 | Ion Torrent PGM |
| Pneumoviridae | RSV | Amplicon | KP317934 | ERR303259 | 7275032 | Illumina MiSeq |
| Pneumoviridae | RSV | Amplicon | KP317922 | ERR303260 | 9278070 | Illumina MiSeq |
| Pneumoviridae | RSV | Amplicon | KP317946 | ERR303261 | 11111114 | Illumina MiSeq |
| Pneumoviridae | RSV | Amplicon | KP317923 | ERR303262 | 13293226 | Illumina MiSeq |
| Pneumoviridae | RSV | Amplicon | KP317952 | ERR303263 | 15237848 | Illumina MiSeq |
| Family | Virus | GenBank ID: | Length (nt) |
|---|---|---|---|
| Retroviridae | Human immunodeficiency virus 1 (HXB2) | K03455 | 9179 |
| Caliciviridae | Norovirus | KM198509.1 | 7425 |
| Filoviridae | Zaire ebolavirus | KM034562.1 | 18957 |
| Pneumoviridae | Human orthopneumovirus (Respiratory Syncytial Virus) | KP317934.1 | 15233 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tapinos, A.; Constantinides, B.; Phan, M.V.T.; Kouchaki, S.; Cotten, M.; Robertson, D.L. The Utility of Data Transformation for Alignment, De Novo Assembly and Classification of Short Read Virus Sequences. Viruses 2019, 11, 394. https://doi.org/10.3390/v11050394
Tapinos A, Constantinides B, Phan MVT, Kouchaki S, Cotten M, Robertson DL. The Utility of Data Transformation for Alignment, De Novo Assembly and Classification of Short Read Virus Sequences. Viruses. 2019; 11(5):394. https://doi.org/10.3390/v11050394
Chicago/Turabian StyleTapinos, Avraam, Bede Constantinides, My V. T. Phan, Samaneh Kouchaki, Matthew Cotten, and David L. Robertson. 2019. "The Utility of Data Transformation for Alignment, De Novo Assembly and Classification of Short Read Virus Sequences" Viruses 11, no. 5: 394. https://doi.org/10.3390/v11050394
APA StyleTapinos, A., Constantinides, B., Phan, M. V. T., Kouchaki, S., Cotten, M., & Robertson, D. L. (2019). The Utility of Data Transformation for Alignment, De Novo Assembly and Classification of Short Read Virus Sequences. Viruses, 11(5), 394. https://doi.org/10.3390/v11050394