Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies


Abstract
1. Introduction
2. Latency and the Latent Reservoirs
2.1. The Latent Reservoir Is Constituted of Several Subsets of Immune Cells with Specific Features
2.1.1. The Different Populations of the Reservoir
2.1.2. The Different Compartments of the Reservoir
2.1.3. Establishment and Maintenance of the Reservoir
2.2. The Establishment and Maintenance of the Latency Involves the Hijacking of Different Cellular Processes
2.2.1. HIV Plays a Game of Cat and Mouse
2.2.2. The Hypermutagenicity of the Virus Favors the Latency
2.2.3. The Site of Integration and the Epigenetic Marks Play a Role in HIV Expression
2.2.4. RNA Processing
2.2.5. miRNAs
2.2.6. The Cell Cycle
2.2.7. Apoptosis
2.2.8. Autophagy
2.2.9. Transcription is Linked to Most of the Processes Listed above
3. The HIV Proximal Promoter and its Cognate Transcriptions Factors
3.1. Pre-Initiation Complex (PIC) Formation and the General Transcription Factors (GTFs)
3.1.1. 7SK Complex (P-TEFb)
3.1.2. NF-κB
3.1.3. NFAT1
3.1.4. SP1
3.1.5. RBF-2 (USF1/USF2/TFII-I)
3.2. Viral Auxiliary Proteins
3.2.1. Tat
3.2.2. Vpr
4. HIV Cure/Remission Strategies
4.1. CRISPR/Cas9
4.2. Immunotherapies
4.3. HIV Transcription and Immune Evasion: A Game of Cat and Mouse?
4.4. Shock and Kill
4.5. Block and Lock
4.6. Other Cure/Remission Strategies
5. Conclusions and Perspectives
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Barré-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef]
- Global HIV & AIDS Statistics—2018 Fact Sheet. UNAIDS. Available online: http://www.unaids.org/en/resources/fact-sheet (accessed on 14 January 2019).
- Joint United Nations Programme on HIV/AIDS (UNAIDS). Progress towards the 90-90-90 Targets Ending AIDS GLOBAL AIDS UPDATE. 2017. Available online: http://www.unaids.org/en/resources/campaigns/globalAIDSupdate2017 (accessed on 3 March 2019).
- Seatla, K.K.; Avalos, A.; Moyo, S.; Mine, M.; Diphoko, T.; Mosepele, M.; Gaolatlhe, T.; Rowley, C.F.; Ramaabya, D.; Jarvis, J.N.; et al. Four-class drug-resistant HIV-1 subtype C in a treatment experienced individual on dolutegravir-based antiretroviral therapy in Botswana. AIDS 2018, 32, 1899–1902. [Google Scholar] [CrossRef]
- Jordan, M.R.; Dean, N.E.; World Health Organization. Department of HIV/AIDS; Bill & Melinda Gates Foundation; United States President’s Emergency Plan for AIDS Relief. Global Report on Early Warning Indicators of HIV Drug Resistance: Technical Report; WHO: Geneva, Switzerland, 2016; ISBN 9789241511179. [Google Scholar]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef]
- Chun, T.-W.; Justement, J.S.; Murray, D.; Hallahan, C.W.; Maenza, J.; Collier, A.C.; Sheth, P.M.; Kaul, R.; Ostrowski, M.; Moir, S.; et al. Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: Implications for eradication. AIDS 2010, 24, 2803–2808. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Hartogensis, W.; Bacchetti, P.; Hunt, P.W.; Hatano, H.; Sinclair, E.; Epling, L.; Lee, T.-H.; Busch, M.P.; McCune, J.M.; et al. Antiretroviral therapy initiated within 6 months of HIV infection is associated with lower T-cell activation and smaller HIV reservoir size. J. Infect. Dis. 2013, 208, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef]
- Lee, G.Q.; Lichterfeld, M. Diversity of HIV-1 reservoirs in CD4+ T-cell subpopulations. Curr. Opin. HIV AIDS 2016, 11, 383–387. [Google Scholar] [CrossRef]
- Barton, K.; Winckelmann, A.; Palmer, S. HIV-1 Reservoirs During Suppressive Therapy. Trends Microbiol. 2016, 24, 345–355. [Google Scholar] [CrossRef] [PubMed]
- von Stockenstrom, S.; Odevall, L.; Lee, E.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Shao, W.; Hoh, R.; Ho, T.; et al. Longitudinal Genetic Characterization Reveals That Cell Proliferation Maintains a Persistent HIV Type 1 DNA Pool During Effective HIV Therapy. J. Infect. Dis. 2015, 212, 596–607. [Google Scholar] [CrossRef]
- Wightman, F.; Solomon, A.; Khoury, G.; Green, J.A.; Gray, L.; Gorry, P.R.; Ho, Y.S.; Saksena, N.K.; Hoy, J.; Crowe, S.M.; et al. Both CD31(+) and CD31− naive CD4(+) T cells are persistent HIV type 1-infected reservoirs in individuals receiving antiretroviral therapy. J. Infect. Dis. 2010, 202, 1738–1748. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Sarabia, N.; Bateson, R.E.; Dahl, N.P.; Crooks, A.M.; Kuruc, J.D.; Margolis, D.M.; Archin, N.M. Quantitation of replication-competent HIV-1 in populations of resting CD4+ T cells. J. Virol. 2014, 88, 14070–14077. [Google Scholar] [CrossRef]
- Kovacs, J.A.; Lempicki, R.A.; Sidorov, I.A.; Adelsberger, J.W.; Sereti, I.; Sachau, W.; Kelly, G.; Metcalf, J.A.; Davey, R.T.; Falloon, J.; et al. Induction of prolonged survival of CD4+ T lymphocytes by intermittent IL-2 therapy in HIV-infected patients. J. Clin. Investig. 2005, 115, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Vandergeeten, C.; Fromentin, R.; DaFonseca, S.; Lawani, M.B.; Sereti, I.; Lederman, M.M.; Ramgopal, M.; Routy, J.-P.; Sékaly, R.-P.; Chomont, N. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 2013, 121, 4321–4329. [Google Scholar] [CrossRef]
- Jaafoura, S.; de Goër de Herve, M.G.; Hernandez-Vargas, E.A.; Hendel-Chavez, H.; Abdoh, M.; Mateo, M.C.; Krzysiek, R.; Merad, M.; Seng, R.; Tardieu, M.; et al. Progressive contraction of the latent HIV reservoir around a core of less-differentiated CD4+ memory T Cells. Nat. Commun. 2014, 5, 5407. [Google Scholar] [CrossRef]
- Darshan, R.; Leblois, A.; Hansel, D. Interference and shaping in sensorimotor adaptations with rewards. PLoS Comput. Biol. 2014, 10, e1003377. [Google Scholar] [CrossRef]
- Gosselin, A.; Monteiro, P.; Chomont, N.; Diaz-Griffero, F.; Said, E.A.; Fonseca, S.; Wacleche, V.; El-Far, M.; Boulassel, M.-R.; Routy, J.-P.; et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J. Immunol. 2010, 184, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Muranski, P.; Restifo, N.P. Essentials of Th17 cell commitment and plasticity. Blood 2013, 121, 2402–2414. [Google Scholar] [CrossRef]
- Wei, S.; Zhao, E.; Kryczek, I.; Zou, W. Th17 cells have stem cell-like features and promote long-term immunity. Oncoimmunology 2012, 1, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kim, D.; Li, X.; Kiselinova, M.; Ouyang, Z.; Vandekerckhove, L.; Shang, H.; Rosenberg, E.S.; Yu, X.G.; Lichterfeld, M. Th1/17 Polarization of CD4 T Cells Supports HIV-1 Persistence during Antiretroviral Therapy. J. Virol. 2015, 89, 11284–11293. [Google Scholar] [CrossRef] [PubMed]
- Wacleche, V.S.; Goulet, J.-P.; Gosselin, A.; Monteiro, P.; Soudeyns, H.; Fromentin, R.; Jenabian, M.-A.; Vartanian, S.; Deeks, S.G.; Chomont, N.; et al. New insights into the heterogeneity of Th17 subsets contributing to HIV-1 persistence during antiretroviral therapy. Retrovirology 2016, 13, 59. [Google Scholar] [CrossRef]
- Perreau, M.; Savoye, A.-L.; de Crignis, E.; Corpataux, J.-M.; Cubas, R.; Haddad, E.K.; de Leval, L.; Graziosi, C.; Pantaleo, G. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J. Exp. Med. 2013, 210, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Schmitt, N.; Bentebibel, S.-E.; Ranganathan, R.; Bourdery, L.; Zurawski, G.; Foucat, E.; Dullaers, M.; Oh, S.; Sabzghabaei, N.; et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011, 34, 108–121. [Google Scholar] [CrossRef]
- Schultz, B.T.; Teigler, J.E.; Pissani, F.; Oster, A.F.; Kranias, G.; Alter, G.; Marovich, M.; Eller, M.A.; Dittmer, U.; Robb, M.L.; et al. Circulating HIV-Specific Interleukin-21(+)CD4(+) T Cells Represent Peripheral Tfh Cells with Antigen-Dependent Helper Functions. Immunity 2016, 44, 167–178. [Google Scholar] [CrossRef]
- Pallikkuth, S.; Sharkey, M.; Babic, D.Z.; Gupta, S.; Stone, G.W.; Fischl, M.A.; Stevenson, M.; Pahwa, S. Peripheral T Follicular Helper Cells Are the Major HIV Reservoir within Central Memory CD4 T Cells in Peripheral Blood from Chronically HIV-Infected Individuals on Combination Antiretroviral Therapy. J. Virol. 2015, 90, 2718–2728. [Google Scholar] [CrossRef]
- García, M.; Górgolas, M.; Cabello, A.; Estrada, V.; Ligos, J.M.; Fernández-Guerrero, M.; Barros, C.; López-Bernaldo, J.C.; De La Hera, F.J.; Montoya, M.; et al. Peripheral T follicular helper Cells Make a Difference in HIV Reservoir Size between Elite Controllers and Patients on Successful cART. Sci. Rep. 2017, 7, 16799. [Google Scholar] [CrossRef]
- Soriano-Sarabia, N.; Archin, N.M.; Bateson, R.; Dahl, N.P.; Crooks, A.M.; Kuruc, J.D.; Garrido, C.; Margolis, D.M. Peripheral Vγ9Vδ2 T Cells Are a Novel Reservoir of Latent HIV Infection. PLoS Pathog. 2015, 11, e1005201. [Google Scholar] [CrossRef]
- Maddon, P.J.; Dalgleish, A.G.; McDougal, J.S.; Clapham, P.R.; Weiss, R.A.; Axel, R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 1986, 47, 333–348. [Google Scholar] [CrossRef]
- Lifson, J.D.; Feinberg, M.B.; Reyes, G.R.; Rabin, L.; Banapour, B.; Chakrabarti, S.; Moss, B.; Wong-Staal, F.; Steimer, K.S.; Engleman, E.G. Induction of CD4-dependent cell fusion by the HTLV-III/LAV envelope glycoprotein. Nature 1986, 323, 725–728. [Google Scholar] [CrossRef]
- Koenig, S.; Gendelman, H.E.; Orenstein, J.M.; Dal Canto, M.C.; Pezeshkpour, G.H.; Yungbluth, M.; Janotta, F.; Aksamit, A.; Martin, M.A.; Fauci, A.S. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 1986, 233, 1089–1093. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Orenstein, J.M.; Martin, M.A.; Ferrua, C.; Mitra, R.; Phipps, T.; Wahl, L.A.; Lane, H.C.; Fauci, A.S.; Burke, D.S. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 1988, 167, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Muthui, D.; Holte, S.; Nickle, D.; Feng, F.; Brodie, S.; Hwangbo, Y.; Mullins, J.I.; Corey, L. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J. Virol. 2002, 76, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Swingler, S.; Brichacek, B.; Jacque, J.-M.; Ulich, C.; Zhou, J.; Stevenson, M. HIV-1 Nef intersects the macrophage CD40L signalling pathway to promote resting-cell infection. Nature 2003, 424, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Beddall, M.H.; Yu, D.; Iyer, S.R.; Marsh, J.W.; Wu, Y. Human macrophages support persistent transcription from unintegrated HIV-1 DNA. Virology 2008, 372, 300–312. [Google Scholar] [CrossRef]
- Churchill, M.J.; Gorry, P.R.; Cowley, D.; Lal, L.; Sonza, S.; Purcell, D.F.J.; Thompson, K.A.; Gabuzda, D.; McArthur, J.C.; Pardo, C.A.; et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 2006, 12, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; van Lunzen, J.; Tenner-Racz, K.; Grossschupff, G.; Racz, P.; Schmitz, H.; Dietrich, M.; Hufert, F.T. Follicular dendritic cells retain HIV-1 particles on their plasma membrane, but are not productively infected in asymptomatic patients with follicular hyperplasia. J. Immunol. 1994, 153, 1352–1359. [Google Scholar]
- Heath, S.L.; Tew, J.G.; Tew, J.G.; Szakal, A.K.; Burton, G.F. Follicular dendritic cells and human immunodeficiency virus infectivity. Nature 1995, 377, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Sathaliyawala, T.; Kubota, M.; Yudanin, N.; Turner, D.; Camp, P.; Thome, J.J.C.; Bickham, K.L.; Lerner, H.; Goldstein, M.; Sykes, M.; et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 2013, 38, 187–197. [Google Scholar] [CrossRef]
- Noto, A.; Procopio, F.A.; Banga, R.; Suffiotti, M.; Corpataux, J.-M.; Cavassini, M.; Riva, A.; Fenwick, C.; Gottardo, R.; Perreau, M.; et al. CD32+ and PD-1+ Lymph Node CD4 T Cells Support Persistent HIV-1 Transcription in Treated Aviremic Individuals. J. Virol. 2018, 92, e00901-18. [Google Scholar] [CrossRef]
- Chun, T.-W.; Nickle, D.C.; Justement, J.S.; Meyers, J.H.; Roby, G.; Hallahan, C.W.; Kottilil, S.; Moir, S.; Mican, J.M.; Mullins, J.I.; et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J. Infect. Dis. 2008, 197, 714–720. [Google Scholar] [CrossRef]
- Clark, R.A.; Watanabe, R.; Teague, J.E.; Schlapbach, C.; Tawa, M.C.; Adams, N.; Dorosario, A.A.; Chaney, K.S.; Cutler, C.S.; Leboeuf, N.R.; et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci. Transl. Med. 2012, 4, 117ra7. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Gehad, A.; Yang, C.; Scott, L.L.; Teague, J.E.; Schlapbach, C.; Elco, C.P.; Huang, V.; Matos, T.R.; Kupper, T.S.; et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med. 2015, 7, 279ra39. [Google Scholar] [CrossRef] [PubMed]
- Schnell, G.; Spudich, S.; Harrington, P.; Price, R.W.; Swanstrom, R. Compartmentalized human immunodeficiency virus type 1 originates from long-lived cells in some subjects with HIV-1-associated dementia. PLoS Pathog. 2009, 5, e1000395. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Noto, A.; Pollakis, G.; Cavassini, M.; Ohmiti, K.; Corpataux, J.-M.; de Leval, L.; Pantaleo, G.; Perreau, M. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 2016, 22, 754–761. [Google Scholar] [CrossRef]
- Boritz, E.A.; Darko, S.; Swaszek, L.; Wolf, G.; Wells, D.; Wu, X.; Henry, A.R.; Laboune, F.; Hu, J.; Ambrozak, D.; et al. Multiple Origins of Virus Persistence during Natural Control of HIV Infection. Cell 2016, 166, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Schacker, T.; Little, S.; Connick, E.; Gebhard, K.; Zhang, Z.Q.; Krieger, J.; Pryor, J.; Havlir, D.; Wong, J.K.; Schooley, R.T.; et al. Productive infection of T cells in lymphoid tissues during primary and early human immunodeficiency virus infection. J. Infect. Dis. 2001, 183, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Di Mascio, M.; Srinivasula, S.; Bhattacharjee, A.; Cheng, L.; Martiniova, L.; Herscovitch, P.; Lertora, J.; Kiesewetter, D. Antiretroviral tissue kinetics: In vivo imaging using positron emission tomography. Antimicrob. Agents Chemother. 2009, 53, 4086–4095. [Google Scholar] [CrossRef] [PubMed]
- Roulet, V.; Satie, A.-P.; Ruffault, A.; Le Tortorec, A.; Denis, H.; Guist’hau, O.; Patard, J.-J.; Rioux-Leclerq, N.; Gicquel, J.; Jégou, B.; et al. Susceptibility of human testis to human immunodeficiency virus-1 infection in situ and in vitro. Am. J. Pathol. 2006, 169, 2094–2103. [Google Scholar] [CrossRef]
- Huang, Y.; Hoque, M.T.; Jenabian, M.-A.; Vyboh, K.; Whyte, S.-K.; Sheehan, N.L.; Brassard, P.; Bélanger, M.; Chomont, N.; Fletcher, C.V.; et al. Antiretroviral drug transporters and metabolic enzymes in human testicular tissue: Potential contribution to HIV-1 sanctuary site. J. Antimicrob. Chemother. 2016, 71, 1954–1965. [Google Scholar] [CrossRef]
- Macallan, D.C.; Borghans, J.A.M.; Asquith, B. Human T Cell Memory: A Dynamic View. Vaccines 2017, 5, 5. [Google Scholar] [CrossRef]
- Hammarlund, E.; Lewis, M.W.; Hansen, S.G.; Strelow, L.I.; Nelson, J.A.; Sexton, G.J.; Hanifin, J.M.; Slifka, M.K. Duration of antiviral immunity after smallpox vaccination. Nat. Med. 2003, 9, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Agosto, L.M.; Herring, M.B.; Mothes, W.; Henderson, A.J. HIV-1-Infected CD4+ T Cells Facilitate Latent Infection of Resting CD4+ T Cells through Cell-Cell Contact. Cell Rep. 2018, 24, 2088–2100. [Google Scholar] [CrossRef]
- Josefsson, L.; von Stockenstrom, S.; Faria, N.R.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Tan, A.; Ho, T.; Lemey, P.; et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc. Natl. Acad. Sci. USA 2013, 110, E4987–E4996. [Google Scholar] [CrossRef]
- Kieffer, T.L.; Finucane, M.M.; Nettles, R.E.; Quinn, T.C.; Broman, K.W.; Ray, S.C.; Persaud, D.; Siliciano, R.F. Genotypic analysis of HIV-1 drug resistance at the limit of detection: Virus production without evolution in treated adults with undetectable HIV loads. J. Infect. Dis. 2004, 189, 1452–1465. [Google Scholar] [CrossRef]
- Mens, H.; Pedersen, A.G.; Jørgensen, L.B.; Hue, S.; Yang, Y.; Gerstoft, J.; Katzenstein, T.L. Investigating signs of recent evolution in the pool of proviral HIV type 1 DNA during years of successful HAART. AIDS Res. Hum. Retrovir. 2007, 23, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Bosque, A.; Famiglietti, M.; Weyrich, A.S.; Goulston, C.; Planelles, V. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog. 2011, 7, e1002288. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Eisele, E.E.; Rabi, S.A.; Lai, J.; Chioma, S.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS Pathog. 2013, 9, e1003398. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Hosmane, N.N.; Kwon, K.J.; Bruner, K.M.; Capoferri, A.A.; Beg, S.; Rosenbloom, D.I.S.; Keele, B.F.; Ho, Y.-C.; Siliciano, J.D.; Siliciano, R.F. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017, 214, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Buzon, M.J.; Sun, H.; Li, C.; Shaw, A.; Seiss, K.; Ouyang, Z.; Martin-Gayo, E.; Leng, J.; Henrich, T.J.; Li, J.Z.; et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat. Med. 2014, 20, 139–142. [Google Scholar] [CrossRef]
- Carr, J.M.; Hocking, H.; Li, P.; Burrell, C.J. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 1999, 265, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Hübner, W.; Spinelli, M.A.; Chen, B.K. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 2007, 81, 12582–12595. [Google Scholar] [CrossRef] [PubMed]
- Massanella, M.; Richman, D.D. Measuring the latent reservoir in vivo. J. Clin. Investig. 2016, 126, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Fun, A.; Mok, H.P.; Wills, M.R.; Lever, A.M. A highly reproducible quantitative viral outgrowth assay for the measurement of the replication-competent latent HIV-1 reservoir. Sci. Rep. 2017, 7, 43231. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Eriksson, S.; Graf, E.H.; Dahl, V.; Strain, M.C.; Yukl, S.A.; Lysenko, E.S.; Bosch, R.J.; Lai, J.; Chioma, S.; Emad, F.; et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog. 2013, 9, e1003174. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.E.; Niessl, J.; Fromentin, R.; Richard, J.; Porichis, F.; Massanella, M.; Brassard, N.; Alsahafi, N.; Routy, J.-P.; Finzi, A.; et al. Multiparametric characterization of rare HIV-infected cells using an RNA-flow FISH technique. Nat. Protoc. 2017, 12, 2029–2049. [Google Scholar] [CrossRef]
- Calvanese, V.; Chavez, L.; Laurent, T.; Ding, S.; Verdin, E. Dual-color HIV reporters trace a population of latently infected cells and enable their purification. Virology 2013, 446, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Kok, Y.L.; Schmutz, S.; Inderbitzin, A.; Neumann, K.; Kelley, A.; Jörimann, L.; Shilaih, M.; Vongrad, V.; Kouyos, R.D.; Günthard, H.F.; et al. Spontaneous reactivation of latent HIV-1 promoters is linked to the cell cycle as revealed by a genetic-insulators-containing dual-fluorescence HIV-1-based vector. Sci. Rep. 2018, 8, 10204. [Google Scholar] [CrossRef] [PubMed]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Tojal Da Silva, I.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.C.; Pillai, S.K.; et al. Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4+ T cells. Elife 2018, 7, e34655. [Google Scholar] [CrossRef]
- Lee, S.Y.; Choi, B.-S.; Yoon, C.-H.; Kang, C.; Kim, K.; Kim, K.-C. Selection of biomarkers for HIV-1 latency by integrated analysis. Genomics 2018, in press. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Sari, D.; Boussiotis, V.A. PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 2012, 11, 4305–4309. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, M.; Fontenot, A.P.; Mack, D.G.; Lozupone, C.; Dillon, S.; Meditz, A.; Wilson, C.C.; Connick, E.; Palmer, B.E. Programmed death 1 expression on HIV-specific CD4+ T cells is driven by viral replication and associated with T cell dysfunction. J. Immunol. 2007, 179, 1979–1987. [Google Scholar] [CrossRef]
- Kaufmann, D.E.; Kavanagh, D.G.; Pereyra, F.; Zaunders, J.J.; Mackey, E.W.; Miura, T.; Palmer, S.; Brockman, M.; Rathod, A.; Piechocka-Trocha, A.; et al. Upregulation of CTLA-4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 2007, 8, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.-R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Evans, V.A.; van der Sluis, R.M.; Solomon, A.; Dantanarayana, A.; McNeil, C.; Garsia, R.; Palmer, S.; Fromentin, R.; Chomont, N.; Sékaly, R.-P.; et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS 2018, 32, 1491–1497. [Google Scholar] [CrossRef]
- Douek, D.C.; Brenchley, J.M.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Okamoto, Y.; Casazza, J.P.; Kuruppu, J.; Kunstman, K.; Wolinsky, S.; et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature 2002, 417, 95–98. [Google Scholar] [CrossRef]
- Hiener, B.; Horsburgh, B.A.; Eden, J.-S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4+ T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef]
- Cuevas, J.M.; Geller, R.; Garijo, R.; López-Aldeguer, J.; Sanjuán, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef] [PubMed]
- Abram, M.E.; Ferris, A.L.; Shao, W.; Alvord, W.G.; Hughes, S.H. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J. Virol. 2010, 84, 9864–9878. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Van Opijnen, T.; Boerlijst, M.C.; Berkhout, B. Effects of random mutations in the human immunodeficiency virus type 1 transcriptional promoter on viral fitness in different host cell environments. J. Virol. 2006, 80, 6678–6685. [Google Scholar] [CrossRef]
- Kearney, M.F.; Spindler, J.; Shao, W.; Yu, S.; Anderson, E.M.; O’Shea, A.; Rehm, C.; Poethke, C.; Kovacs, N.; Mellors, J.W.; et al. Lack of Detectable HIV-1 Molecular Evolution during Suppressive Antiretroviral Therapy. PLoS Pathog. 2014, 10, e1004010. [Google Scholar] [CrossRef]
- Simonetti, F.R.; Sobolewski, M.D.; Fyne, E.; Shao, W.; Spindler, J.; Hattori, J.; Anderson, E.M.; Watters, S.A.; Hill, S.; Wu, X.; et al. Clonally expanded CD4+ T cells can produce infectious HIV-1 in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1883–1888. [Google Scholar] [CrossRef]
- Singh, P.K.; Plumb, M.R.; Ferris, A.L.; Iben, J.R.; Wu, X.; Fadel, H.J.; Luke, B.T.; Esnault, C.; Poeschla, E.M.; Hughes, S.H.; et al. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 2015, 29, 2287–2297. [Google Scholar] [CrossRef]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV integration site selection: Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007, 17, 1186–1194. [Google Scholar] [CrossRef]
- Serrao, E.; Krishnan, L.; Shun, M.-C.; Li, X.; Cherepanov, P.; Engelman, A.; Maertens, G.N. Integrase residues that determine nucleotide preferences at sites of HIV-1 integration: Implications for the mechanism of target DNA binding. Nucleic Acids Res. 2014, 42, 5164–5176. [Google Scholar] [CrossRef] [PubMed]
- Demeulemeester, J.; De Rijck, J.; Gijsbers, R.; Debyser, Z. Retroviral integration: Site matters: Mechanisms and consequences of retroviral integration site selection. Bioessays 2015, 37, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Schröder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Sowd, G.A.; Serrao, E.; Wang, H.; Wang, W.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, E1054–E1063. [Google Scholar] [CrossRef]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef]
- Albanese, A.; Arosio, D.; Terreni, M.; Cereseto, A. HIV-1 pre-integration complexes selectively target decondensed chromatin in the nuclear periphery. PLoS ONE 2008, 3, e2413. [Google Scholar] [CrossRef]
- Eidahl, J.O.; Crowe, B.L.; North, J.A.; McKee, C.J.; Shkriabai, N.; Feng, L.; Plumb, M.; Graham, R.L.; Gorelick, R.J.; Hess, S.; et al. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013, 41, 3924–3936. [Google Scholar] [CrossRef]
- Sherrill-Mix, S.; Lewinski, M.K.; Famiglietti, M.; Bosque, A.; Malani, N.; Ocwieja, K.E.; Berry, C.C.; Looney, D.; Shan, L.; Agosto, L.M.; et al. HIV latency and integration site placement in five cell-based models. Retrovirology 2013, 10, 90. [Google Scholar] [CrossRef]
- Jordan, A.; Bisgrove, D.; Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003, 22, 1868–1877. [Google Scholar] [CrossRef]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.-C.; O’Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, R.F. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D. V Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Sherrill-Mix, S.; Ocwieja, K.E.; Bushman, F.D. Gene activity in primary T cells infected with HIV89.6: Intron retention and induction of genomic repeats. Retrovirology 2015, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, Z.; Hull, R. Can the HIV-1 splicing machinery be targeted for drug discovery? HIV AIDS (Auckl.) 2017, 9, 63–75. [Google Scholar] [CrossRef]
- Exline, C.M.; Feng, Z.; Stoltzfus, C.M. Negative and positive mRNA splicing elements act competitively to regulate human immunodeficiency virus type 1 vif gene expression. J. Virol. 2008, 82, 3921–3931. [Google Scholar] [CrossRef]
- Tranell, A.; Fenyö, E.M.; Schwartz, S. Serine- and arginine-rich proteins 55 and 75 (SRp55 and SRp75) induce production of HIV-1 vpr mRNA by inhibiting the 5′-splice site of exon 3. J. Biol. Chem. 2010, 285, 31537–31547. [Google Scholar] [CrossRef]
- Shkreta, L.; Blanchette, M.; Toutant, J.; Wilhelm, E.; Bell, B.; Story, B.A.; Balachandran, A.; Cochrane, A.; Cheung, P.K.; Harrigan, P.R.; et al. Modulation of the splicing regulatory function of SRSF10 by a novel compound that impairs HIV-1 replication. Nucleic Acids Res. 2017, 45, 4051–4067. [Google Scholar] [CrossRef]
- Jacquenet, S.; Méreau, A.; Bilodeau, P.S.; Damier, L.; Stoltzfus, C.M.; Branlant, C. A second exon splicing silencer within human immunodeficiency virus type 1 tat exon 2 represses splicing of Tat mRNA and binds protein hnRNP H. J. Biol. Chem. 2001, 276, 40464–40475. [Google Scholar] [CrossRef]
- Asai, K.; Platt, C.; Cochrane, A. Control of HIV-1 env RNA splicing and transport: Investigating the role of hnRNP A1 in exon splicing silencer (ESS3a) function. Virology 2003, 314, 229–242. [Google Scholar] [CrossRef]
- Berro, R.; Kehn, K.; de la Fuente, C.; Pumfery, A.; Adair, R.; Wade, J.; Colberg-Poley, A.M.; Hiscott, J.; Kashanchi, F. Acetylated Tat regulates human immunodeficiency virus type 1 splicing through its interaction with the splicing regulator p32. J. Virol. 2006, 80, 3189–3204. [Google Scholar] [CrossRef]
- Baeyens, A.; Naessens, E.; Van Nuffel, A.; Weening, K.E.; Reilly, A.-M.; Claeys, E.; Trypsteen, W.; Vandekerckhove, L.; Eyckerman, S.; Gevaert, K.; et al. HIV-1 Vpr N-terminal tagging affects alternative splicing of the viral genome. Sci. Rep. 2016, 6, 34573. [Google Scholar] [CrossRef]
- McLaren, M.; Asai, K.; Cochrane, A. A novel function for Sam68: Enhancement of HIV-1 RNA 3′ end processing. RNA 2004, 10, 1119–1129. [Google Scholar] [CrossRef]
- Woolaway, K.; Asai, K.; Emili, A.; Cochrane, A. hnRNP E1 and E2 have distinct roles in modulating HIV-1 gene expression. Retrovirology 2007, 4, 28. [Google Scholar] [CrossRef]
- Graf, M.; Bojak, A.; Deml, L.; Bieler, K.; Wolf, H.; Wagner, R. Concerted action of multiple cis-acting sequences is required for Rev dependence of late human immunodeficiency virus type 1 gene expression. J. Virol. 2000, 74, 10822–10826. [Google Scholar] [CrossRef]
- Rao, S.; Amorim, R.; Niu, M.; Temzi, A.; Mouland, A.J. The RNA surveillance proteins UPF1, UPF2 and SMG6 affect HIV-1 reactivation at a post-transcriptional level. Retrovirology 2018, 15, 42. [Google Scholar] [CrossRef]
- Telwatte, S.; Lee, S.; Somsouk, M.; Hatano, H.; Baker, C.; Kaiser, P.; Kim, P.; Chen, T.-H.; Milush, J.; Hunt, P.W.; et al. Gut and blood differ in constitutive blocks to HIV transcription, suggesting tissue-specific differences in the mechanisms that govern HIV latency. PLoS Pathog. 2018, 14, e1007357. [Google Scholar] [CrossRef]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef]
- Golumbeanu, M.; Cristinelli, S.; Rato, S.; Munoz, M.; Cavassini, M.; Beerenwinkel, N.; Ciuffi, A. Single-Cell RNA-Seq Reveals Transcriptional Heterogeneity in Latent and Reactivated HIV-Infected Cells. Cell Rep. 2018, 23, 942–950. [Google Scholar] [CrossRef]
- Shan, C.-X.; Qiu, N.-C.; Liu, M.-E.; Zha, S.-L.; Song, X.; Du, Z.-P.; Rao, W.-S.; Jiang, D.-Z.; Zhang, W.; Qiu, M. Effects of Diet on Bile Acid Metabolism and Insulin Resistance in Type 2 Diabetic Rats after Roux-en-Y Gastric Bypass. Obes. Surg. 2018, 28, 3044–3053. [Google Scholar] [CrossRef]
- Sarracino, A.; Gharu, L.; Kula, A.; Pasternak, A.O.; Avettand-Fenoel, V.; Rouzioux, C.; Bardina, M.; De Wit, S.; Benkirane, M.; Berkhout, B.; et al. Posttranscriptional Regulation of HIV-1 Gene Expression during Replication and Reactivation from Latency by Nuclear Matrix Protein MATR3. MBio 2018, 9, e02158-18. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Hariharan, M.; Scaria, V.; Pillai, B.; Brahmachari, S.K. Targets for human encoded microRNAs in HIV genes. Biochem. Biophys. Res. Commun. 2005, 337, 1214–1218. [Google Scholar] [CrossRef]
- Ahluwalia, J.K.; Khan, S.Z.; Soni, K.; Rawat, P.; Gupta, A.; Hariharan, M.; Scaria, V.; Lalwani, M.; Pillai, B.; Mitra, D.; et al. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology 2008, 5, 117. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Sung, T.-L.; Rice, A.P. Regulation of cyclin T1 and HIV-1 Replication by microRNAs in resting CD4+ T lymphocytes. J. Virol. 2012, 86, 3244–3252. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Triboulet, R.; Mari, B.; Lin, Y.-L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef]
- Sung, T.-L.; Rice, A.P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef]
- Bennasser, Y.; Le, S.-Y.; Benkirane, M.; Jeang, K.-T. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 2005, 22, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Chable-Bessia, C.; Meziane, O.; Latreille, D.; Triboulet, R.; Zamborlini, A.; Wagschal, A.; Jacquet, J.-M.; Reynes, J.; Levy, Y.; Saib, A.; et al. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 2009, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Fujii, Y.R. Regulation of human immunodeficiency virus 1 transcription by nef microRNA. J. Gen. Virol. 2005, 86, 751–755. [Google Scholar] [CrossRef]
- Klase, Z.; Kale, P.; Winograd, R.; Gupta, M.V.; Heydarian, M.; Berro, R.; McCaffrey, T.; Kashanchi, F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol. Biol. 2007, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, M.; Geng, G.; Liu, B.; Huang, Z.; Luo, H.; Zhou, J.; Guo, X.; Cai, W.; Zhang, H. A novel HIV-1-encoded microRNA enhances its viral replication by targeting the TATA box region. Retrovirology 2014, 11, 23. [Google Scholar] [CrossRef]
- Yeung, M.L.; Bennasser, Y.; Myers, T.G.; Jiang, G.; Benkirane, M.; Jeang, K.-T. Changes in microRNA expression profiles in HIV-1-transfected human cells. Retrovirology 2005, 2, 81. [Google Scholar] [CrossRef]
- Nathans, R.; Chu, C.-Y.; Serquina, A.K.; Lu, C.-C.; Cao, H.; Rana, T.M. Cellular microRNA and P bodies modulate host-HIV-1 interactions. Mol. Cell 2009, 34, 696–709. [Google Scholar] [CrossRef]
- Reynoso, R.; Laufer, N.; Hackl, M.; Skalicky, S.; Monteforte, R.; Turk, G.; Carobene, M.; Quarleri, J.; Cahn, P.; Werner, R.; et al. MicroRNAs differentially present in the plasma of HIV elite controllers reduce HIV infection in vitro. Sci. Rep. 2014, 4, 5915. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.J.; Adams, K.L.; Daher, A.; Gatignol, A. Effective inhibition of HIV-1 production by short hairpin RNAs and small interfering RNAs targeting a highly conserved site in HIV-1 Gag RNA is optimized by evaluating alternative length formats. Antimicrob. Agents Chemother. 2015, 59, 5297–5305. [Google Scholar] [CrossRef] [PubMed]
- Coburn, G.A.; Cullen, B.R. siRNAs: A new wave of RNA-based therapeutics. J. Antimicrob. Chemother. 2003, 51, 753–756. [Google Scholar] [CrossRef]
- Coburn, G.A.; Cullen, B.R. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J. Virol. 2002, 76, 9225–9231. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.I.; Stanwick, T.L.; Dempsey, M.P.; Stevenson, M. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 1991, 254, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Korin, Y.D.; Zack, J.A. Progression to the G1b phase of the cell cycle is required for completion of human immunodeficiency virus type 1 reverse transcription in T cells. J. Virol. 1998, 72, 3161–3168. [Google Scholar]
- Budhiraja, S.; Famiglietti, M.; Bosque, A.; Planelles, V.; Rice, A.P. Cyclin T1 and CDK9 T-loop phosphorylation are downregulated during establishment of HIV-1 latency in primary resting memory CD4+ T cells. J. Virol. 2013, 87, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Budhiraja, S.; Ramakrishnan, R.; Rice, A.P. Phosphatase PPM1A negatively regulates P-TEFb function in resting CD4(+) T cells and inhibits HIV-1 gene expression. Retrovirology 2012, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sansó, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Stivahtis, G.L.; Soares, M.A.; Vodicka, M.A.; Hahn, B.H.; Emerman, M. Conservation and host specificity of Vpr-mediated cell cycle arrest suggest a fundamental role in primate lentivirus evolution and biology. J. Virol. 1997, 71, 4331–4338. [Google Scholar]
- Rogel, M.E.; Wu, L.I.; Emerman, M. The human immunodeficiency virus type 1 vpr gene prevents cell proliferation during chronic infection. J. Virol. 1995, 69, 882–888. [Google Scholar] [PubMed]
- Re, F.; Braaten, D.; Franke, E.K.; Luban, J. Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 1995, 69, 6859–6864. [Google Scholar] [PubMed]
- Gummuluru, S.; Emerman, M. Cell cycle- and Vpr-mediated regulation of human immunodeficiency virus type 1 expression in primary and transformed T-cell lines. J. Virol. 1999, 73, 5422–5430. [Google Scholar]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef]
- Shen, A.; Siliciano, J.D.; Pierson, T.C.; Buck, C.B.; Siliciano, R.F. Establishment of latent HIV-1 infection of resting CD4(+) T lymphocytes does not require inactivation of Vpr. Virology 2000, 278, 227–233. [Google Scholar] [CrossRef]
- Zahoor, M.A.; Xue, G.; Sato, H.; Murakami, T.; Takeshima, S.; Aida, Y. HIV-1 Vpr induces interferon-stimulated genes in human monocyte-derived macrophages. PLoS ONE 2014, 9, e106418. [Google Scholar] [CrossRef]
- Aillet, F.; Masutani, H.; Elbim, C.; Raoul, H.; Chêne, L.; Nugeyre, M.T.; Paya, C.; Barré-Sinoussi, F.; Gougerot-Pocidalo, M.A.; Israël, N. Human immunodeficiency virus induces a dual regulation of Bcl-2, resulting in persistent infection of CD4(+) T- or monocytic cell lines. J. Virol. 1998, 72, 9698–9705. [Google Scholar] [PubMed]
- Berro, R.; de la Fuente, C.; Klase, Z.; Kehn, K.; Parvin, L.; Pumfery, A.; Agbottah, E.; Vertes, A.; Nekhai, S.; Kashanchi, F. Identifying the membrane proteome of HIV-1 latently infected cells. J. Biol. Chem. 2007, 282, 8207–8218. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Wang, X.; Devadas, K.; Zhao, J.; Zhang, P.; Hewlett, I. Some mechanisms of FLIP expression in inhibition of HIV-1 replication in Jurkat cells, CD4+ T cells and PBMCs. J. Cell. Physiol. 2013, 228, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ragupathy, V.; Zhao, J.; Hewlett, I. Molecules from apoptotic pathways modulate HIV-1 replication in Jurkat cells. Biochem. Biophys. Res. Commun. 2011, 414, 20–24. [Google Scholar] [CrossRef]
- Piette, J.; Legrand-Poels, S. HIV-1 reactivation after an oxidative stress mediated by different reactive oxygen species. Chem. Biol. Interact. 1994, 91, 79–89. [Google Scholar] [CrossRef]
- Khan, S.Z.; Hand, N.; Zeichner, S.L. Apoptosis-induced activation of HIV-1 in latently infected cell lines. Retrovirology 2015, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.-H.; Ahmad, R.; Lee, G.Q.; Gao, C.; Chen, H.-R.; Ouyang, Z.; Szucs, M.J.; Kim, D.; Tsibris, A.; Chun, T.-W.; et al. Anti-apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4+ T Cells. Immunity 2018, 48, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Piekna-Przybylska, D.; Maggirwar, S.B. CD4+ memory T cells infected with latent HIV-1 are susceptible to drugs targeting telomeres. Cell Cycle 2018, 17, 2187–2203. [Google Scholar] [CrossRef]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.K.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef]
- Cesana, D.; Santoni de Sio, F.R.; Rudilosso, L.; Gallina, P.; Calabria, A.; Beretta, S.; Merelli, I.; Bruzzesi, E.; Passerini, L.; Nozza, S.; et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat. Commun. 2017, 8, 498. [Google Scholar] [CrossRef]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Proudfoot, N.J. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 2009, 136, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Paz, S.; Krainer, A.R.; Caputi, M. HIV-1 transcription is regulated by splicing factor SRSF1. Nucleic Acids Res. 2014, 42, 13812–13823. [Google Scholar] [CrossRef]
- Mueller, N.; Pasternak, A.O.; Klaver, B.; Cornelissen, M.; Berkhout, B.; Das, A.T. The HIV-1 Tat Protein Enhances Splicing at the Major Splice Donor Site. J. Virol. 2018, 92, e01855-17. [Google Scholar] [CrossRef]
- Bohne, J.; Schambach, A.; Zychlinski, D. New way of regulating alternative splicing in retroviruses: The promoter makes a difference. J. Virol. 2007, 81, 3652–3656. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; De Clercq, E. HIV Genome-Wide Protein Associations: A Review of 30 Years of Research. Microbiol. Mol. Biol. Rev. 2016, 80, 679–731. [Google Scholar] [CrossRef]
- Wilhelm, E.; Bell, B. Selective recognition of viral promoters by host cell transcription complexes: Challenges and opportunities to control latency. Curr. Opin. Virol. 2013, 3, 380–386. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Ooms, M.; Malcolm, T.; Simon, V.; Sadowski, I. Identification and functional analysis of a second RBF-2 binding site within the HIV-1 promoter. Virology 2011, 418, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, E.; Doyle, M.-C.; Nzaramba, I.; Magdzinski, A.; Dumais, N.; Bell, B. CTGC motifs within the HIV core promoter specify Tat-responsive pre-initiation complexes. Retrovirology 2012, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.A.; Bentley, K.; Peeters, A.; Churchill, M.J.; Deacon, N.J. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000, 28, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Taube, R.; Peterlin, M. Lost in transcription: Molecular mechanisms that control HIV latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef]
- Hultquist, J.F.; Schumann, K.; Woo, J.M.; Manganaro, L.; McGregor, M.J.; Doudna, J.; Simon, V.; Krogan, N.J.; Marson, A. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 2016, 17, 1438–1452. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, E.; Takacs, C.; Bell, B. Probing endogenous RNA polymerase II pre-initiation complexes by electrophoretic mobility shift assay. Methods Mol. Biol. 2012, 809, 63–74. [Google Scholar]
- Sainsbury, S.; Bernecky, C.; Cramer, P. Structural basis of transcription initiation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 129–143. [Google Scholar] [CrossRef]
- Basehoar, A.D.; Zanton, S.J.; Pugh, B.F.F. Identification and Distinct Regulation of Yeast TATA Box-Containing Genes. Cell 2004, 116, 699–709. [Google Scholar] [CrossRef]
- Xiao, H.; Lis, J.T.; Jeang, K.T. Promoter activity of Tat at steps subsequent to TATA-binding protein recruitment. Mol. Cell. Biol. 1997, 17, 6898–6905. [Google Scholar] [CrossRef]
- van Opijnen, T.; Kamoschinski, J.; Jeeninga, R.E.; Berkhout, B. The human immunodeficiency virus type 1 promoter contains a CATA box instead of a TATA box for optimal transcription and replication. J. Virol. 2004, 78, 6883–6890. [Google Scholar] [CrossRef]
- Kashanchi, F.; Khleif, S.N.; Duvall, J.F.; Sadaie, M.R.; Radonovich, M.F.; Cho, M.; Martin, M.A.; Chen, S.Y.; Weinmann, R.; Brady, J.N. Interaction of human immunodeficiency virus type 1 Tat with a unique site of TFIID inhibits negative cofactor Dr1 and stabilizes the TFIID-TFIIA complex. J. Virol. 1996, 70, 5503–5510. [Google Scholar] [PubMed]
- Veschambre, P.; Roisin, A.; Jalinot, P. Biochemical and functional interaction of the human immunodeficiency virus type 1 Tat transactivator with the general transcription factor TFIIB. J. Gen. Virol. 1997, 78 Pt 9, 2235–2245. [Google Scholar] [CrossRef]
- Fairley, J.A.; Evans, R.; Hawkes, N.A.; Roberts, S.G.E. Core promoter-dependent TFIIB conformation and a role for TFIIB conformation in transcription start site selection. Mol. Cell. Biol. 2002, 22, 6697–6705. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Kashanchi, F.; Jiang, H.; Ge, H.; Brady, J.N. Phosphorylation of the RAP74 subunit of TFIIF correlates with Tat-activated transcription of the HIV-1 long terminal repeat. Virology 2000, 268, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Abbott, K.L.; Archambault, J.; Xiao, H.; Nguyen, B.D.; Roeder, R.G.; Greenblatt, J.; Omichinski, J.G.; Legault, P. Interactions of the HIV-1 Tat and RAP74 proteins with the RNA polymerase II CTD phosphatase FCP1. Biochemistry 2005, 44, 2716–2731. [Google Scholar] [CrossRef] [PubMed]
- Parada, C.A.; Roeder, R.G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 1996, 384, 375–378. [Google Scholar] [CrossRef]
- Kim, Y.K.; Bourgeois, C.F.; Pearson, R.; Tyagi, M.; West, M.J.; Wong, J.; Wu, S.-Y.; Chiang, C.-M.; Karn, J. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J. 2006, 25, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Bell, B.; Tora, L. Regulation of gene expression by multiple forms of TFIID and other novel TAFII-containing complexes. Exp. Cell Res. 1999, 246, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Raha, T.; Cheng, S.W.G.; Green, M.R. HIV-1 Tat stimulates transcription complex assembly through recruitment of TBP in the absence of TAFs. PLoS Biol. 2005, 3, e44. [Google Scholar] [CrossRef]
- Berkhout, B.; Jeang, K.T. Functional roles for the TATA promoter and enhancers in basal and Tat-induced expression of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 1992, 66, 139–149. [Google Scholar]
- Eilebrecht, S.; Benecke, B.-J.; Benecke, A.G. Latent HIV-1 TAR Regulates 7SK-responsive P-TEFb Target Genes and Targets Cellular Immune Responses in the Absence of Tat. Genom. Proteom. Bioinform. 2017, 15, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Yamaguchi, Y.; Yano, K.; Sugimoto, S.; Chanarat, S.; Wada, T.; Kim, D.; Hasegawa, J.; Omori, M.; Inukai, N.; et al. Human transcription elongation factor NELF: Identification of novel subunits and reconstitution of the functionally active complex. Mol. Cell. Biol. 2003, 23, 1863–1873. [Google Scholar] [CrossRef]
- Egloff, S.; Vitali, P.; Tellier, M.; Raffel, R.; Murphy, S.; Kiss, T. The 7SK snRNP associates with the little elongation complex to promote snRNA gene expression. EMBO J. 2017, 36, 934–948. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Valente, S.T. Role of Host Factors on the Regulation of Tat-Mediated HIV-1 Transcription. Curr. Pharm. Des. 2017, 23, 4079–4090. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Abner, E.; Stoszko, M.; Zeng, L.; Chen, H.-C.; Izquierdo-Bouldstridge, A.; Konuma, T.; Zorita, E.; Fanunza, E.; Zhang, Q.; Mahmoudi, T.; et al. A New Quinoline BRD4 Inhibitor Targets a Distinct Latent HIV-1 Reservoir for Reactivation from Other “Shock” Drugs. J. Virol. 2018, 92, e02056-17. [Google Scholar] [CrossRef]
- Eilebrecht, S.; Brysbaert, G.; Wegert, T.; Urlaub, H.; Benecke, B.-J.; Benecke, A. 7SK small nuclear RNA directly affects HMGA1 function in transcription regulation. Nucleic Acids Res. 2011, 39, 2057–2072. [Google Scholar] [CrossRef] [PubMed]
- Eilebrecht, S.; Wilhelm, E.; Benecke, B.-J.; Bell, B.; Benecke, A.G. HMGA1 directly interacts with TAR to modulate basal and Tat-dependent HIV transcription. RNA Biol. 2013, 10, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Estable, M.C.; Naghavi, M.H.; Kato, H.; Xiao, H.; Qin, J.; Vahlne, A.; Roeder, R.G. MCEF, the newest member of the AF4 family of transcription factors involved in leukemia, is a positive transcription elongation factor-b-associated protein. J. Biomed. Sci. 2002, 9, 234–245. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef]
- Schulze-Gahmen, U.; Upton, H.; Birnberg, A.; Bao, K.; Chou, S.; Krogan, N.J.; Zhou, Q.; Alber, T. The AFF4 scaffold binds human P-TEFb adjacent to HIV Tat. Elife 2013, 2, e00327. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Upton, H.; Bao, K.; Schulze-Gahmen, U.; Samelson, A.J.; He, N.; Nowak, A.; Lu, H.; Krogan, N.J.; Zhou, Q.; et al. HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc. Natl. Acad. Sci. USA 2013, 110, E123–E131. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Chan, C.K.; Sobhian, B.; Chou, S.; Xue, Y.; Liu, M.; Alber, T.; Benkirane, M.; Zhou, Q. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc. Natl. Acad. Sci. USA 2011, 108, E636–E645. [Google Scholar] [CrossRef]
- Schulze-Gahmen, U.; Echeverria, I.; Stjepanovic, G.; Bai, Y.; Lu, H.; Schneidman-Duhovny, D.; Doudna, J.A.; Zhou, Q.; Sali, A.; Hurley, J.H. Insights into HIV-1 proviral transcription from integrative structure and dynamics of the Tat:AFF4:P-TEFb:TAR complex. Elife 2016, 5, e15910. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Li, Z.; Schulze-Gahmen, U.; Stjepanovic, G.; Zhou, Q.; Hurley, J.H. Structural basis for ELL2 and AFF4 activation of HIV-1 proviral transcription. Nat. Commun. 2017, 8, 14076. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, H.; Zhou, Q. A Minor Subset of Super Elongation Complexes Plays a Predominant Role in Reversing HIV-1 Latency. Mol. Cell. Biol. 2016, 36, 1194–1205. [Google Scholar] [CrossRef]
- Wu, J.; Ao, M.; Shao, R.; Wang, H.; Yu, D.; Fang, M.; Gao, X.; Wu, Z.; Zhou, Q.; Xue, Y. A chalcone derivative reactivates latent HIV-1 transcription through activating P-TEFb and promoting Tat-SEC interaction on viral promoter. Sci. Rep. 2017, 7, 10657. [Google Scholar] [CrossRef]
- Niedzielski, M.F.; Hopewell, R.; Ismail, Z.; Estable, M.C. MCEF is localized to the nucleus by protein sequences encoded within three distinct exons, where it represses HIV-1 Tat-transactivation of LTR-directed transcription. Int. J. Biol. Sci. 2007, 3, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Garrett, A.S.; De Kumar, B.; Smith, E.R.; Gogol, M.; Seidel, C.; Krumlauf, R.; Shilatifard, A. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev. 2011, 25, 1486–1498. [Google Scholar] [CrossRef]
- Takahashi, H.; Parmely, T.J.; Sato, S.; Tomomori-Sato, C.; Banks, C.A.S.; Kong, S.E.; Szutorisz, H.; Swanson, S.K.; Martin-Brown, S.; Washburn, M.P.; et al. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 2011, 146, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Ghosh, K.; Basu, S.; Roeder, R.G.; Biswas, D. Multivalent Role of Human TFIID in Recruiting Elongation Components at the Promoter-Proximal Region for Transcriptional Control. Cell Rep. 2019, 26, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
- Taki, T.; Kano, H.; Taniwaki, M.; Sako, M.; Yanagisawa, M.; Hayashi, Y. AF5q31, a newly identified AF4-related gene, is fused to MLL in infant acute lymphoblastic leukemia with ins(5;11)(q31;q13q23). Proc. Natl. Acad. Sci. USA 1999, 96, 14535–14540. [Google Scholar] [CrossRef] [PubMed]
- Vijayalingam, S.; Chinnadurai, G. Adenovirus L-E1A activates transcription through mediator complex-dependent recruitment of the super elongation complex. J. Virol. 2013, 87, 3425–3434. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Guest, E.; Garrett, A.S.; Mohaghegh, N.; Swanson, S.; Marshall, S.; Florens, L.; Washburn, M.P.; Shilatifard, A. The super elongation complex family of RNA polymerase II elongation factors: Gene target specificity and transcriptional output. Mol. Cell. Biol. 2012, 32, 2608–2617. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef]
- Ne, E.; Palstra, R.-J.; Mahmoudi, T. Transcription: Insights from the HIV-1 Promoter. Int. Rev. Cell Mol. Biol. 2018, 335, 191–243. [Google Scholar]
- Sadowski, I.; Lourenco, P.; Malcolm, T. Factors controlling chromatin organization and nucleosome positioning for establishment and maintenance of HIV latency. Curr. HIV Res. 2008, 6, 286–295. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, Y.; Yuan, H.; Chen, B.K.; Ip, J.; Ho, D.D. Identification of a replication-competent pathogenic human immunodeficiency virus type 1 with a duplication in the TCF-1alpha region but lacking NF-kappaB binding sites. J. Virol. 1997, 71, 1651–1656. [Google Scholar] [PubMed]
- Chae, C.-S.; Kim, G.-C.; Park, E.S.; Lee, C.-G.; Verma, R.; Cho, H.-L.; Jun, C.-D.; Yoo, Y.J.; Im, S.-H. NFAT1 Regulates Systemic Autoimmunity through the Modulation of a Dendritic Cell Property. J. Immunol. 2017, 199, 3051–3062. [Google Scholar] [CrossRef] [PubMed]
- Giffin, M.J.; Stroud, J.C.; Bates, D.L.; von Koenig, K.D.; Hardin, J.; Chen, L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR kappa B element. Nat. Struct. Biol. 2003, 10, 800–806. [Google Scholar] [CrossRef]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science 1986, 232, 755–759. [Google Scholar] [CrossRef]
- Sadowski, I.; Mitchell, D.A. TFII-I and USF (RBF-2) regulate Ras/MAPK-responsive HIV-1 transcription in T cells. Eur. J. Cancer 2005, 41, 2528–2536. [Google Scholar] [CrossRef]
- Bell, B.; Sadowski, I. Ras-responsiveness of the HIV-1 LTR requires RBF-1 and RBF-2 binding sites. Oncogene 1996, 13, 2687–2697. [Google Scholar]
- Estable, M.C.; Bell, B.; Hirst, M.; Sadowski, I. Naturally occurring human immunodeficiency virus type 1 long terminal repeats have a frequently observed duplication that binds RBF-2 and represses transcription. J. Virol. 1998, 72, 6465–6474. [Google Scholar]
- Estable, M.C.; Bell, B.; Merzouki, A.; Montaner, J.S.; O’Shaughnessy, M.V.; Sadowski, I.J. Human immunodeficiency virus type 1 long terminal repeat variants from 42 patients representing all stages of infection display a wide range of sequence polymorphism and transcription activity. J. Virol. 1996, 70, 4053–4062. [Google Scholar]
- Chen, J.; Malcolm, T.; Estable, M.C.; Roeder, R.G.; Sadowski, I. TFII-I regulates induction of chromosomally integrated human immunodeficiency virus type 1 long terminal repeat in cooperation with USF. J. Virol. 2005, 79, 4396–4406. [Google Scholar] [CrossRef]
- Malcolm, T.; Kam, J.; Pour, P.S.; Sadowski, I. Specific interaction of TFII-I with an upstream element on the HIV-1 LTR regulates induction of latent provirus. FEBS Lett. 2008, 582, 3903–3908. [Google Scholar] [CrossRef]
- Malcolm, T.; Chen, J.; Chang, C.; Sadowski, I. Induction of chromosomally integrated HIV-1 LTR requires RBF-2 (USF/TFII-I) and Ras/MAPK signaling. Virus Genes 2007, 35, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Estable, M.C. In search of a function for the most frequent naturally-occurring length polymorphism (MFNLP) of the HIV-1 LTR: Retaining functional coupling, of Nef and RBF-2, at RBEIII? Int. J. Biol. Sci. 2007, 3, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Kao, SY.; Calman, AF.; Luciw, PA. Peterlin, Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature. 1987, 330, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Ensoli, B.; Barillari, G.; Salahuddin, S.Z.; Gallo, R.C.; Wong-Staal, F. Tat protein of HIV-1 stimulates growth of cells derived from Kaposi’s sarcoma lesions of AIDS patients. Nature 1990, 345, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Demarchi, F.; d’Adda di Fagagna, F.; Falaschi, A.; Giacca, M. Activation of transcription factor NF-kappaB by the Tat protein of human immunodeficiency virus type 1. J. Virol. 1996, 70, 4427–4437. [Google Scholar] [PubMed]
- Li, L.; Li, H.S.; Pauza, C.D.; Bukrinsky, M.; Zhao, R.Y. Roles of HIV-1 auxiliary proteins in viral pathogenesis and host-pathogen interactions. Cell Res. 2005, 15, 923–934. [Google Scholar] [CrossRef]
- Liu, R.; Lin, Y.; Jia, R.; Geng, Y.; Liang, C.; Tan, J.; Qiao, W. HIV-1 Vpr stimulates NF-κB and AP-1 signaling by activating TAK1. Retrovirology 2014, 11, 45. [Google Scholar] [CrossRef]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müssig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef]
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas based antiviral strategies against HIV-1. Virus Res. 2018, 244, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Koup, R.A.; Safrit, J.T.; Cao, Y.; Andrews, C.A.; McLeod, G.; Borkowsky, W.; Farthing, C.; Ho, D.D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994, 68, 4650–4655. [Google Scholar] [PubMed]
- Schmitz, J.E.; Kuroda, M.J.; Santra, S.; Sasseville, V.G.; Simon, M.A.; Lifton, M.A.; Racz, P.; Tenner-Racz, K.; Dalesandro, M.; Scallon, B.J.; et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 1999, 283, 857–860. [Google Scholar] [CrossRef]
- McBrien, J.B.; Kumar, N.A.; Silvestri, G. Mechanisms of CD8+ T cell-mediated suppression of HIV/SIV replication. Eur. J. Immunol. 2018, 48, 898–914. [Google Scholar] [CrossRef]
- Cartwright, E.K.; Spicer, L.; Smith, S.A.; Lee, D.; Fast, R.; Paganini, S.; Lawson, B.O.; Nega, M.; Easley, K.; Schmitz, J.E.; et al. CD8(+) Lymphocytes Are Required for Maintaining Viral Suppression in SIV-Infected Macaques Treated with Short-Term Antiretroviral Therapy. Immunity 2016, 45, 656–668. [Google Scholar] [CrossRef]
- Cao, Y.; Cartwright, E.K.; Silvestri, G.; Perelson, A.S. CD8+ lymphocyte control of SIV infection during antiretroviral therapy. PLoS Pathog. 2018, 14, e1007350. [Google Scholar] [CrossRef]
- Kuhlmann, A.-S.; Peterson, C.W.; Kiem, H.-P. Chimeric antigen receptor T-cell approaches to HIV cure. Curr. Opin. HIV AIDS 2018, 13, 446–453. [Google Scholar] [CrossRef]
- Wagner, T.A. Quarter Century of Anti-HIV CAR T Cells. Curr. HIV/AIDS Rep. 2018, 15, 147–154. [Google Scholar] [CrossRef]
- Riddell, S.R.; Elliott, M.; Lewinsohn, D.A.; Gilbert, M.J.; Wilson, L.; Manley, S.A.; Lupton, S.D.; Overell, R.W.; Reynolds, T.C.; Corey, L.; et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat. Med. 1996, 2, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.; Peterson, C.W.; Carrillo, M.A.; Reddy, S.S.; Youn, C.S.; Lam, B.B.; Chang, N.Y.; Martin, H.A.; Rick, J.W.; Kim, J.; et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. 2017, 13, e1006753. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, J.; Clotet, B.; Blanco, J. Antibodies and Antibody Derivatives: New Partners in HIV Eradication Strategies. Front. Immunol. 2018, 9, 2429. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, Y.; Gruell, H.; Schoofs, T.; Pai, J.A.; Nogueira, L.; Butler, A.L.; Millard, K.; Lehmann, C.; Suárez, I.; Oliveira, T.Y.; et al. Safety and antiviral activity of combination HIV-1 broadly neutralizing antibodies in viremic individuals. Nat. Med. 2018, 24, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Gautam, R.; Chun, T.-W.; Sadjadpour, R.; Foulds, K.E.; Shingai, M.; Klein, F.; Gazumyan, A.; Golijanin, J.; Donaldson, M.; et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature 2017, 543, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, P.; Gruell, H.; Nogueira, L.; Pai, J.A.; Butler, A.L.; Millard, K.; Lehmann, C.; Suárez, I.; Oliveira, T.Y.; Lorenzi, J.C.C.; et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 2018, 561, 479–484. [Google Scholar] [CrossRef]
- Fabozzi, G.; Pegu, A.; Koup, R.A.; Petrovas, C. Bispecific antibodies: Potential immunotherapies for HIV treatment. Methods 2019, 154, 118–124. [Google Scholar] [CrossRef]
- Padte, N.N.; Yu, J.; Huang, Y.; Ho, D.D. Engineering multi-specific antibodies against HIV-1. Retrovirology 2018, 15, 60. [Google Scholar] [CrossRef]
- Pace, C.S.; Song, R.; Ochsenbauer, C.; Andrews, C.D.; Franco, D.; Yu, J.; Oren, D.A.; Seaman, M.S.; Ho, D.D. Bispecific antibodies directed to CD4 domain 2 and HIV envelope exhibit exceptional breadth and picomolar potency against HIV-1. Proc. Natl. Acad. Sci. USA 2013, 110, 13540–13545. [Google Scholar] [CrossRef]
- Pegu, A.; Asokan, M.; Wu, L.; Wang, K.; Hataye, J.; Casazza, J.P.; Guo, X.; Shi, W.; Georgiev, I.; Zhou, T.; et al. Activation and lysis of human CD4 cells latently infected with HIV-1. Nat. Commun. 2015, 6, 8447. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yu, J.; Lanzi, A.; Yao, X.; Andrews, C.D.; Tsai, L.; Gajjar, M.R.; Sun, M.; Seaman, M.S.; Padte, N.N.; et al. Engineered Bispecific Antibodies with Exquisite HIV-1-Neutralizing Activity. Cell 2016, 165, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Gazumyan, A.; Seaman, M.S.; Nussenzweig, M.C.; Ravetch, J. V Bispecific Anti-HIV-1 Antibodies with Enhanced Breadth and Potency. Cell 2016, 165, 1609–1620. [Google Scholar] [CrossRef]
- Xu, L.; Pegu, A.; Rao, E.; Doria-Rose, N.; Beninga, J.; McKee, K.; Lord, D.M.; Wei, R.R.; Deng, G.; Louder, M.; et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science 2017, 358, 85–90. [Google Scholar] [CrossRef]
- Sloan, D.D.; Lam, C.-Y.K.; Irrinki, A.; Liu, L.; Tsai, A.; Pace, C.S.; Kaur, J.; Murry, J.P.; Balakrishnan, M.; Moore, P.A.; et al. Targeting HIV Reservoir in Infected CD4 T Cells by Dual-Affinity Re-targeting Molecules (DARTs) that Bind HIV Envelope and Recruit Cytotoxic T Cells. PLoS Pathog. 2015, 11, e1005233. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.A.M.; Pickeral, J.; Liu, L.; Stanfield-Oakley, S.A.; Lam, C.-Y.K.; Garrido, C.; Pollara, J.; LaBranche, C.; Bonsignori, M.; Moody, M.A.; et al. Dual-Affinity Re-Targeting proteins direct T cell-mediated cytolysis of latently HIV-infected cells. J. Clin. Investig. 2015, 125, 4077–4090. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.V.; Miller, A.D. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 1991, 350, 508–511. [Google Scholar] [CrossRef]
- Desrosiers, R.C. Strategies used by human immunodeficiency virus that allow persistent viral replication. Nat. Med. 1999, 5, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.; Deeks, S.G. HIV control: Is getting there the same as staying there? PLoS Pathog. 2018, 14, e1007222. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.D.; Meyer, L.; Prins, M.; Thiebaut, R.; Gurdasani, D.; Guiguet, M.; Chaix, M.-L.; Amornkul, P.; Babiker, A.; Sandhu, M.S.; et al. An evaluation of HIV elite controller definitions within a large seroconverter cohort collaboration. PLoS ONE 2014, 9, e86719. [Google Scholar] [CrossRef]
- Ilyinskii, P.O.; Simon, M.A.; Czajak, S.C.; Lackner, A.A.; Desrosiers, R.C. Induction of AIDS by simian immunodeficiency virus lacking NF-kappaB and SP1 binding elements. J. Virol. 1997, 71, 1880–1887. [Google Scholar]
- Blancou, P.; Chenciner, N.; Cumont, M.C.; Wain-Hobson, S.; Hurtrel, B.; Cheynier, R. The infiltration kinetics of simian immunodeficiency virus-specific T cells drawn to sites of high antigenic stimulation determines local in vivo viral escape. Proc. Natl. Acad. Sci. USA 2001, 98, 13237–13242. [Google Scholar] [CrossRef] [PubMed]
- Blazek, D.; Teque, F.; Mackewicz, C.; Peterlin, M.; Levy, J.A. The CD8+ cell non-cytotoxic antiviral response affects RNA polymerase II-mediated human immunodeficiency virus transcription in infected CD4+ cells. J. Gen. Virol. 2016, 97, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Shridhar, V.; Chen, Y.; Gupta, P. The CD8 antiviral factor (CAF) can suppress HIV-1 transcription from the long terminal repeat (LTR) promoter in the absence of elements upstream of the CATATAA box. Virol. J. 2014, 11, 130. [Google Scholar] [CrossRef]
- Jean, M.J.; Fiches, G.; Hayashi, T.; Zhu, J. Current Strategies for Elimination of HIV-1 Latent Reservoirs Using Chemical Compounds Targeting Host and Viral Factors. AIDS Res. Hum. Retrovir. 2019, 35, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Das, A.T.; Berkhout, B. Tackling HIV Persistence: Pharmacological versus CRISPR-Based Shock Strategies. Viruses 2018, 10, 157. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef]
- Imperial College of London. Wighton Kate First Randomized Trial of “Kick and Kill” Approach to HIV Cure Leaves Puzzles to Be Solved—ScienceDaily. Available online: https://www.sciencedaily.com/releases/2018/07/180724110046.htm (accessed on 12 December 2018).
- Moron-Lopez, S.; Kim, P.; SøGaard, O.S.; Tolstrup, M.; Wong, J.K.; Yukl, S.A. Characterization of the HIV-1 transcription profile after romidepsin administration in ART-suppressed individuals. AIDS 2019, 33, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; Spina, C.A.; Wiegand, A.; Landay, A.L.; Coombs, R.W.; Richman, D.D.; Mellors, J.W.; et al. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet 2005, 366, 549–555. [Google Scholar] [CrossRef]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retrovir. 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef] [PubMed]
- Kulkosky, J.; Culnan, D.M.; Roman, J.; Dornadula, G.; Schnell, M.; Boyd, M.R.; Pomerantz, R.J. Prostratin: Activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 2001, 98, 3006–3015. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock”; Strategy for HIV-1 Treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Does HIV-1 Tat induce a change in viral initiation rights? Cell 1993, 73, 417–420. [Google Scholar] [CrossRef]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. MBio 2019, 10, e02662-18. [Google Scholar] [CrossRef]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. MBio 2015, 6, e00465. [Google Scholar] [CrossRef]
- Wagschal, A.; Rousset, E.; Basavarajaiah, P.; Contreras, X.; Harwig, A.; Laurent-Chabalier, S.; Nakamura, M.; Chen, X.; Zhang, K.; Meziane, O.; et al. Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell 2012, 150, 1147–1157. [Google Scholar] [CrossRef]
- Contreras, X.; Salifou, K.; Sanchez, G.; Helsmoortel, M.; Beyne, E.; Bluy, L.; Pelletier, S.; Rousset, E.; Rouquier, S.; Kiernan, R. Nuclear RNA surveillance complexes silence HIV-1 transcription. PLoS Pathog. 2018, 14, e1006950. [Google Scholar] [CrossRef] [PubMed]
- Campos, N.; Myburgh, R.; Garcel, A.; Vautrin, A.; Lapasset, L.; Nadal, E.S.; Mahuteau-Betzer, F.; Najman, R.; Fornarelli, P.; Tantale, K.; et al. Long lasting control of viral rebound with a new drug ABX464 targeting Rev - mediated viral RNA biogenesis. Retrovirology 2015, 12, 30. [Google Scholar] [CrossRef]
- Steens, J.-M.; Scherrer, D.; Gineste, P.; Barrett, P.N.; Khuanchai, S.; Winai, R.; Ruxrungtham, K.; Tazi, J.; Murphy, R.; Ehrlich, H. Safety, Pharmacokinetics, and Antiviral Activity of a Novel HIV Antiviral, ABX464, in Treatment-Naive HIV-Infected Subjects in a Phase 2 Randomized, Controlled Study. Antimicrob. Agents Chemother. 2017, 61, e00545-17. [Google Scholar] [CrossRef]
- Vautrin, A.; Manchon, L.; Garcel, A.; Campos, N.; Lapasset, L.; Laaref, A.M.; Bruno, R.; Gislard, M.; Dubois, E.; Scherrer, D.; et al. Both anti-inflammatory and antiviral properties of novel drug candidate ABX464 are mediated by modulation of RNA splicing. Sci. Rep. 2019, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Ntie-Kang, F.; Tietjen, I. Natural product-derived compounds in HIV suppression, remission, and eradication strategies. Antiviral Res. 2018, 158, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Jean, M.J.; Hayashi, T.; Huang, H.; Brennan, J.; Simpson, S.; Purmal, A.; Gurova, K.; Keefer, M.C.; Kobie, J.J.; Santoso, N.G.; et al. Curaxin CBL0100 Blocks HIV-1 Replication and Reactivation through Inhibition of Viral Transcriptional Elongation. Front. Microbiol. 2017, 8, 2007. [Google Scholar] [CrossRef]
- Hayashi, T.; Jean, M.; Huang, H.; Simpson, S.; Santoso, N.G.; Zhu, J. Screening of an FDA-approved compound library identifies levosimendan as a novel anti-HIV-1 agent that inhibits viral transcription. Antiviral Res. 2017, 146, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Méndez, C.; Ledger, S.; Petoumenos, K.; Ahlenstiel, C.; Kelleher, A.D. RNA-induced epigenetic silencing inhibits HIV-1 reactivation from latency. Retrovirology 2018, 15, 67. [Google Scholar] [CrossRef]
- Leoz, M.; Kukanja, P.; Luo, Z.; Huang, F.; Cary, D.C.; Peterlin, B.M.; Fujinaga, K. HEXIM1-Tat chimera inhibits HIV-1 replication. PLoS Pathog. 2018, 14, e1007402. [Google Scholar] [CrossRef]
- Rutsaert, S.; Steens, J.-M.; Gineste, P.; Cole, B.; Kint, S.; Barrett, P.N.; Tazi, J.; Scherrer, D.; Ehrlich, H.J.; Vandekerckhove, L. Safety, tolerability and impact on viral reservoirs of the addition to antiretroviral therapy of ABX464, an investigational antiviral drug, in individuals living with HIV-1: A Phase IIa randomised controlled study. J. Virus Erad. 2019, 5, 10–22. [Google Scholar]
- Turner, L.S.; Tsygankov, A.Y.; Henderson, E.E. StpC-based gene therapy targeting latent reservoirs of HIV-1. Antiviral Res. 2006, 72, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, N.; Sandstrom, T.S.; Burke Schinkel, S.C.; Côté, S.C.; Angel, J.B. The Oncolytic Virus MG1 Targets and Eliminates Cells Latently Infected With HIV-1: Implications for an HIV Cure. J. Infect. Dis. 2018, 217, 721–730. [Google Scholar] [CrossRef]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Mujib, S.; Saiyed, A.; Fadel, S.; Bozorgzad, A.; Aidarus, N.; Yue, F.Y.; Benko, E.; Kovacs, C.; Emert-Sedlak, L.A.; Smithgall, T.E.; et al. Pharmacologic HIV-1 Nef blockade promotes CD8 T cell-mediated elimination of latently HIV-1-infected cells in vitro. JCI Insight 2017, 2, 93684. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, R.J. Managing Expectations of an HIV Cure: What Should We Expect? AIDS Res. Hum. Retrovir. 2018, 34, 1. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Epigenetics and Genetics of Viral Latency. Cell Host Microbe 2016, 19, 619–628. [Google Scholar] [CrossRef]
- Salgame, P.; Geadas, C.; Collins, L.; Jones-López, E.; Ellner, J.J. Latent tuberculosis infection—Revisiting and revising concepts. Tuberculosis 2015, 95, 373–384. [Google Scholar] [CrossRef]





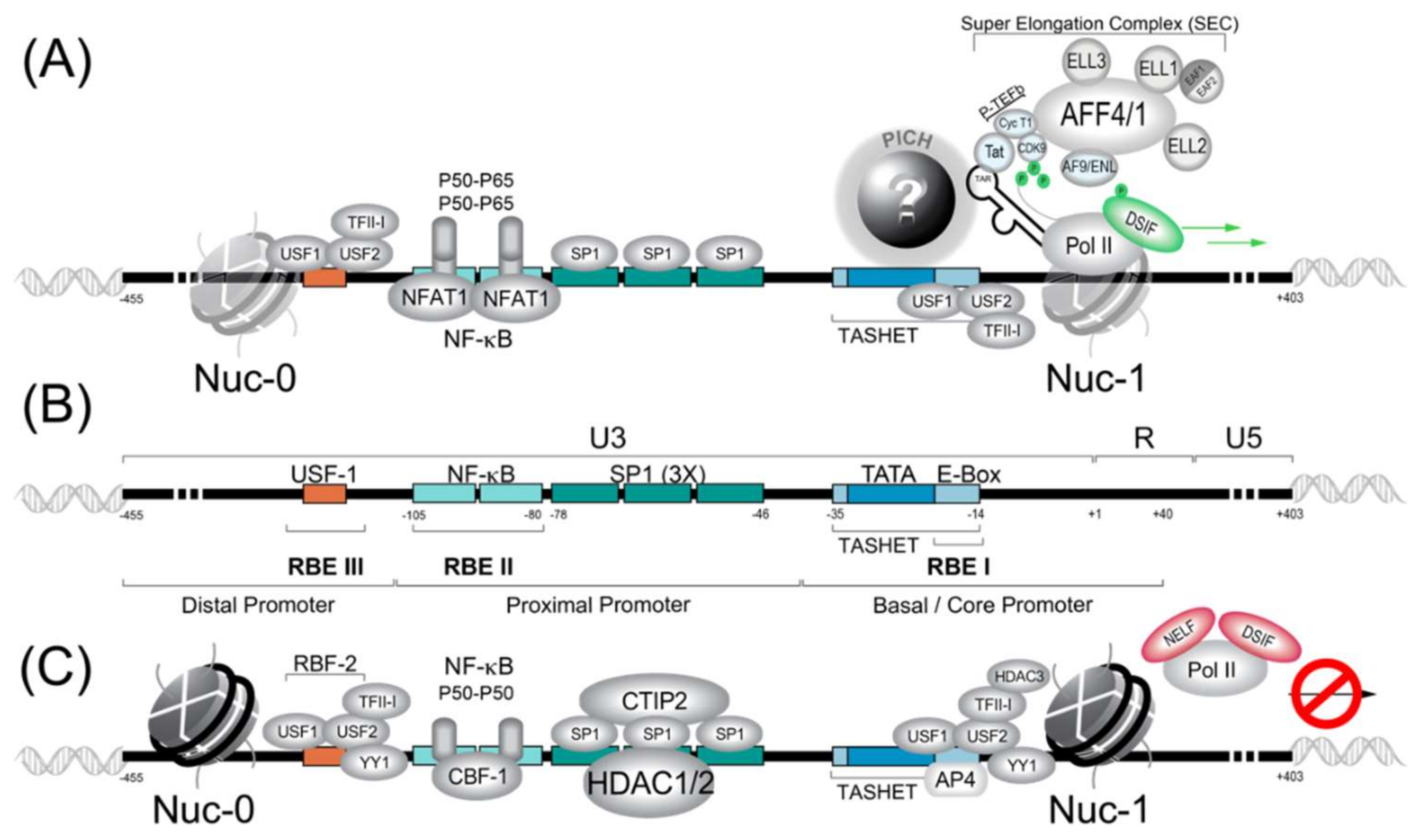{kind=link}
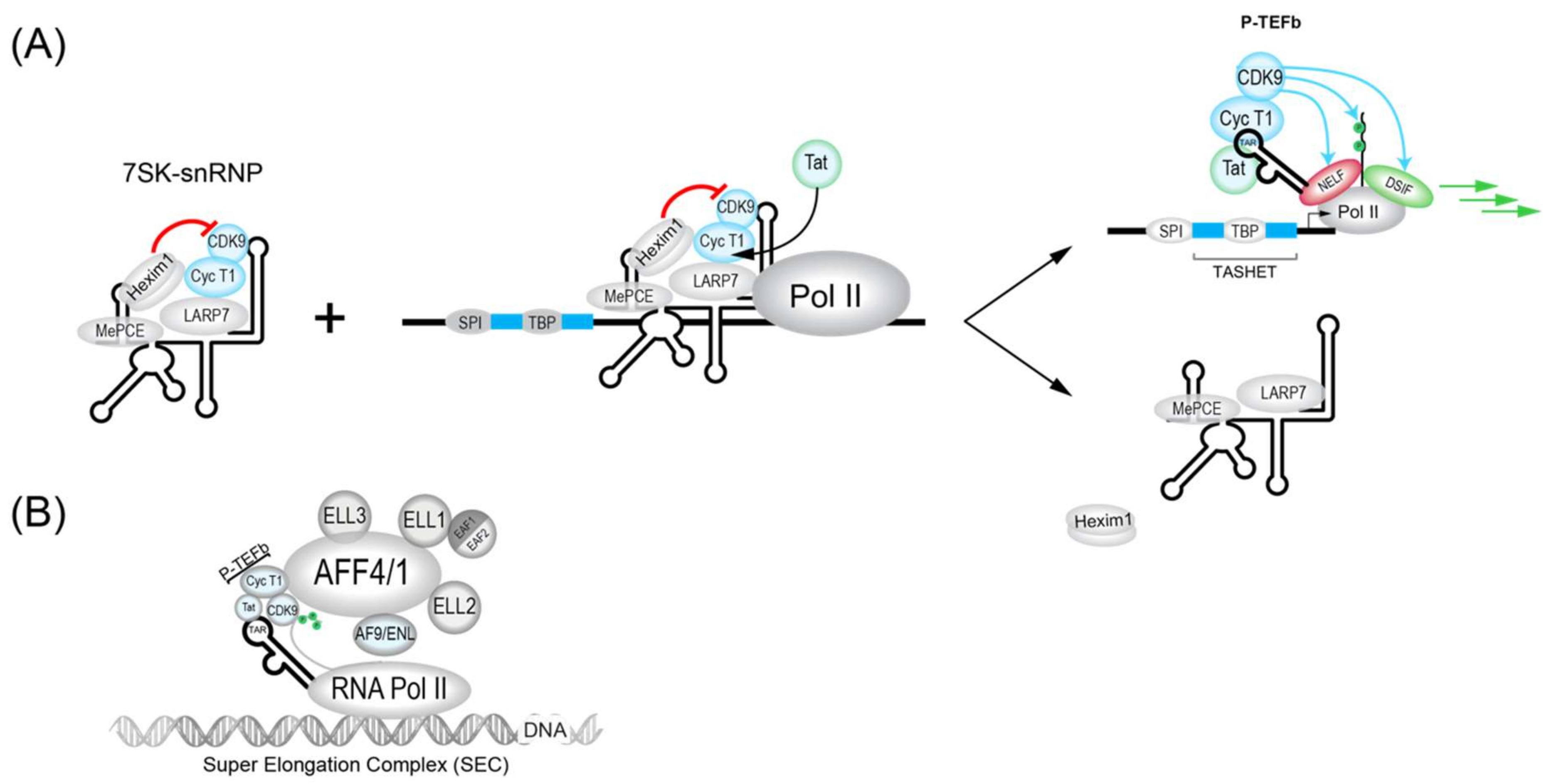{kind=link}
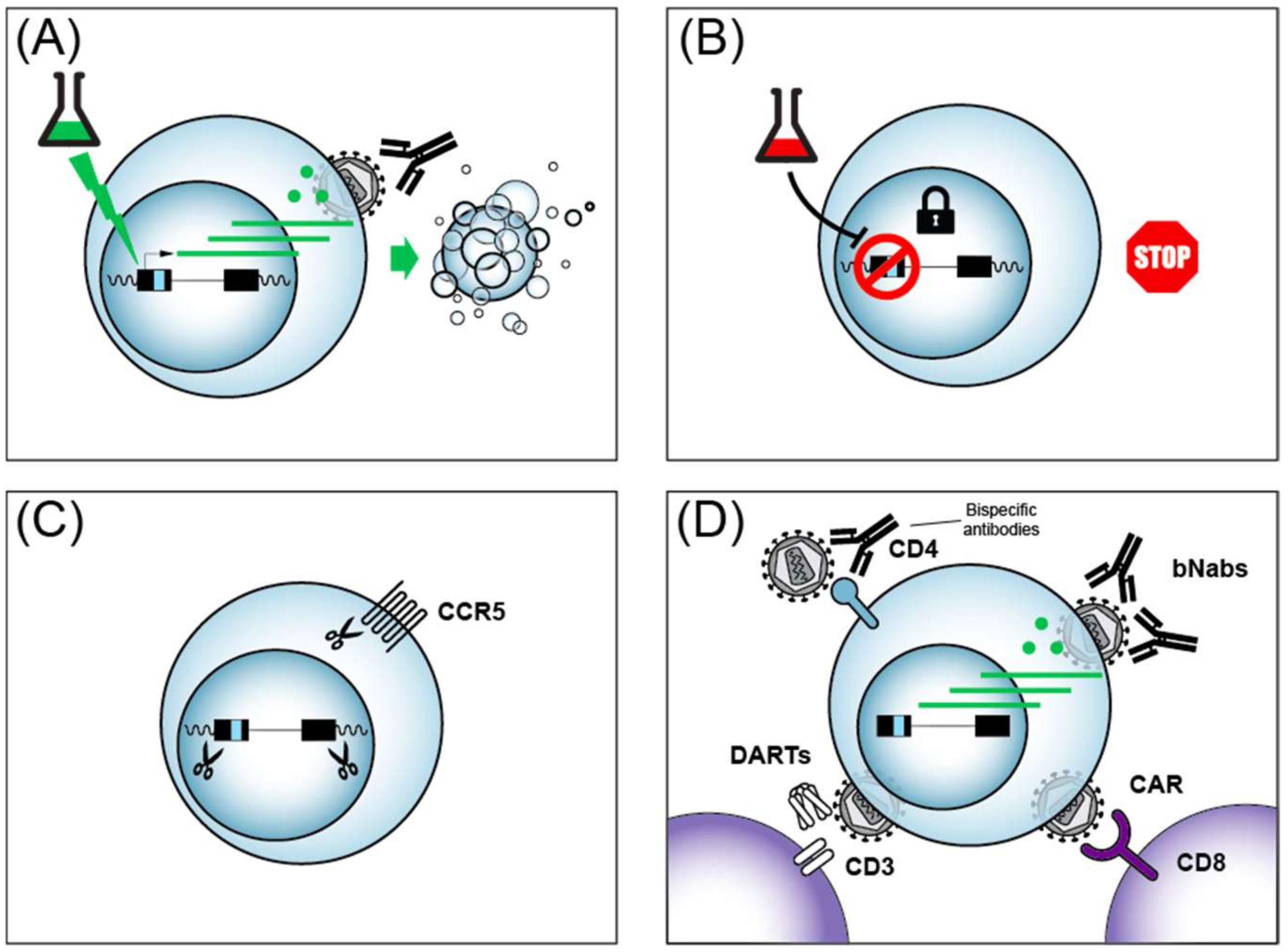{kind=link}
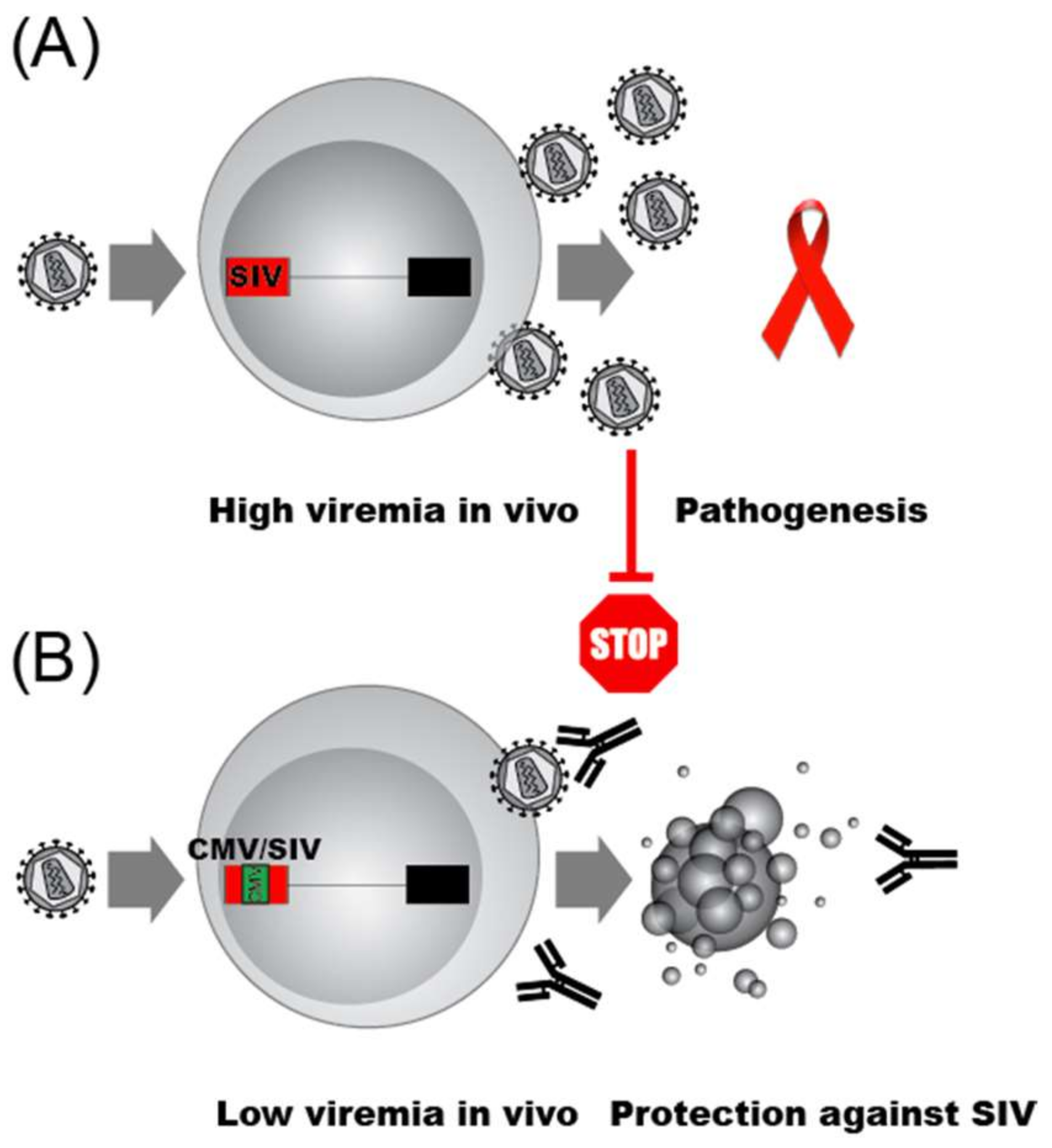{kind=link}
| Latency Reversing Agent (LRA) * | LRA Class (Target/Pathway) | Reference (s) |
|---|---|---|
| Valproic acid | HDAC inhibitor | [284] |
| SAHA | HDAC inhibitor | [285] |
| Romidepsin | HDAC inhibitor | [286] |
| JQ1 | BET domain inhibitor/BRD4 | [199] |
| MMQO | BET domain inhibitor/BRD4 | [201] |
| Prostratin | Stimulates PKC/NF-κB | [287] |
| BIX01294 | Methyltransferase inhibitor | [288] |
| Latency Promoting Agent (LPA) | LPA Mode of Action | Animal Model Data | Clinical Trial Data |
|---|---|---|---|
| dAC | Binds HIV tat to inhibit transcription [292] | Represses viral rebound in BLT humanized mice [289] | N.A. |
| ABX464 | HIV Rev inhibitor [296]; HIV splicing enhancer [298] | Delayed viral rebound in NOG humanized mice [297] | Good safety and tolerability; measurable reduction in HIV DNA [304] |
| Curaxin (CBL0100) | FACT inhibitor [300] | N.A. | N.A. |
| levosimendan | Inhibits HIV transcription [301] | N.A. | N.A. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delannoy, A.; Poirier, M.; Bell, B. Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses 2019, 11, 269. https://doi.org/10.3390/v11030269
Delannoy A, Poirier M, Bell B. Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses. 2019; 11(3):269. https://doi.org/10.3390/v11030269
Chicago/Turabian StyleDelannoy, Aurélie, Mikaël Poirier, and Brendan Bell. 2019. "Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies" Viruses 11, no. 3: 269. https://doi.org/10.3390/v11030269
APA StyleDelannoy, A., Poirier, M., & Bell, B. (2019). Cat and Mouse: HIV Transcription in Latency, Immune Evasion and Cure/Remission Strategies. Viruses, 11(3), 269. https://doi.org/10.3390/v11030269