Manipulation of Epithelial Differentiation by HPV Oncoproteins


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Differentiation-Dependent HPV Biology
3. Keratinocyte Differentiation and Models of HPV Replication
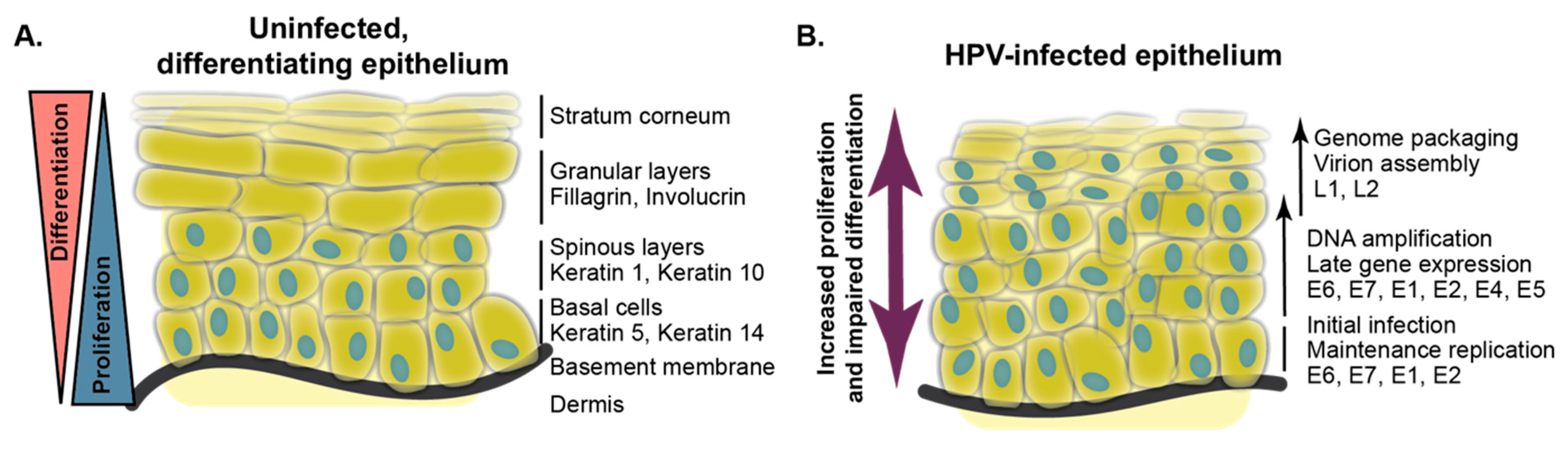
3.1. Stages of Keratinocyte Differentiation
3.2. Differentiation Models
4. HPV Carcinogenic Activity and the E6 and E7 Oncoproteins
5. HPV-Associated Cancer and Differentiation
6. High-Risk HPV Oncoproteins Alter Epithelial Differentiation
6.1. Delay of Differentiation in Organotypic Culture
6.2. Downregulation of Differentiation-Related Cellular Gene Expression
6.3. HPV E6 and Differentiation
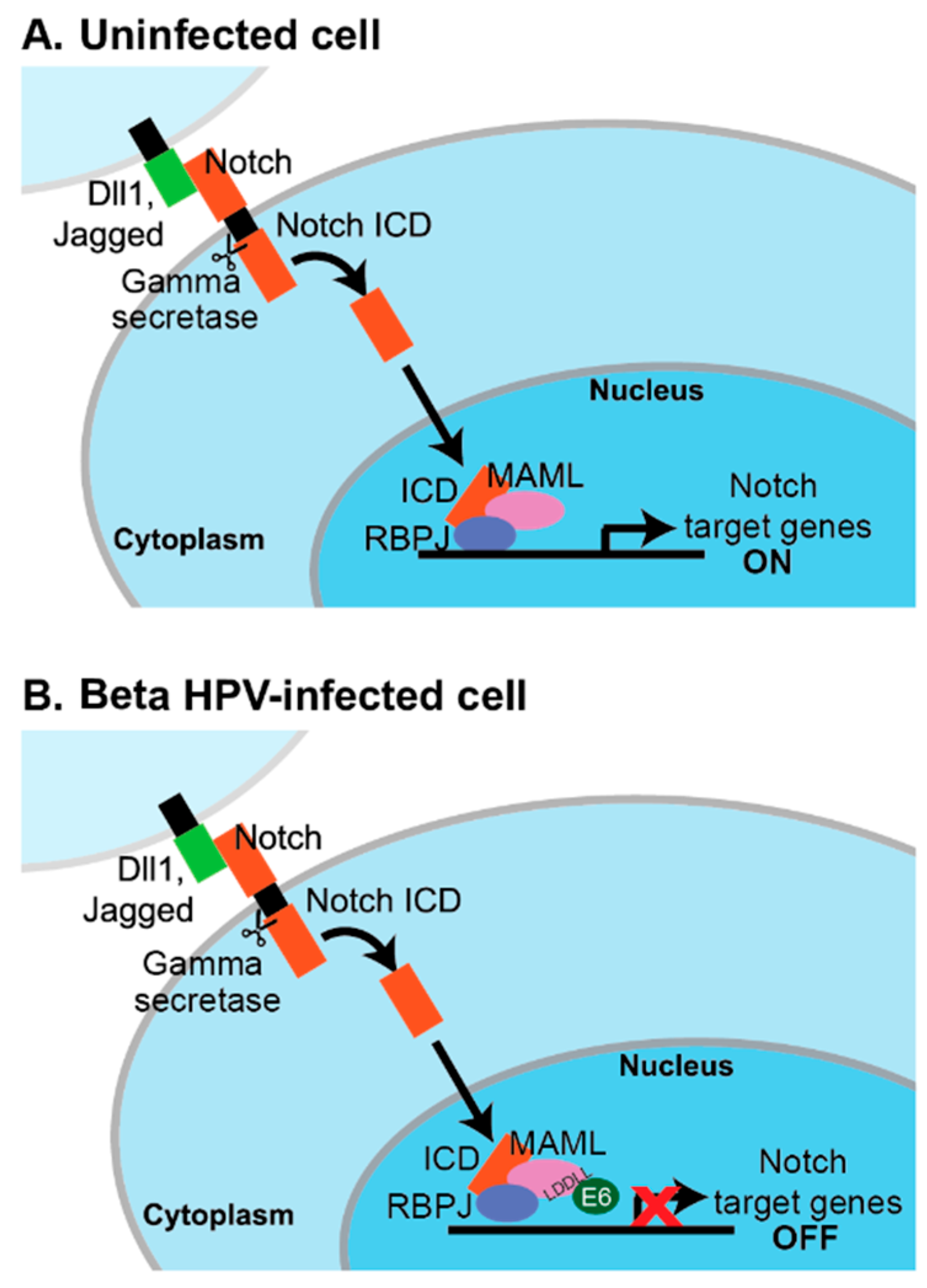
6.3.1. MAML1, Notch, and Genus Beta HPV E6
6.3.2. Genus Alpha HPV E6
6.3.3. Notch and SCC
6.3.4. TGFβ
6.4. HPV E7 and Differentiation
6.4.1. Inhibition of Differentiation by HPV E7
6.4.2. Differentiation Pathways Altered by HPV E7
6.4.3. HPV E7 and PTPN14
6.5. HPV E5 and Differentiation
7. Differentiation Therapy
8. Conclusions and Open Questions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Schiller, J.T.; Lowy, D.R. Papillomaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2013; pp. 1662–1703. [Google Scholar]
- Graham, S.V. Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation. Viruses 2017, 9, 245. [Google Scholar] [CrossRef]
- Moody, C. Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses 2017, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Livingston, D.M. Small DNA tumor viruses: Large contributors to biomedical sciences. Virology 2009, 384, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Tan, Q.; Xirasagar, S.; Bandaru, S.; Gopalan, V.; Mohamoud, Y.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A central resource for papillomavirus sequence data and analysis. Nucleic Acids Res 2013, 41, D571–D578. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Gopalan, V.; Bandaru, S.; Mahmoud, Y.; Tan, Q.; Xirasaga, S.; McBride, A.A. Papillomavirus Episteme (PaVE). Available online: http://pave.niaid.nih.gov/ (accessed on 25 February 2019).
- Campos, S.K. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses 2017, 9, 370. [Google Scholar] [CrossRef]
- Aksoy, P.; Gottschalk, E.Y.; Meneses, P.I. HPV entry into cells. Mutat. Res. Rev. Mutat. Res. 2017, 772, 13–22. [Google Scholar] [CrossRef]
- Zhang, P.; Monteiro da Silva, G.; Deatherage, C.; Burd, C.; DiMaio, D. Cell-Penetrating Peptide Mediates Intracellular Membrane Passage of Human Papillomavirus L2 Protein to Trigger Retrograde Trafficking. Cell 2018, 174, 1465–1476.e13. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, K.; Rapp, B.; Maschek, H.; Petry, K.U.; Iftner, T. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J. Virol. 1996, 70, 2339–2349. [Google Scholar]
- Wang, X.; Liu, H.; Ge, H.; Ajiro, M.; Sharma, N.R.; Meyers, C.; Morozov, P.; Tuschl, T.; Klar, A.; Court, D.; et al. Viral DNA Replication Orientation and hnRNPs Regulate Transcription of the Human Papillomavirus 18 Late Promoter. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Meyers, C. Genetic analysis of the human papillomavirus type 31 differentiation-dependent late promoter. J. Virol. 2005, 79, 3309–3321. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Meyers, C.; Wang, H.K.; Chow, L.T.; Zheng, Z.M. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J. Virol. 2011, 85, 8080–8092. [Google Scholar] [CrossRef]
- Brant, A.C.; Majerciak, V.; Moreira, M.A.M.; Zheng, Z.M. HPV18 Utilizes Two Alternative Branch Sites for E6*I Splicing to Produce E7 Protein. Virol. Sin. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Tao, M.; McCoy, J.P., Jr.; Zheng, Z.M. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of HPV transcription. Clinics 2018, 73, e486s. [Google Scholar] [CrossRef]
- Biryukov, J.; Myers, J.C.; McLaughlin-Drubin, M.E.; Griffin, H.M.; Milici, J.; Doorbar, J.; Meyers, C. Mutations in HPV18 E1^E4 Impact Virus Capsid Assembly, Infectivity Competence, and Maturation. Viruses 2017, 9, 385. [Google Scholar] [CrossRef]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef]
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef]
- Fuchs, E.; Green, H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell 1980, 19, 1033–1042. [Google Scholar] [CrossRef]
- Gibbs, S.; Ponec, M. Intrinsic regulation of differentiation markers in human epidermis, hard palate and buccal mucosa. Arch. Oral Biol. 2000, 45, 149–158. [Google Scholar] [CrossRef]
- Baek, J.H.; Lee, G.; Kim, S.N.; Kim, J.M.; Kim, M.; Chung, S.C.; Min, B.M. Common genes responsible for differentiation and senescence of human mucosal and epidermal keratinocytes. Int. J. Mol. Med. 2003, 12, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T. Model systems to study the life cycle of human papillomaviruses and HPV-associated cancers. Virol. Sin. 2015, 30, 92–100. [Google Scholar] [CrossRef]
- Fehrmann, F.; Laimins, L.A. Human papillomavirus type 31 life cycle: Methods for study using tissue culture models. Methods Mol. Biol. 2005, 292, 317–330. [Google Scholar]
- McLaughlin-Drubin, M.E.; Meyers, C. Propagation of infectious, high-risk HPV in organotypic “raft” culture. Methods Mol. Med. 2005, 119, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Ingle, A.; Ghim, S.; Joh, J.; Chepkoech, I.; Bennett Jenson, A.; Sundberg, J.P. Novel laboratory mouse papillomavirus (MusPV) infection. Vet. Pathol. 2011, 48, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Mueller-Decker, K.; Kloz, U.; Aengeneyndt, B.; Kopp-Schneider, A.; Grone, H.J.; Gheit, T.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. E6 and E7 from beta HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog. 2011, 7, e1002125. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.F. Transgenic Mouse Models of Tumor Virus Action. Annu. Rev. Virol. 2016, 3, 473–489. [Google Scholar] [CrossRef]
- Meyers, J.M.; Uberoi, A.; Grace, M.; Lambert, P.F.; Munger, K. Cutaneous HPV8 and MmuPV1 E6 Proteins Target the NOTCH and TGF-beta Tumor Suppressors to Inhibit Differentiation and Sustain Keratinocyte Proliferation. PLoS Pathog. 2017, 13, e1006171. [Google Scholar] [CrossRef]
- Santos, C.; Vilanova, M.; Medeiros, R.; Gil da Costa, R.M. HPV-transgenic mouse models: Tools for studying the cancer-associated immune response. Virus Res. 2017, 235, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Tuong, Z.K.; Noske, K.; Kuo, P.; Bashaw, A.A.; Teoh, S.M.; Frazer, I.H. Murine HPV16 E7-expressing transgenic skin effectively emulates the cellular and molecular features of human high-grade squamous intraepithelial lesions. Papillomavirus Res. 2018, 5, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.M.; Alam, S.; Meyers, C. Regulation of human papillomavirus type 31 late promoter activation and genome amplification by protein kinase C. Virology 2006, 348, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Frattini, M.G.; Hudson, J.B.; Laimins, L.A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992, 257, 971–973. [Google Scholar] [CrossRef]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef]
- Van Dyne, E.A.; Henley, S.J.; Saraiya, M.; Thomas, C.C.; Markowitz, L.E.; Benard, V.B. Trends in Human Papillomavirus–Associated Cancers—United States, 1999–2015. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 918–924. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high- and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef]
- Durst, M.; Gissmann, L.; Ikenberg, H.; zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef]
- Boshart, M.; Gissmann, L.; Ikenberg, H.; Kleinheinz, A.; Scheurlen, W.; zur Hausen, H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. EMBO J. 1984, 3, 1151–1157. [Google Scholar] [CrossRef]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar]
- Schneider-Gadicke, A.; Schwarz, E. Different human cervical carcinoma cell lines show similar transcription patterns of human papillomavirus type 18 early genes. EMBO J. 1986, 5, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef]
- Smotkin, D.; Wettstein, F.O. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc. Natl. Acad. Sci. USA 1986, 83, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Phelps, W.C.; Bubb, V.; Howley, P.M.; Schlegel, R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J. Virol. 1989, 63, 4417–4421. [Google Scholar]
- Bedell, M.A.; Jones, K.H.; Laimins, L.A. The E6-E7 region of human papillomavirus type 18 is sufficient for transformation of NIH 3T3 and rat-1 cells. J. Virol. 1987, 61, 3635–3640. [Google Scholar]
- Bedell, M.A.; Jones, K.H.; Grossman, S.R.; Laimins, L.A. Identification of human papillomavirus type 18 transforming genes in immortalized and primary cells. J. Virol. 1989, 63, 1247–1255. [Google Scholar] [PubMed]
- Yee, C.; Krishnan-Hewlett, I.; Baker, C.C.; Schlegel, R.; Howley, P.M. Presence and expression of human papillomavirus sequences in human cervical carcinoma cell lines. Am. J. Pathol. 1985, 119, 361–366. [Google Scholar]
- Dyson, N.; Guida, P.; Munger, K.; Harlow, E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J. Virol. 1992, 66, 6893–6902. [Google Scholar] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Zerfass-Thome, K.; Zwerschke, W.; Mannhardt, B.; Tindle, R.; Botz, J.W.; Jansen-Durr, P. Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 1996, 13, 2323–2330. [Google Scholar] [PubMed]
- Jones, D.L.; Alani, R.M.; Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Demers, G.W.; Halbert, C.L.; Galloway, D.A. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene. Virology 1994, 198, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef]
- Duensing, S.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar]
- Ueno, T.; Sasaki, K.; Yoshida, S.; Kajitani, N.; Satsuka, A.; Nakamura, H.; Sakai, H. Molecular mechanisms of hyperplasia induction by human papillomavirus E7. Oncogene 2006, 25, 4155–4164. [Google Scholar] [CrossRef][Green Version]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Munger, K.; Jones, D.L. Human papillomavirus carcinogenesis: An identity crisis in the retinoblastoma tumor suppressor pathway. J. Virol. 2015, 89, 4708–4711. [Google Scholar] [CrossRef] [PubMed]
- Storey, A.; Osborn, K.; Crawford, L. Co-transformation by human papillomavirus types 6 and 11. J. Gen. Virol. 1990, 71 Pt 1, 165–171. [Google Scholar] [CrossRef]
- Ciccolini, F.; Di Pasquale, G.; Carlotti, F.; Crawford, L.; Tommasino, M. Functional studies of E7 proteins from different HPV types. Oncogene 1994, 9, 2633–2638. [Google Scholar]
- Ibaraki, T.; Satake, M.; Kurai, N.; Ichijo, M.; Ito, Y. Transacting activities of the E7 genes of several types of human papillomavirus. Virus Genes 1993, 7, 187–196. [Google Scholar] [CrossRef]
- Phelps, W.C.; Munger, K.; Yee, C.L.; Barnes, J.A.; Howley, P.M. Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J. Virol. 1992, 66, 2418–2427. [Google Scholar] [PubMed]
- Jewers, R.J.; Hildebrandt, P.; Ludlow, J.W.; Kell, B.; McCance, D.J. Regions of human papillomavirus type 16 E7 oncoprotein required for immortalization of human keratinocytes. J. Virol. 1992, 66, 1329–1335. [Google Scholar]
- White, E.A.; Kramer, R.E.; Hwang, J.H.; Pores Fernando, A.T.; Naetar, N.; Hahn, W.C.; Roberts, T.M.; Schaffhausen, B.S.; Livingston, D.M.; Howley, P.M. Papillomavirus e7 oncoproteins share functions with polyomavirus small T antigens. J. Virol. 2015, 89, 2857–2865. [Google Scholar] [CrossRef]
- Helt, A.M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef]
- Banks, L.; Edmonds, C.; Vousden, K.H. Ability of the HPV16 E7 protein to bind RB and induce DNA synthesis is not sufficient for efficient transforming activity in NIH3T3 cells. Oncogene 1990, 5, 1383–1389. [Google Scholar]
- Huh, K.W.; DeMasi, J.; Ogawa, H.; Nakatani, Y.; Howley, P.M.; Munger, K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc. Natl. Acad. Sci. USA 2005, 102, 11492–11497. [Google Scholar] [CrossRef]
- Strati, K.; Lambert, P.F. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res. 2007, 67, 11585–11593. [Google Scholar] [CrossRef] [PubMed]
- Balsitis, S.; Dick, F.; Dyson, N.; Lambert, P.F. Critical roles for non-pRb targets of human papillomavirus type 16 E7 in cervical carcinogenesis. Cancer Res. 2006, 66, 9393–9400. [Google Scholar] [CrossRef] [PubMed]
- Balsitis, S.; Dick, F.; Lee, D.; Farrell, L.; Hyde, R.K.; Griep, A.E.; Dyson, N.; Lambert, P.F. Examination of the pRb-dependent and pRb-independent functions of E7 in vivo. J. Virol. 2005, 79, 11392–11402. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Munger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef]
- Soto, D.R.; Barton, C.; Munger, K.; McLaughlin-Drubin, M.E. KDM6A addiction of cervical carcinoma cell lines is triggered by E7 and mediated by p21CIP1 suppression of replication stress. PLoS Pathog. 2017, 13, e1006661. [Google Scholar] [CrossRef]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W.; et al. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174.e6. [Google Scholar] [CrossRef]
- White, E.A.; Munger, K. Crowd Control: E7 Conservation Is the Key to Cancer. Cell 2017, 170, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.R.; Duensing, S.; Brake, T.; Munger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871. [Google Scholar]
- Mendelsohn, A.H.; Lai, C.K.; Shintaku, I.P.; Elashoff, D.A.; Dubinett, S.M.; Abemayor, E.; St John, M.A. Histopathologic findings of HPV and p16 positive HNSCC. Laryngoscope 2010, 120, 1788–1794. [Google Scholar] [CrossRef]
- Pai, S.I.; Westra, W.H. Molecular pathology of head and neck cancer: Implications for diagnosis, prognosis, and treatment. Annu. Rev. Pathol. 2009, 4, 49–70. [Google Scholar] [CrossRef]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef]
- Harden, M.E.; Prasad, N.; Griffiths, A.; Munger, K. Modulation of microRNA-mRNA Target Pairs by Human Papillomavirus 16 Oncoproteins. mBio 2017, 8. [Google Scholar] [CrossRef]
- Roland, N.J.; Rowley, H.; Scraggs, M.; Johnson, P.; Jones, A.S. MIB-1 and involucrin expression in laryngeal squamous carcinoma: The relationship to host and tumour factors and survival. Clin. Otolaryngol. Allied Sci. 1996, 21, 429–438. [Google Scholar] [CrossRef]
- Keck, M.K.; Zuo, Z.; Khattri, A.; Stricker, T.P.; Brown, C.D.; Imanguli, M.; Rieke, D.; Endhardt, K.; Fang, P.; Bragelmann, J.; et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin. Cancer Res. 2015, 21, 870–881. [Google Scholar] [CrossRef]
- Santoro, A.; Pannone, G.; Ninivaggi, R.; Petruzzi, M.; Santarelli, A.; Russo, G.M.; Lepore, S.; Pietrafesa, M.; Laurenzana, I.; Leonardi, R.; et al. Relationship between CK19 expression, deregulation of normal keratinocyte differentiation pattern and high risk-human papilloma virus infection in oral and oropharyngeal squamous cell carcinoma. Infect. Agents Cancer 2015, 10, 46. [Google Scholar] [CrossRef]
- Kopan, R.; Traska, G.; Fuchs, E. Retinoids as important regulators of terminal differentiation: Examining keratin expression in individual epidermal cells at various stages of keratinization. J. Cell Biol. 1987, 105, 427–440. [Google Scholar] [CrossRef]
- Wu, Y.J.; Rheinwald, J.G. A new small (40 kd) keratin filament protein made by some cultured human squamous cell carcinomas. Cell 1981, 25, 627–635. [Google Scholar] [CrossRef]
- McCance, D.J.; Kopan, R.; Fuchs, E.; Laimins, L.A. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc. Natl. Acad. Sci. USA 1988, 85, 7169–7173. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 2000, 74, 6622–6631. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Demers, G.W.; Galloway, D.A. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J. Virol. 1992, 66, 2125–2134. [Google Scholar]
- Hudson, J.B.; Bedell, M.A.; McCance, D.J.; Laiminis, L.A. Immortalization and altered differentiation of human keratinocytes in vitro by the E6 and E7 open reading frames of human papillomavirus type 18. J. Virol. 1990, 64, 519–526. [Google Scholar]
- Woodworth, C.D.; Cheng, S.; Simpson, S.; Hamacher, L.; Chow, L.T.; Broker, T.R.; DiPaolo, J.A. Recombinant retroviruses encoding human papillomavirus type 18 E6 and E7 genes stimulate proliferation and delay differentiation of human keratinocytes early after infection. Oncogene 1992, 7, 619–626. [Google Scholar]
- Schutze, D.M.; Snijders, P.J.; Bosch, L.; Kramer, D.; Meijer, C.J.; Steenbergen, R.D. Differential in vitro immortalization capacity of eleven (probable) [corrected] high-risk human papillomavirus types. J. Virol. 2014, 88, 1714–1724. [Google Scholar] [CrossRef]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Sattler, C.A.; Lambert, P.F. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology 1999, 262, 344–354. [Google Scholar] [CrossRef][Green Version]
- Blanton, R.A.; Coltrera, M.D.; Gown, A.M.; Halbert, C.L.; McDougall, J.K. Expression of the HPV16 E7 gene generates proliferation in stratified squamous cell cultures which is independent of endogenous p53 levels. Cell Growth Differ. 1992, 3, 791–802. [Google Scholar]
- Bergner, S.; Halec, G.; Schmitt, M.; Aubin, F.; Alonso, A.; Auvinen, E. Individual and Complementary Effects of Human Papillomavirus Oncogenes on Epithelial Cell Proliferation and Differentiation. Cells Tissues Organs 2016, 201, 97–108. [Google Scholar] [CrossRef]
- Lee, C.; Laimins, L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004, 78, 12366–12377. [Google Scholar] [CrossRef] [PubMed]
- Zehbe, I.; Richard, C.; DeCarlo, C.A.; Shai, A.; Lambert, P.F.; Lichtig, H.; Tommasino, M.; Sherman, L. Human papillomavirus 16 E6 variants differ in their dysregulation of human keratinocyte differentiation and apoptosis. Virology 2009, 383, 69–77. [Google Scholar] [CrossRef][Green Version]
- Nees, M.; Geoghegan, J.M.; Munson, P.; Prabhu, V.; Liu, Y.; Androphy, E.; Woodworth, C.D. Human papillomavirus type 16 E6 and E7 proteins inhibit differentiation-dependent expression of transforming growth factor-beta2 in cervical keratinocytes. Cancer Res. 2000, 60, 4289–4298. [Google Scholar] [PubMed]
- Duffy, C.L.; Phillips, S.L.; Klingelhutz, A.J. Microarray analysis identifies differentiation-associated genes regulated by human papillomavirus type 16 E6. Virology 2003, 314, 196–205. [Google Scholar] [CrossRef][Green Version]
- Wan, F.; Miao, X.; Quraishi, I.; Kennedy, V.; Creek, K.E.; Pirisi, L. Gene expression changes during HPV-mediated carcinogenesis: A comparison between an in vitro cell model and cervical cancer. Int. J. Cancer 2008, 123, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Zhan, F.; Bignotti, E.; Siegel, E.R.; Cane, S.; Bellone, S.; Palmieri, M.; Anfossi, S.; Thomas, M.; Burnett, A.; et al. Gene expression profiles of primary HPV16- and HPV18-infected early stage cervical cancers and normal cervical epithelium: Identification of novel candidate molecular markers for cervical cancer diagnosis and therapy. Virology 2005, 331, 269–291. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.F.; Cheung, T.H.; Tsao, G.S.; Lo, K.W.; Yim, S.F.; Wang, V.W.; Heung, M.M.; Chan, S.C.; Chan, L.K.; Ho, T.W.; et al. Genome-wide gene expression profiling of cervical cancer in Hong Kong women by oligonucleotide microarray. Int. J. Cancer 2006, 118, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Gyongyosi, E.; Szalmas, A.; Ferenczi, A.; Poliska, S.; Konya, J.; Veress, G. Transcriptional regulation of genes involved in keratinocyte differentiation by human papillomavirus 16 oncoproteins. Arch. Virol. 2015, 160, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, T.; Gu, Q.; Herbert, I.; Stevenson, A.; Iliev, V.; Watkins, G.; Pollock, C.; Bhatia, R.; Cuschieri, K.; Herzyk, P.; et al. RNASeq analysis of differentiated keratinocytes reveals a massive response to late events during human papillomavirus type 16 infection, including loss of epithelial barrier function. J. Virol. 2017. [Google Scholar] [CrossRef]
- Gyongyosi, E.; Szalmas, A.; Ferenczi, A.; Konya, J.; Gergely, L.; Veress, G. Effects of human papillomavirus (HPV) type 16 oncoproteins on the expression of involucrin in human keratinocytes. Virol. J. 2012, 9, 36. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Pfister, H.J. Beta genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef]
- Tan, M.J.; White, E.A.; Sowa, M.E.; Harper, J.W.; Aster, J.C.; Howley, P.M. Cutaneous beta-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1473–1480. [Google Scholar] [CrossRef]
- Brimer, N.; Lyons, C.; Wallberg, A.E.; Vande Pol, S.B. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31, 4639–4646. [Google Scholar] [CrossRef] [PubMed]
- Neary, K.; DiMaio, D. Open reading frames E6 and E7 of bovine papillomavirus type 1 are both required for full transformation of mouse C127 cells. J. Virol. 1989, 63, 259–266. [Google Scholar] [PubMed]
- Sarver, N.; Rabson, M.S.; Yang, Y.C.; Byrne, J.C.; Howley, P.M. Localization and analysis of bovine papillomavirus type 1 transforming functions. J. Virol. 1984, 52, 377–388. [Google Scholar] [PubMed]
- Tong, X.; Howley, P.M. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 1997, 94, 4412–4417. [Google Scholar] [CrossRef]
- Tong, X.; Salgia, R.; Li, J.L.; Griffin, J.D.; Howley, P.M. The bovine papillomavirus E6 protein binds to the LD motif repeats of paxillin and blocks its interaction with vinculin and the focal adhesion kinase. J. Biol. Chem. 1997, 272, 33373–33376. [Google Scholar] [CrossRef]
- White, E.A.; Walther, J.; Javanbakht, H.; Howley, P.M. Genus beta human papillomavirus E6 proteins vary in their effects on the transactivation of p53 target genes. J. Virol. 2014, 88, 8201–8212. [Google Scholar] [CrossRef]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef]
- Nomine, Y.; Masson, M.; Charbonnier, S.; Zanier, K.; Ristriani, T.; Deryckere, F.; Sibler, A.P.; Desplancq, D.; Atkinson, R.A.; Weiss, E.; et al. Structural and functional analysis of E6 oncoprotein: Insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol. Cell 2006, 21, 665–678. [Google Scholar] [CrossRef]
- Zanier, K.; Charbonnier, S.; Sidi, A.O.; McEwen, A.G.; Ferrario, M.G.; Poussin-Courmontagne, P.; Cura, V.; Brimer, N.; Babah, K.O.; Ansari, T.; et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 2013, 339, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Bohl, J.; Das, K.; Dasgupta, B.; Vande Pol, S.B. Competitive binding to a charged leucine motif represses transformation by a papillomavirus E6 oncoprotein. Virology 2000, 271, 163–170. [Google Scholar] [CrossRef]
- Chen, J.J.; Hong, Y.; Rustamzadeh, E.; Baleja, J.D.; Androphy, E.J. Identification of an alpha helical motif sufficient for association with papillomavirus E6. J. Biol. Chem. 1998, 273, 13537–13544. [Google Scholar] [CrossRef]
- McCormack, S.J.; Brazinski, S.E.; Moore, J.L., Jr.; Werness, B.A.; Goldstein, D.J. Activation of the focal adhesion kinase signal transduction pathway in cervical carcinoma cell lines and human genital epithelial cells immortalized with human papillomavirus type 18. Oncogene 1997, 15, 265–274. [Google Scholar] [CrossRef]
- Vande Pol, S.B.; Brown, M.C.; Turner, C.E. Association of Bovine Papillomavirus Type 1 E6 oncoprotein with the focal adhesion protein paxillin through a conserved protein interaction motif. Oncogene 1998, 16, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Drews, C.M.; Vande Pol, S.B. Association of papillomavirus E6 proteins with either MAML1 or E6AP clusters E6 proteins by structure, function, and evolutionary relatedness. PLoS Pathog. 2017, 13, e1006781. [Google Scholar] [CrossRef]
- McElhinny, A.S.; Li, J.L.; Wu, L. Mastermind-like transcriptional co-activators: Emerging roles in regulating cross talk among multiple signaling pathways. Oncogene 2008, 27, 5138–5147. [Google Scholar] [CrossRef] [PubMed]
- Saint Just Ribeiro, M.; Wallberg, A.E. Transcriptional mechanisms by the coregulator MAML1. Curr. Protein Pept. Sci. 2009, 10, 570–576. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.E.; Macdonald, R.J. Notch-independent functions of CSL. Curr. Top. Dev. Biol. 2011, 97, 55–74. [Google Scholar] [CrossRef]
- Shao, H.; Huang, Q.; Liu, Z.J. Targeting Notch signaling for cancer therapeutic intervention. Adv. Pharmacol. 2012, 65, 191–234. [Google Scholar] [CrossRef]
- Meyers, J.M.; Spangle, J.M.; Munger, K. The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J. Virol. 2013, 87, 4762–4767. [Google Scholar] [CrossRef]
- Wu, L.; Aster, J.C.; Blacklow, S.C.; Lake, R.; Artavanis-Tsakonas, S.; Griffin, J.D. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 2000, 26, 484–489. [Google Scholar] [CrossRef]
- Liang, Y.; Ganem, D. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi’s sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 2004, 78, 6818–6826. [Google Scholar] [CrossRef]
- Robertson, E.S.; Grossman, S.; Johannsen, E.; Miller, C.; Lin, J.; Tomkinson, B.; Kieff, E. Epstein-Barr virus nuclear protein 3C modulates transcription through interaction with the sequence-specific DNA-binding protein J kappa. J. Virol. 1995, 69, 3108–3116. [Google Scholar]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar] [CrossRef]
- Grossman, S.R.; Johannsen, E.; Tong, X.; Yalamanchili, R.; Kieff, E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 7568–7572. [Google Scholar] [CrossRef]
- Zimber-Strobl, U.; Strobl, L.J.; Meitinger, C.; Hinrichs, R.; Sakai, T.; Furukawa, T.; Honjo, T.; Bornkamm, G.W. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J. 1994, 13, 4973–4982. [Google Scholar] [CrossRef]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Ansieau, S.; Strobl, L.J.; Leutz, A. Activation of the Notch-regulated transcription factor CBF1/RBP-Jkappa through the 13SE1A oncoprotein. Genes Dev. 2001, 15, 380–385. [Google Scholar] [CrossRef][Green Version]
- Sherman, L.; Schlegel, R. Serum- and calcium-induced differentiation of human keratinocytes is inhibited by the E6 oncoprotein of human papillomavirus type 16. J. Virol. 1996, 70, 3269–3279. [Google Scholar]
- Pei, X.F.; Sherman, L.; Sun, Y.H.; Schlegel, R. HPV-16 E7 protein bypasses keratinocyte growth inhibition by serum and calcium. Carcinogenesis 1998, 19, 1481–1486. [Google Scholar] [CrossRef]
- Lichtig, H.; Algrisi, M.; Botzer, L.E.; Abadi, T.; Verbitzky, Y.; Jackman, A.; Tommasino, M.; Zehbe, I.; Sherman, L. HPV16 E6 natural variants exhibit different activities in functional assays relevant to the carcinogenic potential of E6. Virology 2006, 350, 216–227. [Google Scholar] [CrossRef]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007, 21, 562–577. [Google Scholar] [CrossRef]
- Dotto, G.P. Crosstalk of Notch with p53 and p63 in cancer growth control. Nat. Rev. Cancer 2009, 9, 587–595. [Google Scholar] [CrossRef]
- Ben Khalifa, Y.; Teissier, S.; Tan, M.K.; Phan, Q.T.; Daynac, M.; Wong, W.Q.; Thierry, F. The human papillomavirus E6 oncogene represses a cell adhesion pathway and disrupts focal adhesion through degradation of TAp63beta upon transformation. PLoS Pathog. 2011, 7, e1002256. [Google Scholar] [CrossRef][Green Version]
- Yugawa, T.; Handa, K.; Narisawa-Saito, M.; Ohno, S.; Fujita, M.; Kiyono, T. Regulation of Notch1 gene expression by p53 in epithelial cells. Mol. Cell. Biol. 2007, 27, 3732–3742. [Google Scholar] [CrossRef]
- Kranjec, C.; Holleywood, C.; Libert, D.; Griffin, H.; Mahmood, R.; Isaacson, E.; Doorbar, J. Modulation of basal cell fate during productive and transforming HPV-16 infection is mediated by progressive E6-driven depletion of Notch. J. Pathol. 2017, 242, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Kuncharin, Y.; Sangphech, N.; Kueanjinda, P.; Bhattarakosol, P.; Palaga, T. MAML1 regulates cell viability via the NF-kappaB pathway in cervical cancer cell lines. Exp. Cell Res. 2011, 317, 1830–1840. [Google Scholar] [CrossRef]
- Ramdass, B.; Maliekal, T.T.; Lakshmi, S.; Rehman, M.; Rema, P.; Nair, P.; Mukherjee, G.; Reddy, B.K.; Krishna, S.; Radhakrishna Pillai, M. Coexpression of Notch1 and NF-kappaB signaling pathway components in human cervical cancer progression. Gynecol. Oncol. 2007, 104, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Rong, C.; Feng, Y.; Ye, Z. Notch is a critical regulator in cervical cancer by regulating Numb splicing. Oncol. Lett. 2017, 13, 2465–2470. [Google Scholar] [CrossRef]
- Tripathi, R.; Rath, G.; Hussain, S.; Jawanjal, P.; Bandil, K.; Sharma, V.; Bharadwaj, M.; Mehrotra, R. Jagged-1 induced molecular alterations in HPV associated invasive squamous cell and adenocarcinoma of the human uterine cervix. Sci. Rep. 2018, 8, 9359. [Google Scholar] [CrossRef]
- Vazquez-Ulloa, E.; Ramos-Cruz, A.C.; Prada, D.; Aviles-Salas, A.; Chavez-Blanco, A.D.; Herrera, L.A.; Lizano, M.; Contreras-Paredes, A. Loss of nuclear NOTCH1, but not its negative regulator NUMB, is an independent predictor of cervical malignancy. Oncotarget 2018, 9, 18916–18928. [Google Scholar] [CrossRef]
- Zagouras, P.; Stifani, S.; Blaumueller, C.M.; Carcangiu, M.L.; Artavanis-Tsakonas, S. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc. Natl. Acad. Sci. USA 1995, 92, 6414–6418. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Fukusumi, T.; Califano, J.A. The NOTCH Pathway in Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2018, 97, 645–653. [Google Scholar] [CrossRef]
- Rettig, E.M.; Chung, C.H.; Bishop, J.A.; Howard, J.D.; Sharma, R.; Li, R.J.; Douville, C.; Karchin, R.; Izumchenko, E.; Sidransky, D.; et al. Cleaved NOTCH1 Expression Pattern in Head and Neck Squamous Cell Carcinoma Is Associated with NOTCH1 Mutation, HPV Status, and High-Risk Features. Cancer Prev. Res. 2015, 8, 287–295. [Google Scholar] [CrossRef]
- Meyers, J.M.; Grace, M.; Uberoi, A.; Lambert, P.F.; Munger, K. Inhibition of TGF-beta and NOTCH Signaling by Cutaneous Papillomaviruses. Front. Microbiol. 2018, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Fleming, N.I.; Jorissen, R.N.; Mouradov, D.; Christie, M.; Sakthianandeswaren, A.; Palmieri, M.; Day, F.; Li, S.; Tsui, C.; Lipton, L.; et al. SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res. 2013, 73, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Schonleben, F.; Li, X.; Su, G.H. Disruption of transforming growth factor beta-Smad signaling pathway in head and neck squamous cell carcinoma as evidenced by mutations of SMAD2 and SMAD4. Cancer Lett. 2007, 245, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Kanuma, T.; Mizunuma, H.; Takama, F.; Ibuki, Y.; Wake, N.; Mogi, A.; Shitara, Y.; Takenoshita, S. Analysis of specific gene mutations in the transforming growth factor-beta signal transduction pathway in human ovarian cancer. Cancer Res. 2000, 60, 4507–4512. [Google Scholar] [PubMed]
- Valcourt, U.; Carthy, J.; Okita, Y.; Alcaraz, L.; Kato, M.; Thuault, S.; Bartholin, L.; Moustakas, A. Analysis of Epithelial-Mesenchymal Transition Induced by Transforming Growth Factor beta. Methods Mol. Biol. 2016, 1344, 147–181. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.A.; Jacob, Y.; Cassonnet, P.; Favre, M. Human papillomavirus type 5 E6 oncoprotein represses the transforming growth factor beta signaling pathway by binding to SMAD3. J. Virol. 2006, 80, 12420–12424. [Google Scholar] [CrossRef]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef]
- Collins, A.S.; Nakahara, T.; Do, A.; Lambert, P.F. Interactions with pocket proteins contribute to the role of human papillomavirus type 16 E7 in the papillomavirus life cycle. J. Virol. 2005, 79, 14769–14780. [Google Scholar] [CrossRef]
- Westphal, K.; Akgul, B.; Storey, A.; Nindl, I. Cutaneous human papillomavirus E7 type-specific effects on differentiation and proliferation of organotypic skin cultures. Anal. Cell. Pathol. 2009, 31, 213–226. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef]
- Balsitis, S.J.; Sage, J.; Duensing, S.; Munger, K.; Jacks, T.; Lambert, P.F. Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Mol. Cell. Biol. 2003, 23, 9094–9103. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Stein, R.W.; Moran, E.; Yaciuk, P.; Schlegel, R.; Lyons, R.M.; Pittelkow, M.R.; Munger, K.; Howley, P.M.; Moses, H.L. TGF-beta 1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell 1990, 61, 777–785. [Google Scholar] [CrossRef]
- Lee, D.K.; Kim, B.C.; Kim, I.Y.; Cho, E.A.; Satterwhite, D.J.; Kim, S.J. The human papilloma virus E7 oncoprotein inhibits transforming growth factor-beta signaling by blocking binding of the Smad complex to its target sequence. J. Biol. Chem. 2002, 277, 38557–38564. [Google Scholar] [CrossRef]
- Soares, E.; Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 2017. [Google Scholar] [CrossRef]
- Melar-New, M.; Laimins, L.A. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 2010, 84, 5212–5221. [Google Scholar] [CrossRef]
- Mighty, K.K.; Laimins, L.A. p63 is necessary for the activation of human papillomavirus late viral functions upon epithelial differentiation. J. Virol. 2011, 85, 8863–8869. [Google Scholar] [CrossRef]
- Eldakhakhny, S.; Zhou, Q.; Crosbie, E.J.; Sayan, B.S. Human papillomavirus E7 induces p63 expression to modulate DNA damage response. Cell Death Dis. 2018, 9, 127. [Google Scholar] [CrossRef]
- Wang, T.Y.; Chen, B.F.; Yang, Y.C.; Chen, H.; Wang, Y.; Cviko, A.; Quade, B.J.; Sun, D.; Yang, A.; McKeon, F.D.; et al. Histologic and immunophenotypic classification of cervical carcinomas by expression of the p53 homologue p63: A study of 250 cases. Hum. Pathol. 2001, 32, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Szalmas, A.; Tomaic, V.; Basukala, O.; Massimi, P.; Mittal, S.; Konya, J.; Banks, L. The PTPN14 Tumor Suppressor Is a Degradation Target of Human Papillomavirus E7. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Munger, K.; Howley, P.M. High-Risk Human Papillomavirus E7 Proteins Target PTPN14 for Degradation. mBio 2016, 7. [Google Scholar] [CrossRef]
- Hansen, C.G.; Moroishi, T.; Guan, K.L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Nagatomo, I.; Suzuki, E.; Mizuno, T.; Kumagai, T.; Berezov, A.; Zhang, H.; Karlan, B.; Greene, M.I.; Wang, Q. YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene 2013, 32, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, N.; Figel, S.A.; Wilson, K.E.; Morrison, C.D.; Gelman, I.H.; Zhang, J. PTPN14 interacts with and negatively regulates the oncogenic function of YAP. Oncogene 2013, 32, 1266–1273. [Google Scholar] [CrossRef]
- Michaloglou, C.; Lehmann, W.; Martin, T.; Delaunay, C.; Hueber, A.; Barys, L.; Niu, H.; Billy, E.; Wartmann, M.; Ito, M.; et al. The tyrosine phosphatase PTPN14 is a negative regulator of YAP activity. PLoS ONE 2013, 8, e61916. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, J.; Wang, X.; Yuan, J.; Li, X.; Feng, L.; Park, J.I.; Chen, J. PTPN14 is required for the density-dependent control of YAP1. Genes Dev. 2012, 26, 1959–1971. [Google Scholar] [CrossRef]
- Zhang, H.; Pasolli, H.A.; Fuchs, E. Yes-associated protein (YAP) transcriptional coactivator functions in balancing growth and differentiation in skin. Proc. Natl. Acad. Sci. USA 2011, 108, 2270–2275. [Google Scholar] [CrossRef]
- Wasson, C.W.; Morgan, E.L.; Muller, M.; Ross, R.L.; Hartley, M.; Roberts, S.; Macdonald, A. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget 2017, 8, 103581–103600. [Google Scholar] [CrossRef]
- De Thé, H. Differentiation therapy revisited. Nat. Rev. Cancer 2018, 18, 117–127. [Google Scholar] [CrossRef]
- Ablain, J.; de Thé, H. Retinoic acid signaling in cancer: The parable of acute promyelocytic leukemia. Int. J. Cancer 2014, 135, 2262–2272. [Google Scholar] [CrossRef]
- Yan, M.; Liu, Q. Differentiation therapy: A promising strategy for cancer treatment. Chin. J. Cancer 2016, 35, 3. [Google Scholar] [CrossRef]
- Cheepala, S.B.; Yin, W.; Syed, Z.; Gill, J.N.; McMillian, A.; Kleiner, H.E.; Lynch, M.; Loganantharaj, R.; Trutschl, M.; Cvek, U.; et al. Identification of the B-Raf/Mek/Erk MAP kinase pathway as a target for all-trans retinoic acid during skin cancer promotion. Mol. Cancer 2009, 8, 27. [Google Scholar] [CrossRef]
- Verma, A.K. Inhibition of both stage I and stage II mouse skin tumour promotion by retinoic acid and the dependence of inhibition of tumor promotion on the duration of retinoic acid treatment. Cancer Res. 1987, 47, 5097–5101. [Google Scholar]
- Zhang, M.L.; Tao, Y.; Zhou, W.Q.; Ma, P.C.; Cao, Y.P.; He, C.D.; Wei, J.; Li, L.J. All-trans retinoic acid induces cell-cycle arrest in human cutaneous squamous carcinoma cells by inhibiting the mitogen-activated protein kinase-activated protein 1 pathway. Clin. Exp. Dermatol. 2014, 39, 354–360. [Google Scholar] [CrossRef]
- Arany, I.; Ember, I.A.; Tyring, S.K. All-trans-retinoic acid activates caspase-1 in a dose-dependent manner in cervical squamous carcinoma cells. Anticancer Res. 2003, 23, 471–473. [Google Scholar]
- Arany, I.; Whitehead, W.E.; Ember, I.A.; Tyring, S.K. Dose-dependent activation of p21WAF1 transcription by all-trans-acid in cervical squamous carcinoma cells. Anticancer Res. 2003, 23, 495–497. [Google Scholar]
- Ruffin, M.T.; Bailey, J.M.; Normolle, D.P.; Michael, C.W.; Bieniasz, M.E.; Kmak, D.C.; Unger, E.R.; Brenner, D.E. Low-dose topical delivery of all-trans retinoic acid for cervical intraepithelial neoplasia II and III. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2148–2152. [Google Scholar]
- Wahlberg, P.; Einarsdottir, M.; Fex, G.; Rydell, R. The additive antiproliferative effect of all-trans retinoic acid and interferon-alpha2a on human cervical carcinoma cell lines is not associated with increased expression of retinoid receptors. Anti-Cancer Drugs 1997, 8, 522–528. [Google Scholar] [CrossRef]
- Weiss, G.R.; Liu, P.Y.; Alberts, D.S.; Peng, Y.M.; Fisher, E.; Xu, M.J.; Scudder, S.A.; Baker, L.H., Jr.; Moore, D.F.; Lippman, S.M. 13-cis-retinoic acid or all-trans-retinoic acid plus interferon-alpha in recurrent cervical cancer: A Southwest Oncology Group phase II randomized trial. Gynecol. Oncol. 1998, 71, 386–390. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.K.; Li, Y.; Hafner, M.; Banerjee, N.S.; Tang, S.; Briskin, D.; Meyers, C.; Chow, L.T.; Xie, X.; et al. microRNAs are biomarkers of oncogenic human papillomavirus infections. Proc. Natl. Acad. Sci. USA 2014, 111, 4262–4267. [Google Scholar] [CrossRef] [PubMed]
- Freije, A.; Molinuevo, R.; Ceballos, L.; Cagigas, M.; Alonso-Lecue, P.; Rodriguez, R.; Menendez, P.; Aberdam, D.; De Diego, E.; Gandarillas, A. Inactivation of p53 in Human Keratinocytes Leads to Squamous Differentiation and Shedding via Replication Stress and Mitotic Slippage. Cell Rep. 2014, 9, 1349–1360. [Google Scholar] [CrossRef]
- Ying, Z.; Sandoval, M.; Beronja, S. Oncogenic activation of PI3K induces progenitor cell differentiation to suppress epidermal growth. Nat. Cell Biol. 2018, 20, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Gandarillas, A.; Watt, F.M. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997, 11, 2869–2882. [Google Scholar] [CrossRef]
- Molinuevo, R.; Freije, A.; de Pedro, I.; Stoll, S.W.; Elder, J.T.; Gandarillas, A. FOXM1 allows human keratinocytes to bypass the oncogene-induced differentiation checkpoint in response to gain of MYC or loss of p53. Oncogene 2017, 36, 956–965. [Google Scholar] [CrossRef]
- Waikel, R.L.; Wang, X.J.; Roop, D.R. Targeted expression of c-Myc in the epidermis alters normal proliferation, differentiation and UV-B induced apoptosis. Oncogene 1999, 18, 4870–4878. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Watt, F.M.; Frye, M.; Benitah, S.A. MYC in mammalian epidermis: How can an oncogene stimulate differentiation? Nat. Rev. Cancer 2008, 8, 234–242. [Google Scholar] [CrossRef]
- Gebhardt, A.; Frye, M.; Herold, S.; Benitah, S.A.; Braun, K.; Samans, B.; Watt, F.M.; Elsasser, H.P.; Eilers, M. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. J. Cell Biol. 2006, 172, 139–149. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Kemp, C.J.; Donehower, L.A.; Bradley, A.; Balmain, A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 1993, 74, 813–822. [Google Scholar] [CrossRef]


© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, E.A. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses 2019, 11, 369. https://doi.org/10.3390/v11040369
White EA. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses. 2019; 11(4):369. https://doi.org/10.3390/v11040369
Chicago/Turabian StyleWhite, Elizabeth A. 2019. "Manipulation of Epithelial Differentiation by HPV Oncoproteins" Viruses 11, no. 4: 369. https://doi.org/10.3390/v11040369
APA StyleWhite, E. A. (2019). Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses, 11(4), 369. https://doi.org/10.3390/v11040369