Ebola Virus Entry: From Molecular Characterization to Drug Discovery

 ,
, 
 ,
, 

Abstract
1. Introduction
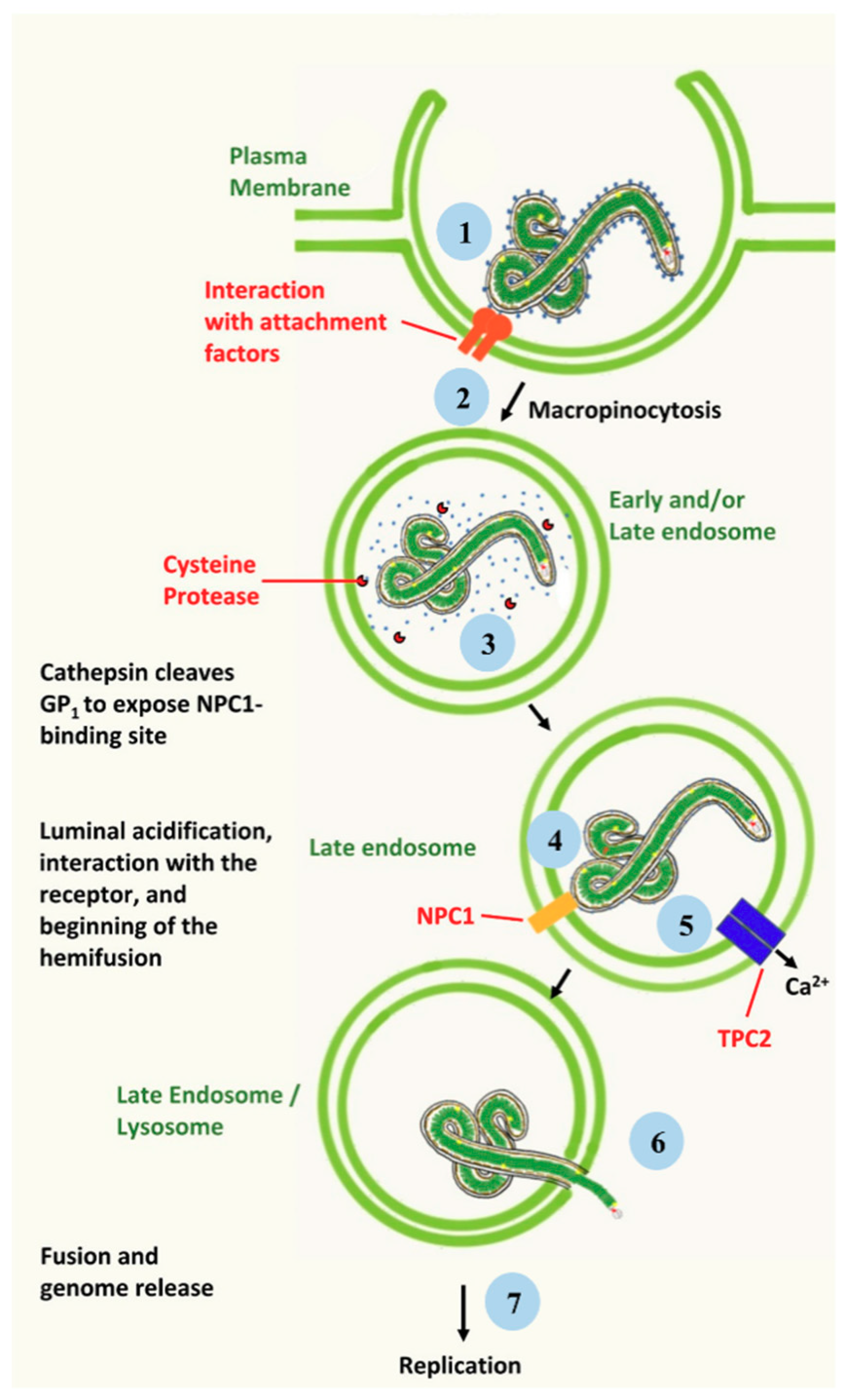
2. Ebola Virus Infection of Target Cells
3. Viral Models for Drug Discovery That Can Be Handled in BSL-2 Facilities: Targeting the Entry Step
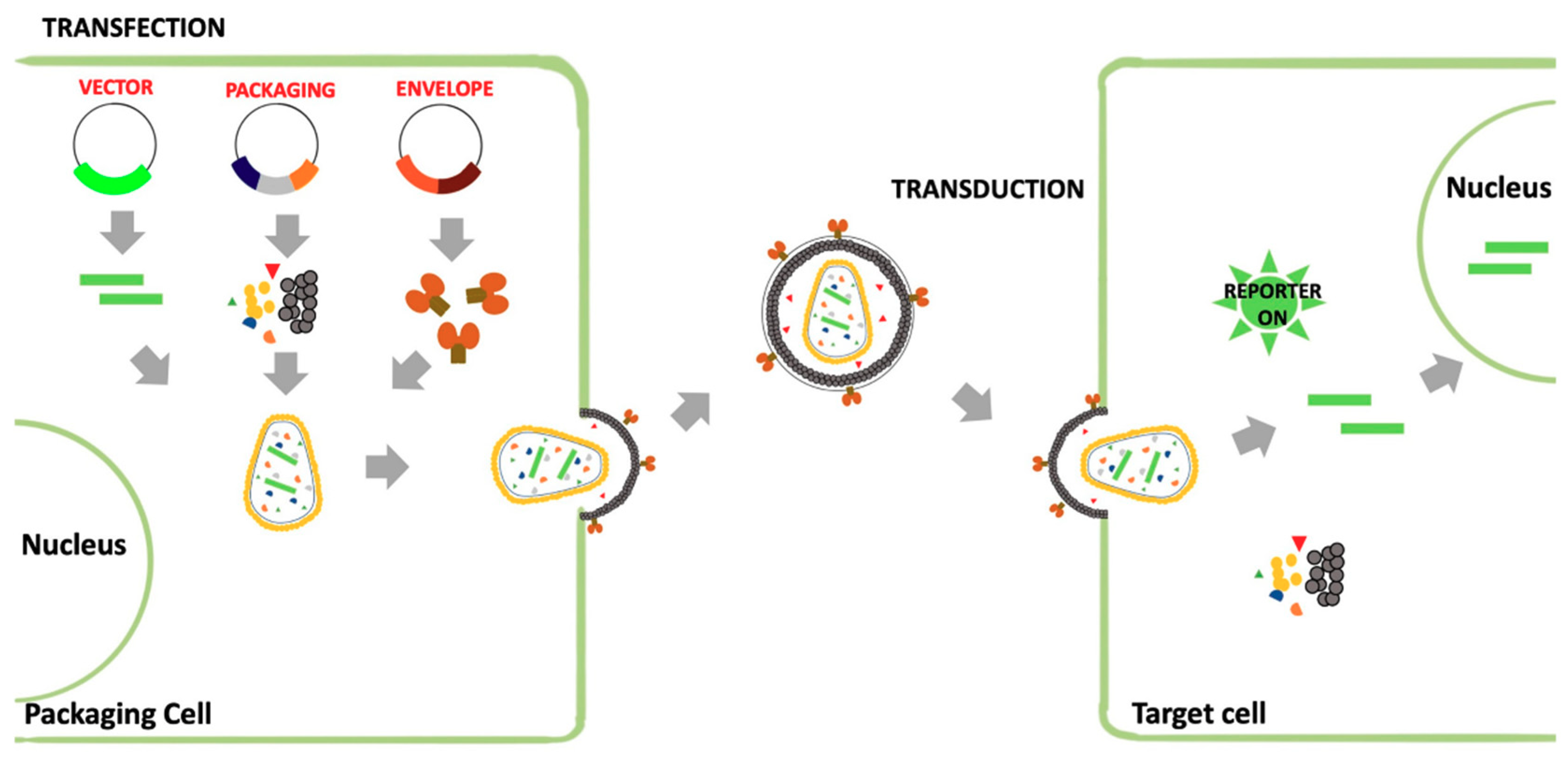
3.1. Viral Pseudotypes
3.1.1. Recombinant Indiana Vesiculovirus
3.1.2. Retroviral Vectors (RVs)
3.2. Ebola Virus-Like Particles (eVLPs)
4. Ebola Virus Entry Inhibitors
4.1. Ion Channel Inhibitors
4.2. Antimicrobial Agents
4.2.1. Antiparasitic Drugs
4.2.2. Antibiotics and Antifungal Drugs
4.3. Psychoactive Drugs
4.4. Selective Estrogen Receptor Modulators
4.5. Protein Kinase Inhibitors
4.6. Miscellaneous Compounds That Inhibit EBOV Entry
5. Conclusions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class &Compound | Viral Model | Validation with EBOV 1 | Reference |
|---|---|---|---|
| Ion channel inhibitors | |||
| Amiodarone | RV, VSV, VLP | YES | [62,63,64] |
| Bepridil | RV, VSV | YES | [69] |
| Dronedarone | RV | YES | [62] |
| Noricumazole A | RV, VSV | N/A | [72] |
| Tetrandrine | VSV | YES | [33] |
| Verapamil | RV | YES | [62] |
| Antimicrobial agents | |||
| Albendazole | VLP | N/A | [82] |
| Aminoquinoline | VSV | YES | [75] |
| Amodiaquine | VSV | YES | [75] |
| Azithromycin | VLP | N/A | [82] |
| Chloroquine | VSV | YES | [75] |
| Emetine | VLP | YES | [81] |
| Ferroquine | VLP | N/A | [79] |
| Hydroxychloroquine | VSV | YES | [75] |
| Mebendazole | VLP | N/A | [82] |
| Suramin | VSV | YES | [80] |
| Teicoplanin | RV, VLP | N/A | [83,84] |
| Terconazole | RV, VSV | YES | [69,85] |
| Triparanol | RV, VSV | YES | [85] |
| Psychoactive drugs | |||
| Benztropine | RV | YES | [88] |
| Chlorpromazine | RV | YES | [86] |
| Chlorcyclizine | RV | YES | [89] |
| Diphenhydramine | RV | YES | [89] |
| Diphenhylpyraline | VSV | YES | [75] |
| Imipramine | VSV | YES | [30] |
| Ketotifen | VSV | YES | [75] |
| Maprotiline | VLP | N/A | [82] |
| Promethazine | RV | YES | [88] |
| Sertraline | RV, VSV | YES | [69] |
| Trimipramine | RV | YES | [88] |
| Selective estrogen receptor modulators | |||
| Clomiphene | VLP, VSV | YES | [69,85,91] |
| Toremifene | VLP | YES | [69,91] |
| Protein kinase inhibitors | |||
| Apilimod | RV, VLP | YES | [98] |
| Erlotinib | VSV | YES | [97] |
| Pyridinyl imidazole | VLP | YES | [100] |
| Sunitinib | VSV | YES | [97] |
| 1-Benzyl-3-cetyl-2-methylimidazolium iodide | VSV | N/A | [103] |
| Miscellaneous compounds | |||
| Aloperine derivatives | RV | N/A | [107] |
| Benzodiazepine derivatives | RV | YES | [104] |
| C-peptide | VSV | YES | [113] |
| Calpeptin | VLP | N/A | [79] |
| Cyclo-peptides | VSV | N/A | [114] |
| Dyphyllin derivatives | VSV | YES | [110] |
| Ellagic acid | RV | YES | [108] |
| Imynodyn 17 | VLP | N/A | [79] |
| MBX2254/2270 | RV | YES | [105] |
| ML9 | VLP | N/A | [79] |
| N-acetyl-l-leucyl-l-leucyl-l methional | VLP | N/A | [79] |
| Nobiletin | VLP | N/A | [79] |
| Retro 2 and derivatives | VLP, VSV | YES | [109] |
| U18666A | VSV | YES | [30,31] |
References
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet 2011, 377, 849–862. [Google Scholar] [CrossRef]
- Goldstein, T.; Anthony, S.J.; Gbakima, A.; Bird, B.H.; Bangura, J.; Tremeau-Bravard, A.; Belaganahalli, M.N.; Wells, H.L.; Dhanota, J.K.; Liang, E.; et al. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. Nat. Microbiol. 2018, 3, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-L.; Tan, C.W.; Anderson, D.E.; Jiang, R.-D.; Li, B.; Zhang, W.; Zhu, Y.; Lim, X.F.; Zhou, P.; Liu, X.-L.; et al. Characterization of a filovirus (Měnglà virus) from Rousettus bats in China. Nat. Microbiol. 2019, 4, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Subissi, L.; Keita, M.; Mesfin, S.; Rezza, G.; Diallo, B.; Van Gucht, S.; Musa, E.O.; Yoti, Z.; Keita, S.; Djingarey, M.H.; et al. Ebola Virus Transmission Caused by Persistently Infected Survivors of the 2014–2016 Outbreak in West Africa. J. Infect. Dis. 2018, 218, S287–S291. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.; Bollinger, L.; Johnson, J.C.; Wada, J.; Radoshitzky, S.R.; Palacios, G.; Bavari, S.; Jahrling, P.B.; Kuhn, J.H. Neglected filoviruses. FEMS Microbiol. Rev. 2016, 40, 494–519. [Google Scholar] [CrossRef] [PubMed]
- Baseler, L.; Chertow, D.S.; Johnson, K.M.; Feldmann, H.; Morens, D.M. The Pathogenesis of Ebola Virus Disease. Annu. Rev. Pathol. 2017, 12, 387–418. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular Entry of Ebola Virus Involves Uptake by a Macropinocytosis-Like Mechanism and Subsequent Trafficking through Early and Late Endosomes. PLoS Pathog. 2010, 6, e1001110. [Google Scholar] [CrossRef]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus Is Internalized into Host Cells via Macropinocytosis in a Viral Glycoprotein-Dependent Manner. PLoS Pathog. 2010, 6, e1001121. [Google Scholar] [CrossRef]
- Mulherkar, N.; Raaben, M.; de la Torre, J.C.; Whelan, S.P.; Chandran, K. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 2011, 419, 72–83. [Google Scholar] [CrossRef]
- Takada, A.; Watanabe, S.; Ito, H.; Okazaki, K.; Kida, H.; Kawaoka, Y. Downregulation of β1 Integrins by Ebola Virus Glycoprotein: Implication for Virus Entry. Virology 2000, 278, 20–26. [Google Scholar] [CrossRef]
- Alvarez, C.P.; Lasala, F.; Carrillo, J.; Muñiz, O.; Corbí, A.L.; Delgado, R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002, 76, 6841–6844. [Google Scholar] [CrossRef]
- Usami, K.; Matsuno, K.; Igarashi, M.; Denda-Nagai, K.; Takada, A.; Irimura, T. Involvement of viral envelope GP2 in Ebola virus entry into cells expressing the macrophage galactose-type C-type lectin. Biochem. Biophys. Res. Commun. 2011, 407, 74–78. [Google Scholar] [CrossRef]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. USA 2011, 108, 8426–8431. [Google Scholar] [CrossRef] [PubMed]
- Brudner, M.; Karpel, M.; Lear, C.; Chen, L.; Yantosca, L.M.; Scully, C.; Sarraju, A.; Sokolovska, A.; Zariffard, M.R.; Eisen, D.P.; et al. Lectin-Dependent Enhancement of Ebola Virus Infection via Soluble and Transmembrane C-type Lectin Receptors. PLoS ONE 2013, 8, e60838. [Google Scholar] [CrossRef]
- Simmons, G.; Reeves, J.D.; Grogan, C.C.; Vandenberghe, L.H.; Baribaud, F.; Whitbeck, J.C.; Burke, E.; Buchmeier, M.J.; Soilleux, E.J.; Riley, J.L.; et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 2003, 305, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Takada, A.; Fujioka, K.; Tsuiji, M.; Morikawa, A.; Higashi, N.; Ebihara, H.; Kobasa, D.; Feldmann, H.; Irimura, T.; Kawaoka, Y. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Virol. 2004, 78, 2943–2947. [Google Scholar] [CrossRef]
- Shimojima, M.; Ikeda, Y.; Kawaoka, Y. The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 2007, 196 (Suppl. 2), S259–S263. [Google Scholar] [CrossRef]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Dube, D.; Brecher, M.B.; Delos, S.E.; Rose, S.C.; Park, E.W.; Schornberg, K.L.; Kuhn, J.H.; White, J.M. The primed ebolavirus glycoprotein (19-kilodalton GP1,2): Sequence and residues critical for host cell binding. J. Virol. 2009, 83, 2883–2891. [Google Scholar] [CrossRef]
- Schornberg, K.L.; Shoemaker, C.J.; Dube, D.; Abshire, M.Y.; Delos, S.E.; Bouton, A.H.; White, J.M. α5β1-Integrin controls ebolavirus entry by regulating endosomal cathepsins. Proc. Natl. Acad. Sci. USA 2009, 106, 8003–8008. [Google Scholar] [CrossRef]
- Brindley, M.A.; Hunt, C.L.; Kondratowicz, A.S.; Bowman, J.; Sinn, P.L.; McCray, P.B.; Quinn, K.; Weller, M.L.; Chiorini, J.A.; Maury, W. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 2011, 415, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.L.; Kolokoltsov, A.A.; Davey, R.A.; Maury, W. The Tyro3 Receptor Kinase Axl Enhances Macropinocytosis of Zaire Ebolavirus. J. Virol. 2011, 85, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.E.; Adhikary, S.; Kolokoltsov, A.A.; Davey, R.A. Ebolavirus Requires Acid Sphingomyelinase Activity and Plasma Membrane Sphingomyelin for Infection. J. Virol. 2012, 86, 7473–7483. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef] [PubMed]
- Schornberg, K.; Matsuyama, S.; Kabsch, K.; Delos, S.; Bouton, A.; White, J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. [Google Scholar] [CrossRef] [PubMed]
- Hood, C.L.; Abraham, J.; Boyington, J.C.; Leung, K.; Kwong, P.D.; Nabel, G.J. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: Implications for viral entry and immunogenicity. J. Virol. 2010, 84, 2972–2982. [Google Scholar] [CrossRef] [PubMed]
- Brecher, M.; Schornberg, K.L.; Delos, S.E.; Fusco, M.L.; Saphire, E.O.; White, J.M. Cathepsin Cleavage Potentiates the Ebola Virus Glycoprotein To Undergo a Subsequent Fusion-Relevant Conformational Change. J. Virol. 2012, 86, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Bornholdt, Z.A.; Ndungo, E.; Fusco, M.L.; Bale, S.; Flyak, A.I.; Crowe, J.E.; Chandran, K.; Saphire, E.O. Host-Primed Ebola Virus GP Exposes a Hydrophobic NPC1 Receptor-Binding Pocket, Revealing a Target for Broadly Neutralizing Antibodies. mBio 2016, 7, e02154-15. [Google Scholar] [CrossRef]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann–Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.H.; Obernosterer, G.; Raaben, M.; Herbert, A.S.; Deffieu, M.S.; Krishnan, A.; Ndungo, E.; Sandesara, R.G.; Carette, J.E.; Kuehne, A.I.; et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012, 31, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.-C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.A.; D’Souza, R.S.; Ruas, M.; Galione, A.; Casanova, J.E.; White, J.M. Ebolavirus Glycoprotein Directs Fusion through NPC1+ Endolysosomes. J. Virol. 2016, 90, 605–610. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Schornberg, K.L. A new player in the puzzle of filovirus entry. Nat. Rev. Microbiol. 2012, 10, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Coll, J.M. The glycoprotein G of rhabdoviruses. Arch. Virol. 1995, 140, 827–851. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Feldmann, H. Recombinant Vesicular Stomatitis Virus–Based Vaccines Against Ebola and Marburg Virus Infections. J. Infect. Dis. 2011, 204, S1075–S1081. [Google Scholar] [CrossRef] [PubMed]
- Whitt, M.A. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 2010, 169, 365–374. [Google Scholar] [CrossRef]
- Whitt, M.A.; Geisbert, T.W.; Mire, C.E. Single-Vector, Single-Injection Recombinant Vesicular Stomatitis Virus Vaccines Against High-Containment Viruses. In Methods in Molecular Biology; Clifton, N.J., Ed.; Humana Press: New York, NY, USA, 2016; Volume 1403, pp. 295–311. [Google Scholar]
- Chen, Q.; Tang, K.; Zhang, X.; Chen, P.; Guo, Y. Establishment of pseudovirus infection mouse models for in vivo pharmacodynamics evaluation of filovirus entry inhibitors. Acta Pharm. Sin. B 2018, 8, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Saïb, A. Early steps of retrovirus replicative cycle. Retrovirology 2004, 1, 9. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Krausslich, H.-G. HIV-1 Assembly, Budding, and Maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.E.; Chicaybam, L.; Stein, R.T.; Tanuri, A.; Delgado-Cañedo, A.; Bonamino, M.H. Retroviral vectors and transposons for stable gene therapy: Advances, current challenges and perspectives. J. Transl. Med. 2016, 14, 288. [Google Scholar] [CrossRef]
- Joglekar, A.V.; Sandoval, S. Pseudotyped Lentiviral Vectors: One Vector, Many Guises. Hum. Gene Ther. Methods 2017, 28, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.; Schoehn, G.; Dessen, A.; Forest, E.; Volchkov, V.; Dolnik, O.; Klenk, H.D.; Weissenhorn, W. Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J. Mol. Biol. 2000, 300, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.; Schoehn, G.; Kohlhaas, C.; Klenk, H.-D.; Ruigrok, R.W.H.; Weissenhorn, W. Oligomerization and polymerization of the filovirus matrix protein VP40. Virology 2003, 312, 359–368. [Google Scholar] [CrossRef]
- Bornholdt, Z.A.; Noda, T.; Abelson, D.M.; Halfmann, P.; Wood, M.R.; Kawaoka, Y.; Saphire, E.O. Structural Rearrangement of Ebola Virus VP40 Begets Multiple Functions in the Virus Life Cycle. Cell 2013, 154, 763–774. [Google Scholar] [CrossRef]
- Harty, R.N.; Brown, M.E.; Wang, G.; Huibregtse, J.; Hayes, F.P. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: Implications for filovirus budding. Proc. Natl. Acad. Sci. USA 2000, 97, 13871–13876. [Google Scholar] [CrossRef]
- Jasenosky, L.D.; Neumann, G.; Lukashevich, I.; Kawaoka, Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J. Virol. 2001, 75, 5205–5214. [Google Scholar] [CrossRef]
- Timmins, J.; Scianimanico, S.; Schoehn, G.; Weissenhorn, W. Vesicular Release of Ebola Virus Matrix Protein VP40. Virology 2001, 283, 1–6. [Google Scholar] [CrossRef]
- Hoenen, T.; Groseth, A.; Kolesnikova, L.; Theriault, S.; Ebihara, H.; Hartlieb, B.; Bamberg, S.; Feldmann, H.; Ströher, U.; Becker, S. Infection of naive target cells with virus-like particles: Implications for the function of ebola virus VP24. J. Virol. 2006, 80, 7260–7264. [Google Scholar] [CrossRef]
- Reynard, O.; Nemirov, K.; Page, A.; Mateo, M.; Raoul, H.; Weissenhorn, W.; Volchkov, V.E. Conserved proline-rich region of Ebola virus matrix protein VP40 is essential for plasma membrane targeting and virus-like particle release. J. Infect. Dis. 2011, 204 (Suppl. 3), S884–S891. [Google Scholar] [CrossRef] [PubMed]
- Martinez, O.; Johnson, J.; Manicassamy, B.; Rong, L.; Olinger, G.G.; Hensley, L.E.; Basler, C.F. Zaire Ebola virus entry into human dendritic cells is insensitive to cathepsin L inhibition. Cell. Microbiol. 2010, 12, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Tscherne, D.M.; Manicassamy, B.; García-Sastre, A. An enzymatic virus-like particle assay for sensitive detection of virus entry. J. Virol. Methods 2010, 163, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, T.; Groseth, A.; de Kok-Mercado, F.; Kuhn, J.H.; Wahl-Jensen, V. Minigenomes, transcription and replication competent virus-like particles and beyond: Reverse genetics systems for filoviruses and other negative stranded hemorrhagic fever viruses. Antivir. Res. 2011, 91, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, T.; Feldmann, H. Reverse genetics systems as tools for the development of novel therapies against filoviruses. Expert Rev. Anti-Infect. Ther. 2014, 12, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Watt, A.; Moukambi, F.; Banadyga, L.; Groseth, A.; Callison, J.; Herwig, A.; Ebihara, H.; Feldmann, H.; Hoenen, T. A Novel Life Cycle Modeling System for Ebola Virus Shows a Genome Length-Dependent Role of VP24 in Virus Infectivity. J. Virol. 2014, 88, 10511–10524. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Hoenen, T.; Canard, B.; Decroly, E. Filovirus proteins for antiviral drug discovery: A structure/function analysis of surface glycoproteins and virus entry. Antivir. Res. 2016, 135, 1–14. [Google Scholar] [CrossRef]
- Bixler, S.L.; Duplantier, A.J.; Bavari, S. Discovering Drugs for the Treatment of Ebola Virus. Curr. Treat. Options Infect. Dis. 2017, 9, 299–317. [Google Scholar] [CrossRef]
- Sweiti, H.; Ekwunife, O.; Jaschinski, T.; Lhachimi, S.K. Repurposed Therapeutic Agents Targeting the Ebola Virus: A Systematic Review. Curr. Ther. Res. 2017, 84, 10–21. [Google Scholar] [CrossRef]
- Salata, C.; Calistri, A.; Parolin, C.; Baritussio, A.; Palù, G. Antiviral activity of cationic amphiphilic drugs. Expert Rev. Anti-Infect. Ther. 2017, 15. [Google Scholar] [CrossRef]
- Gehring, G.; Rohrmann, K.; Atenchong, N.; Mittler, E.; Becker, S.; Dahlmann, F.; Pöhlmann, S.; Vondran, F.W.R.; David, S.; Manns, M.P.; et al. The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J. Antimicrob. Chemother. 2014, 69, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Salata, C.; Baritussio, A.; Munegato, D.; Calistri, A.; Ha, H.R.; Bigler, L.; Fabris, F.; Parolin, C.; Palù, G.; Mirazimi, A. Amiodarone and metabolite MDEA inhibit Ebola virus infection by interfering with the viral entry process. Pathog. Dis. 2015, 73. [Google Scholar] [CrossRef] [PubMed]
- Salata, C.; Munegato, D.; Martelli, F.; Parolin, C.; Calistri, A.; Baritussio, A.; Palù, G. Amiodarone affects Ebola virus binding and entry into target cells. New Microbiol. 2018, 41, 162–164. [Google Scholar] [PubMed]
- Madrid, P.B.; Panchal, R.G.; Warren, T.K.; Shurtleff, A.C.; Endsley, A.N.; Green, C.E.; Kolokoltsov, A.; Davey, R.; Manger, I.D.; Gilfillan, L.; et al. Evaluation of Ebola Virus Inhibitors for Drug Repurposing. ACS Infect. Dis. 2015, 1, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Kann, G.; Becker, S.; Stephan, C.; Brodt, H.-R.; de Leuw, P.; Grünewald, T.; Vogl, T.; Kempf, V.A.J.; Keppler, O.T.; et al. Severe Ebola virus disease with vascular leakage and multiorgan failure: Treatment of a patient in intensive care. Lancet 2015, 385, 1428–1435. [Google Scholar] [CrossRef]
- Lanini, S.; Portella, G.; Vairo, F.; Kobinger, G.P.; Pesenti, A.; Langer, M.; Kabia, S.; Brogiato, G.; Amone, J.; Castilletti, C.; et al. Blood kinetics of Ebola virus in survivors and nonsurvivors. J. Clin. Investig. 2015, 125, 4692–4698. [Google Scholar] [CrossRef] [PubMed]
- Dyall, J.; Johnson, J.C.; Hart, B.J.; Postnikova, E.; Cong, Y.; Zhou, H.; Gerhardt, D.M.; Michelotti, J.; Honko, A.N.; Kern, S.; et al. In Vitro and In Vivo Activity of Amiodarone Against Ebola Virus. J. Infect. Dis. 2018, 218, S592–S596. [Google Scholar] [CrossRef]
- Johansen, L.M.; DeWald, L.E.; Shoemaker, C.J.; Hoffstrom, B.G.; Lear-Rooney, C.M.; Stossel, A.; Nelson, E.; Delos, S.E.; Simmons, J.A.; Grenier, J.M.; et al. A screen of approved drugs and molecular probes identifies therapeutics with anti–Ebola virus activity. Sci. Transl. Med. 2015, 7, 290ra89. [Google Scholar] [CrossRef]
- Ren, J.; Zhao, Y.; Fry, E.E.; Stuart, D.I. Target Identification and Mode of Action of Four Chemically Divergent Drugs against Ebolavirus Infection. J. Med. Chem. 2018, 61, 724–733. [Google Scholar] [CrossRef]
- DeWald, L.E.; Dyall, J.; Sword, J.M.; Torzewski, L.; Zhou, H.; Postnikova, E.; Kollins, E.; Alexander, I.; Gross, R.; Cong, Y.; et al. The Calcium Channel Blocker Bepridil Demonstrates Efficacy in the Murine Model of Marburg Virus Disease. J. Infect. Dis. 2018, 218, S588–S591. [Google Scholar] [CrossRef]
- Beck, S.; Henß, L.; Weidner, T.; Herrmann, J.; Müller, R.; Chao, Y.-K.; Grimm, C.; Weber, C.; Sliva, K.; Schnierle, B.S. Identification of entry inhibitors of Ebola virus pseudotyped vectors from a myxobacterial compound library. Antivir. Res. 2016, 132, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Akpovwa, H. Chloroquine could be used for the treatment of filoviral infections and other viral infections that emerge or emerged from viruses requiring an acidic pH for infectivity. Cell Biochem. Funct. 2016, 34, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A. Use of chloroquine in viral diseases. Lancet Infect. Dis. 2011, 11, 653–654. [Google Scholar] [CrossRef]
- Madrid, P.B.; Chopra, S.; Manger, I.D.; Gilfillan, L.; Keepers, T.R.; Shurtleff, A.C.; Green, C.E.; Iyer, L.V.; Dilks, H.H.; Davey, R.A.; et al. A Systematic Screen of FDA-Approved Drugs for Inhibitors of Biological Threat Agents. PLoS ONE 2013, 8, e60579. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, D.; Safronetz, D.; Prescott, J.; Marzi, A.; Feldmann, F.; Feldmann, H. Lack of Protection Against Ebola Virus from Chloroquine in Mice and Hamsters. Emerg. Infect. Dis. 2015, 21, 1065–1067. [Google Scholar] [CrossRef]
- Dowall, S.D.; Bosworth, A.; Watson, R.; Bewley, K.; Taylor, I.; Rayner, E.; Hunter, L.; Pearson, G.; Easterbrook, L.; Pitman, J.; et al. Chloroquine inhibited Ebola virus replication in vitro but failed to protect against infection and disease in the in vivo guinea pig model. J. Gen. Virol. 2015, 96, 3484–3492. [Google Scholar] [CrossRef]
- Gignoux, E.; Azman, A.S.; de Smet, M.; Azuma, P.; Massaquoi, M.; Job, D.; Tiffany, A.; Petrucci, R.; Sterk, E.; Potet, J.; et al. Effect of Artesunate–Amodiaquine on Mortality Related to Ebola Virus Disease. N. Engl. J. Med. 2016, 374, 23–32. [Google Scholar] [CrossRef]
- Lee, N.; Shum, D.; König, A.; Kim, H.; Heo, J.; Min, S.; Lee, J.; Ko, Y.; Choi, I.; Lee, H.; et al. High-throughput drug screening using the Ebola virus transcription- and replication-competent virus-like particle system. Antivir. Res. 2018, 158, 226–237. [Google Scholar] [CrossRef]
- Henß, L.; Beck, S.; Weidner, T.; Biedenkopf, N.; Sliva, K.; Weber, C.; Becker, S.; Schnierle, B.S. Suramin is a potent inhibitor of Chikungunya and Ebola virus cell entry. Virol. J. 2016, 13, 149. [Google Scholar] [CrossRef]
- Yang, S.; Xu, M.; Lee, E.M.; Gorshkov, K.; Shiryaev, S.A.; He, S.; Sun, W.; Cheng, Y.-S.; Hu, X.; Tharappel, A.M.; et al. Emetine inhibits Zika and Ebola virus infections through two molecular mechanisms: Inhibiting viral replication and decreasing viral entry. Cell Discov. 2018, 4, 31. [Google Scholar] [CrossRef]
- Kouznetsova, J.; Sun, W.; Martínez-Romero, C.; Tawa, G.; Shinn, P.; Chen, C.Z.; Schimmer, A.; Sanderson, P.; McKew, J.C.; Zheng, W.; et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg. Microbes Infect. 2014, 3, e84. [Google Scholar] [CrossRef]
- Zhou, N.; Pan, T.; Zhang, J.; Li, Q.; Zhang, X.; Bai, C.; Huang, F.; Peng, T.; Zhang, J.; Liu, C.; et al. Glycopeptide Antibiotics Potently Inhibit Cathepsin L in the Late Endosome/Lysosome and Block the Entry of Ebola Virus, Middle East Respiratory Syndrome Coronavirus (MERS-CoV), and Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV). J. Biol. Chem. 2016, 291, 9218–9232. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, R.; Li, G.; Gao, Q.; Yuan, S.; Altmeyer, R.; Zou, G. Teicoplanin inhibits Ebola pseudovirus infection in cell culture. Antivir. Res. 2016, 125, 1–7. [Google Scholar] [CrossRef]
- Shoemaker, C.J.; Schornberg, K.L.; Delos, S.E.; Scully, C.; Pajouhesh, H.; Olinger, G.G.; Johansen, L.M.; White, J.M. Multiple Cationic Amphiphiles Induce a Niemann-Pick C Phenotype and Inhibit Ebola Virus Entry and Infection. PLoS ONE 2013, 8, e56265. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Warfield, K.L.; Ruthel, G.; Bavari, S.; Aman, M.J.; Hope, T.J. Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology 2010, 401, 18–28. [Google Scholar] [CrossRef]
- Daniel, J.A.; Chau, N.; Abdel-Hamid, M.K.; Hu, L.; von Kleist, L.; Whiting, A.; Krishnan, S.; Maamary, P.; Joseph, S.R.; Simpson, F.; et al. Phenothiazine-Derived Antipsychotic Drugs Inhibit Dynamin and Clathrin-Mediated Endocytosis. Traffic 2015, 16, 635–654. [Google Scholar] [CrossRef]
- Cheng, H.; Lear-Rooney, C.M.; Johansen, L.; Varhegyi, E.; Chen, Z.W.; Olinger, G.G.; Rong, L. Inhibition of Ebola and Marburg Virus Entry by G Protein-Coupled Receptor Antagonists. J. Virol. 2015, 89, 9932–9938. [Google Scholar] [CrossRef]
- Schafer, A.; Cheng, H.; Xiong, R.; Soloveva, V.; Retterer, C.; Mo, F.; Bavari, S.; Thatcher, G.; Rong, L. Repurposing potential of 1st generation H1-specific antihistamines as anti-filovirus therapeutics. Antivir. Res. 2018, 157, 47–56. [Google Scholar] [CrossRef]
- Cheng, H.; Schafer, A.; Soloveva, V.; Gharaibeh, D.; Kenny, T.; Retterer, C.; Zamani, R.; Bavari, S.; Peet, N.P.; Rong, L. Identification of a coumarin-based antihistamine-like small molecule as an anti-filoviral entry inhibitor. Antivir. Res. 2017, 145, 24–32. [Google Scholar] [CrossRef]
- Johansen, L.M.; Brannan, J.M.; Delos, S.E.; Shoemaker, C.J.; Stossel, A.; Lear, C.; Hoffstrom, B.G.; DeWald, L.E.; Schornberg, K.L.; Scully, C.; et al. FDA-Approved Selective Estrogen Receptor Modulators Inhibit Ebola Virus Infection. Sci. Transl. Med. 2013, 5, 190ra79. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, J.; Harlos, K.; Jones, D.M.; Zeltina, A.; Bowden, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef]
- Fan, H.; Du, X.; Zhang, J.; Zheng, H.; Lu, X.; Wu, Q.; Li, H.; Wang, H.; Shi, Y.; Gao, G.; et al. Selective inhibition of Ebola entry with selective estrogen receptor modulators by disrupting the endolysosomal calcium. Sci. Rep. 2017, 7, 41226. [Google Scholar] [CrossRef]
- Nelson, E.; Barnes, A.; Wiehle, R.; Fontenot, G.; Hoenen, T.; White, J. Clomiphene and Its Isomers Block Ebola Virus Particle Entry and Infection with Similar Potency: Potential Therapeutic Implications. Viruses 2016, 8, 206. [Google Scholar] [CrossRef]
- Bekerman, E.; Einav, S. Combating emerging viral threats. Science 2015, 348, 282–283. [Google Scholar] [CrossRef]
- Garcia, M.; Cooper, A.; Shi, W.; Bornmann, W.; Carrion, R.; Kalman, D.; Nabel, G.J. Productive Replication of Ebola Virus Is Regulated by the c-Abl1 Tyrosine Kinase. Sci. Transl. Med. 2012, 4, 123ra24. [Google Scholar] [CrossRef]
- Bekerman, E.; Neveu, G.; Shulla, A.; Brannan, J.; Pu, S.-Y.; Wang, S.; Xiao, F.; Barouch-Bentov, R.; Bakken, R.R.; Mateo, R.; et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J. Clin. Investig. 2017, 127, 1338–1352. [Google Scholar] [CrossRef]
- Nelson, E.A.; Dyall, J.; Hoenen, T.; Barnes, A.B.; Zhou, H.; Liang, J.Y.; Michelotti, J.; Dewey, W.H.; DeWald, L.E.; Bennett, R.S.; et al. The phosphatidylinositol-3-phosphate 5-kinase inhibitor apilimod blocks filoviral entry and infection. PLoS Negl. Trop. Dis. 2017, 11, e0005540. [Google Scholar] [CrossRef]
- Dyall, J.; Nelson, E.A.; DeWald, L.E.; Guha, R.; Hart, B.J.; Zhou, H.; Postnikova, E.; Logue, J.; Vargas, W.M.; Gross, R.; et al. Identification of Combinations of Approved Drugs With Synergistic Activity Against Ebola Virus in Cell Cultures. J. Infect. Dis. 2018, 218, S672–S678. [Google Scholar] [CrossRef]
- Johnson, J.C.; Martinez, O.; Honko, A.N.; Hensley, L.E.; Olinger, G.G.; Basler, C.F. Pyridinyl imidazole inhibitors of p38 MAP kinase impair viral entry and reduce cytokine induction by Zaire ebolavirus in human dendritic cells. Antivir. Res. 2014, 107, 102–109. [Google Scholar] [CrossRef]
- Neveu, G.; Ziv-Av, A.; Barouch-Bentov, R.; Berkerman, E.; Mulholland, J.; Einav, S. AP-2-Associated Protein Kinase 1 and Cyclin G-Associated Kinase Regulate Hepatitis C Virus Entry and Are Potential Drug Targets. J. Virol. 2015, 89, 4387–4404. [Google Scholar] [CrossRef]
- Pu, S.-Y.; Wouters, R.; Schor, S.; Rozenski, J.; Barouch-Bentov, R.; Prugar, L.I.; O’Brien, C.M.; Brannan, J.M.; Dye, J.M.; Herdewijn, P.; et al. Optimization of Isothiazolo[4,3- b ]pyridine-Based Inhibitors of Cyclin G Associated Kinase (GAK) with Broad-Spectrum Antiviral Activity. J. Med. Chem. 2018, 61, 6178–6192. [Google Scholar] [CrossRef] [PubMed]
- Moeschler, S.; Locher, S.; Zimmer, G. 1-Benzyl-3-cetyl-2-methylimidazolium Iodide (NH125) Is a Broad-Spectrum Inhibitor of Virus Entry with Lysosomotropic Features. Viruses 2018, 10, 306. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Li, B.; Mills, D.M.; Panchal, R.G.; Cardinale, S.C.; Butler, M.M.; Peet, N.P.; Majgier-Baranowska, H.; Williams, J.D.; Patel, I.; et al. Identification of a Small-Molecule Entry Inhibitor for Filoviruses. J. Virol. 2011, 85, 3106–3119. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Mills, D.M.; Mitchell, D.; Ndungo, E.; Williams, J.D.; Herbert, A.S.; Dye, J.M.; Moir, D.T.; Chandran, K.; Patterson, J.L.; et al. Novel Small Molecule Entry Inhibitors of Ebola Virus. J. Infect. Dis. 2015, 212, S425–S434. [Google Scholar] [CrossRef] [PubMed]
- Anantpadma, M.; Kouznetsova, J.; Wang, H.; Huang, R.; Kolokoltsov, A.; Guha, R.; Lindstrom, A.R.; Shtanko, O.; Simeonov, A.; Maloney, D.J.; et al. Large-Scale Screening and Identification of Novel Ebola Virus and Marburg Virus Entry Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 4471–4481. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Q.; Zhang, N.; Li, Q.; Liu, Z.; Li, Y.; Gao, L.; Wang, Y.; Deng, H.; Song, D. Discovery and evolution of aloperine derivatives as novel anti-filovirus agents through targeting entry stage. Eur. J. Med. Chem. 2018, 149, 45–55. [Google Scholar] [CrossRef]
- Cui, Q.; Du, R.; Anantpadma, M.; Schafer, A.; Hou, L.; Tian, J.; Davey, R.; Cheng, H.; Rong, L. Identification of Ellagic Acid from Plant Rhodiola rosea L. as an Anti-Ebola Virus Entry Inhibitor. Viruses 2018, 10, 152. [Google Scholar] [CrossRef]
- Shtanko, O.; Sakurai, Y.; Reyes, A.N.; Noël, R.; Cintrat, J.-C.; Gillet, D.; Barbier, J.; Davey, R.A. Retro-2 and its dihydroquinazolinone derivatives inhibit filovirus infection. Antivir. Res. 2018, 149, 154–163. [Google Scholar] [CrossRef]
- Lindstrom, A.; Anantpadma, M.; Baker, L.; Raghavendra, N.M.; Davey, R.; Davisson, V.J. Phenotypic Prioritization of Diphyllin Derivatives That Block Filoviral Cell Entry by Vacuolar (H+)-ATPase Inhibition. ChemMedChem 2018, 13, 2664–2676. [Google Scholar] [CrossRef]
- van der Linden, W.A.; Schulze, C.J.; Herbert, A.S.; Krause, T.B.; Wirchnianski, A.A.; Dye, J.M.; Chandran, K.; Bogyo, M. Cysteine Cathepsin Inhibitors as Anti-Ebola Agents. ACS Infect. Dis. 2016, 2, 173–179. [Google Scholar] [CrossRef]
- Cui, Q.; Cheng, H.; Xiong, R.; Zhang, G.; Du, R.; Anantpadma, M.; Davey, R.; Rong, L. Identification of Diaryl-Quinoline Compounds as Entry Inhibitors of Ebola Virus. Viruses 2018, 10, 678. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.H.; Harrison, J.S.; Radoshitzky, S.R.; Higgins, C.D.; Chi, X.; Dong, L.; Kuhn, J.H.; Bavari, S.; Lai, J.R.; Chandran, K. Inhibition of Ebola Virus Entry by a C-peptide Targeted to Endosomes. J. Biol. Chem. 2011, 286, 15854–15861. [Google Scholar] [CrossRef]
- Li, Q.; Ma, L.; Yi, D.; Wang, H.; Wang, J.; Zhang, Y.; Guo, Y.; Li, X.; Zhou, J.; Shi, Y.; et al. Novel cyclo-peptides inhibit Ebola pseudotyped virus entry by targeting primed GP protein. Antivir. Res. 2018, 155, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; He, S.; Martínez-Romero, C.; Kouznetsova, J.; Tawa, G.; Xu, M.; Shinn, P.; Fisher, E.; Long, Y.; Motabar, O.; et al. Synergistic drug combination effectively blocks Ebola virus infection. Antivir. Res. 2017, 137, 165–172. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salata, C.; Calistri, A.; Alvisi, G.; Celestino, M.; Parolin, C.; Palù, G. Ebola Virus Entry: From Molecular Characterization to Drug Discovery. Viruses 2019, 11, 274. https://doi.org/10.3390/v11030274
Salata C, Calistri A, Alvisi G, Celestino M, Parolin C, Palù G. Ebola Virus Entry: From Molecular Characterization to Drug Discovery. Viruses. 2019; 11(3):274. https://doi.org/10.3390/v11030274
Chicago/Turabian StyleSalata, Cristiano, Arianna Calistri, Gualtiero Alvisi, Michele Celestino, Cristina Parolin, and Giorgio Palù. 2019. "Ebola Virus Entry: From Molecular Characterization to Drug Discovery" Viruses 11, no. 3: 274. https://doi.org/10.3390/v11030274
APA StyleSalata, C., Calistri, A., Alvisi, G., Celestino, M., Parolin, C., & Palù, G. (2019). Ebola Virus Entry: From Molecular Characterization to Drug Discovery. Viruses, 11(3), 274. https://doi.org/10.3390/v11030274