Complexities of Type I Interferon Biology: Lessons from LCMV



Abstract
:
1. Introduction
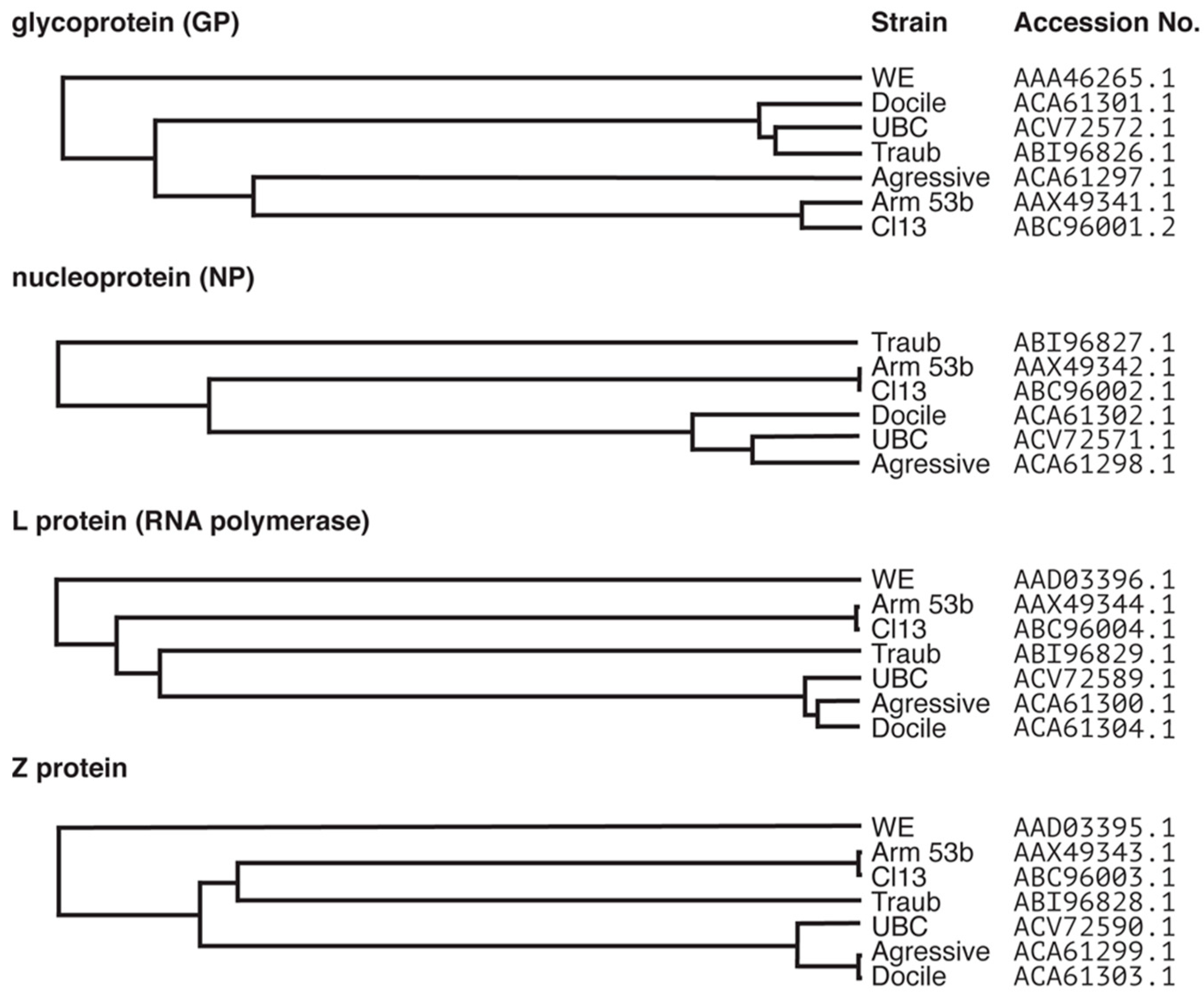
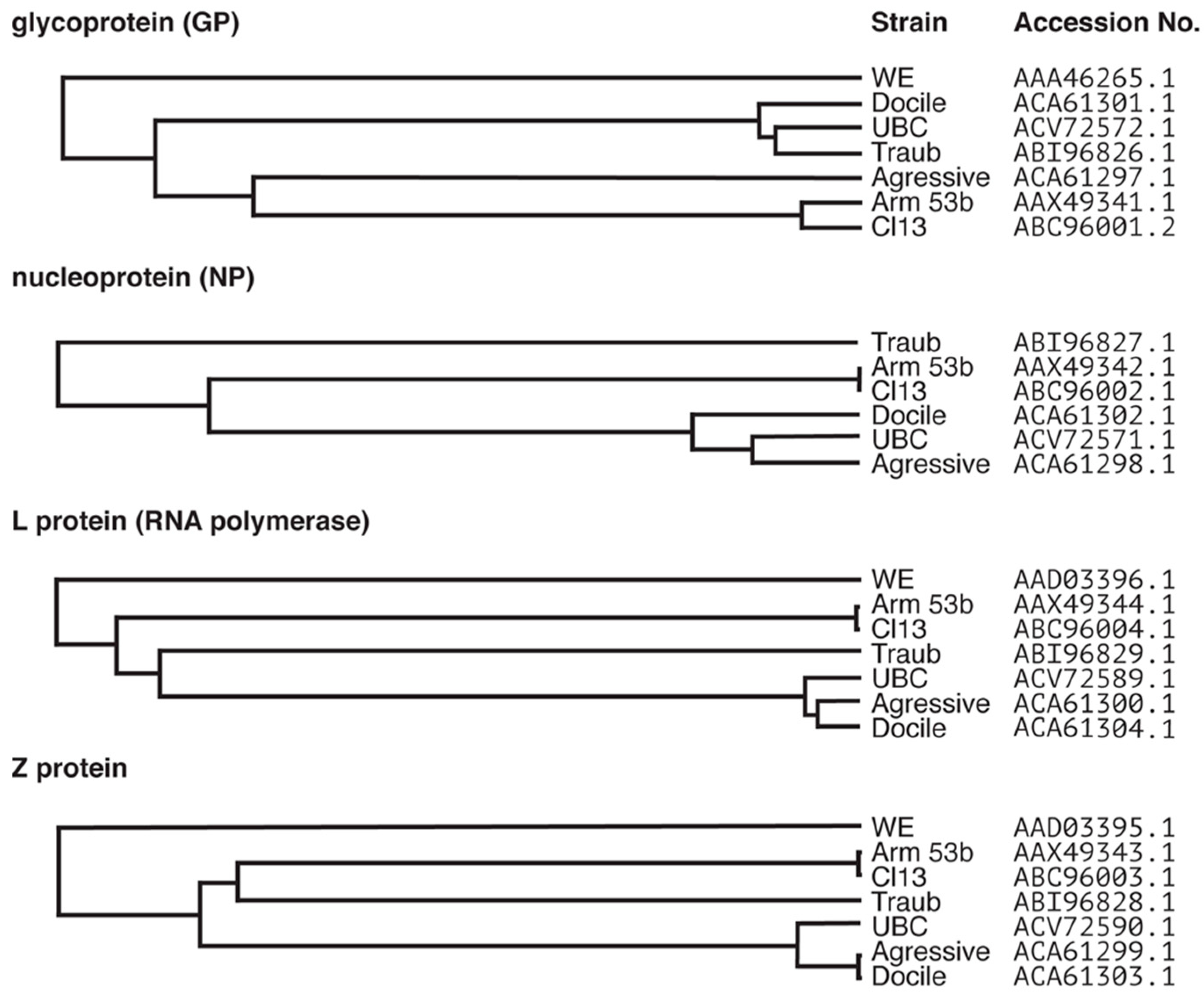
2. Lymphocytic Choriomeningitis Virus (LCMV): One Virus with Many Outcomes
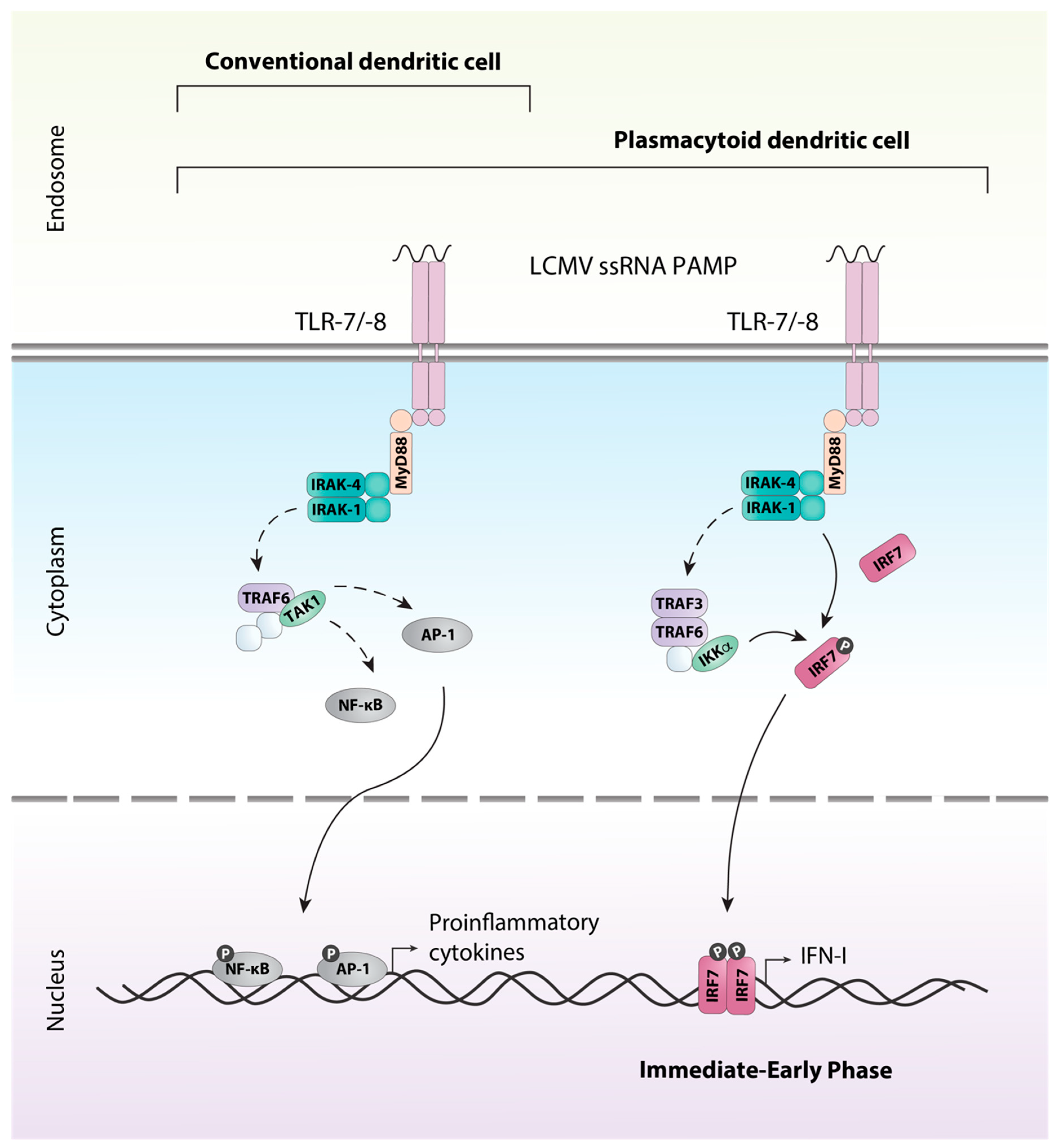
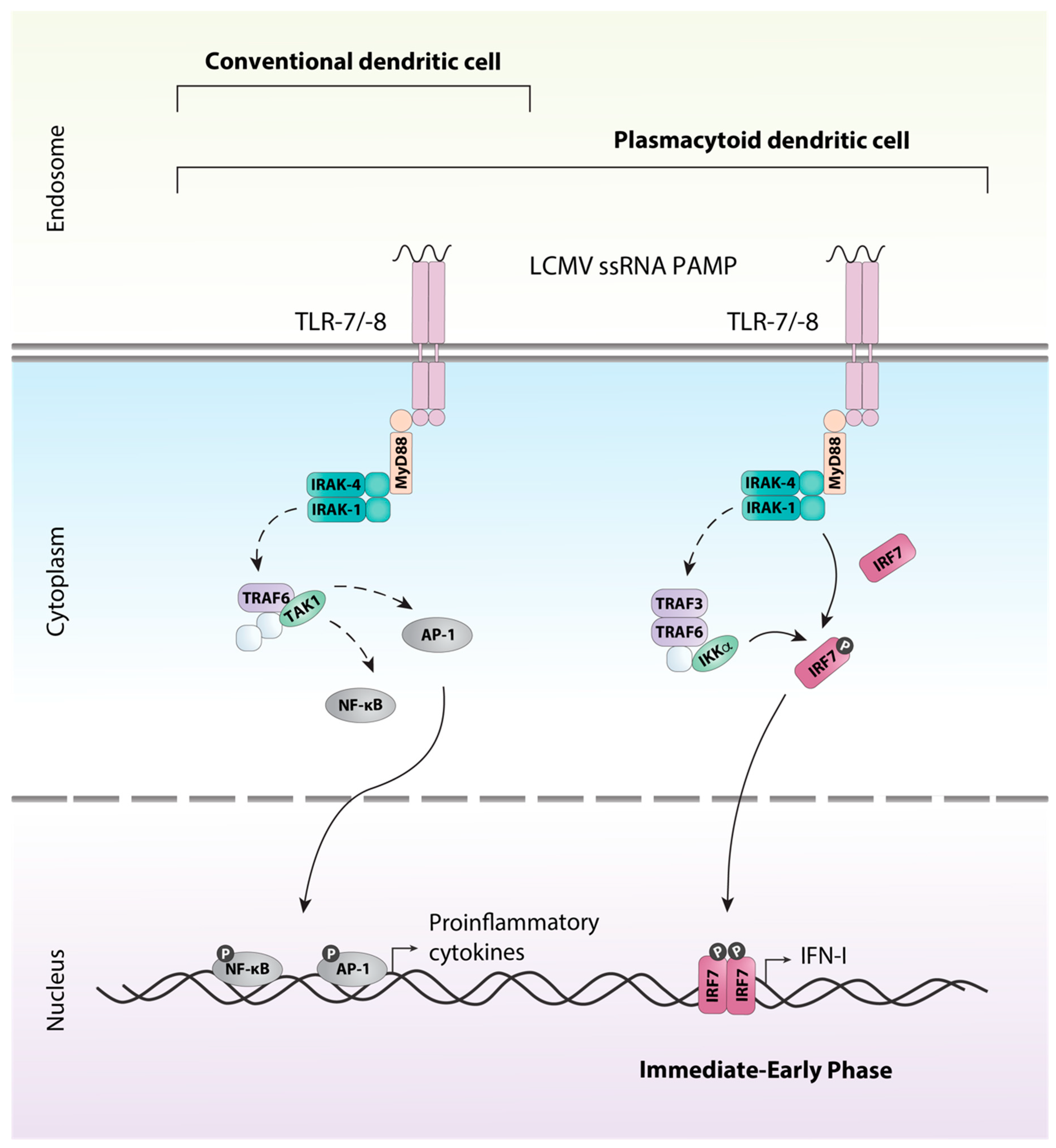
3. Detection of LCMV and Induction of the Antiviral Response
4. Type I Interferons (IFN-Is) are More than a Mediator of the Innate Response
4.1. The Role of IFN-Is in the Outcome of LCMV Infection Varies Between Virus Strains
4.2. IFN-Is Modulate the Innate Immune Response by Regulating Interferon-Stimulated Gene (ISG) Expression and Innate Immune Cells
4.3. IFN-Is are Central Regulators of T and B Cell Activity
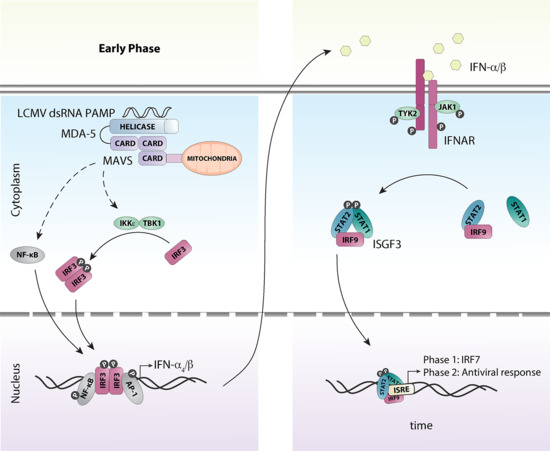
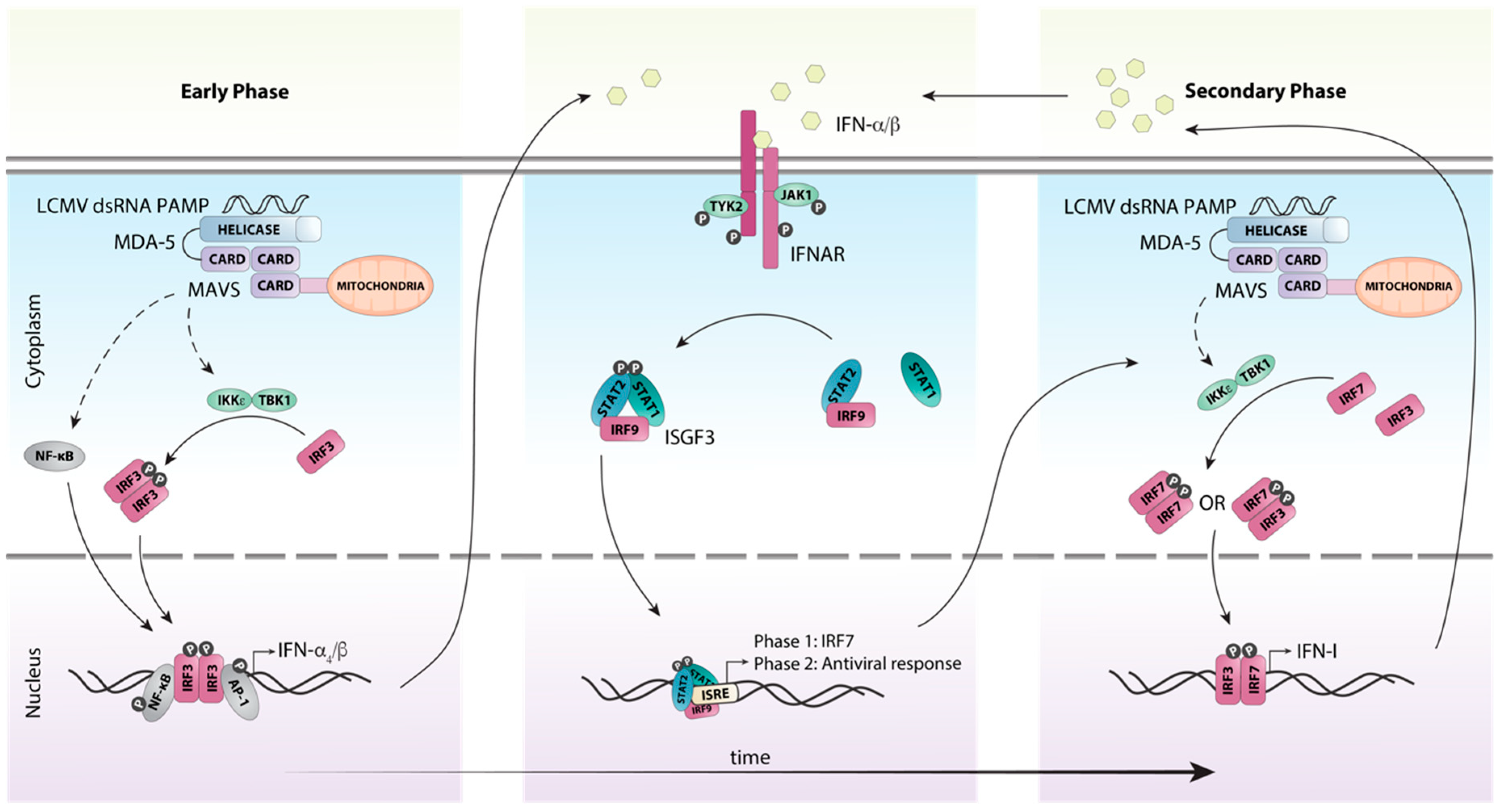
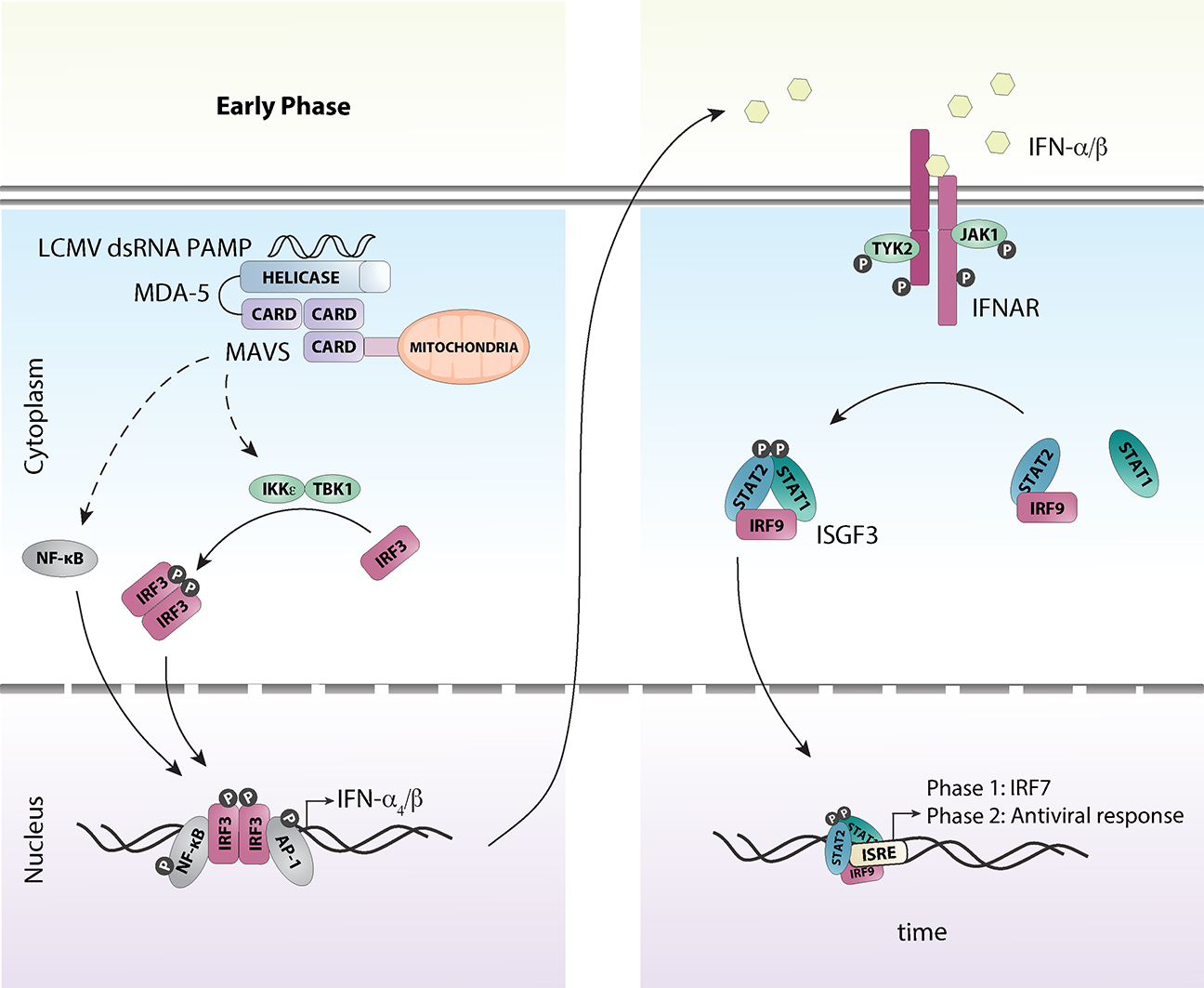
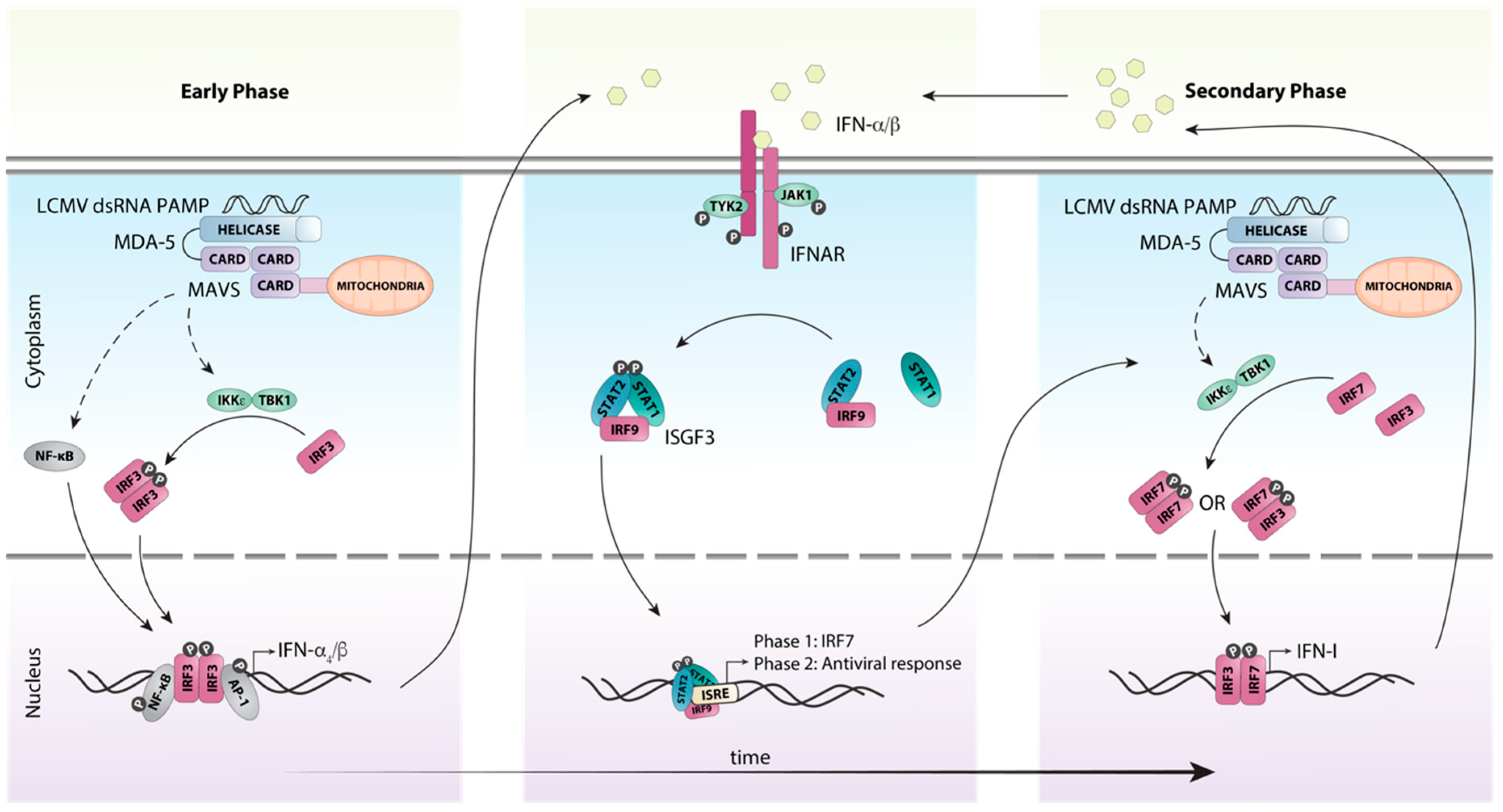
5. A Coordinated Induction of IFN-Is is Critical for Early Antiviral Responses Against LCMV
6. Efficient Antiviral Responses to LCMV Require Canonical IFN-I Signaling Through the ISGF3 Complex
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kang, S.S.; McGavern, D.B. Lymphocytic choriomeningitis infection of the central nervous system. Front. Biosci. 2008, 13, 4529–4543. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Ohsawa, M.; Yamanaka, H.; Matsuda, N.; Sato, H.; Ohsawa, K. Difference of two new LCMV strains in lethality and viral genome load in tissues. Exp. Anim. 2017, 66, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Salmi, A.; Butler, L.D.; Chiller, J.M.; Oldstone, M.B. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 1984, 160, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Traub, E. A FILTERABLE VIRUS RECOVERED FROM WHITE MICE. Science 1935, 81, 298–299. [Google Scholar] [CrossRef] [PubMed]
- Rivers, T.M.; McNair Scott, T.F. MENINGITIS IN MAN CAUSED BY A FILTERABLE VIRUS. Science 1935, 81, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Albarino, C.G.; Palacios, G.; Khristova, M.L.; Erickson, B.R.; Carroll, S.A.; Comer, J.A.; Hui, J.; Briese, T.; St George, K.; Ksiazek, T.G.; et al. High diversity and ancient common ancestry of lymphocytic choriomeningitis virus. Emerg. Infect. Dis. 2010, 16, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Pfau, C.J.; Valenti, J.K.; Pevear, D.C.; Hunt, K.D. Lymphocytic choriomeningitis virus killer T cells are lethal only in weakly disseminated murine infections. J. Exp. Med. 1982, 156, 79–89. [Google Scholar] [CrossRef]
- Buesa-Gomez, J.; Teng, M.N.; Oldstone, C.E.; Oldstone, M.B.; de la Torre, J.C. Variants able to cause growth hormone deficiency syndrome are present within the disease-nil WE strain of lymphocytic choriomeningitis virus. J. Virol. 1996, 70, 8988–8992. [Google Scholar]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef]
- Wang, Y.; Swiecki, M.; Cella, M.; Alber, G.; Schreiber, R.D.; Gilfillan, S.; Colonna, M. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 2012, 11, 631–642. [Google Scholar] [CrossRef]
- Waggoner, S.N.; Cornberg, M.; Selin, L.K.; Welsh, R.M. Natural killer cells act as rheostats modulating antiviral T cells. Nature 2011, 481, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B.A.; Ware, B.C.; Horton, L.E.; Welch, M.J.; Aiolfi, R.; Zarpellon, A.; Ruggeri, Z.M.; Sullivan, B.M. Lymphocytic choriomeningitis virus Clone 13 infection causes either persistence or acute death dependent on IFN-1, cytotoxic T lymphocytes (CTLs), and host genetics. Proc. Natl. Acad. Sci. USA 2018, 115, E7814–E7823. [Google Scholar] [CrossRef] [PubMed]
- Louten, J.; van Rooijen, N.; Biron, C.A. Type 1 IFN deficiency in the absence of normal splenic architecture during lymphocytic choriomeningitis virus infection. J. Immunol. 2006, 177, 3266–3272. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.; Lillie, R.D. Experimental Lymphocytic Choriomeningitis of Monkeys and Mice Produced by a Virus Encountered in Studies of the 1933 St. Louis Encephalitis Epidemic. Public Health Reports (1896–1970) 1934, 49, 1019–1027. [Google Scholar] [CrossRef]
- Dutko, F.J.; Oldstone, M.B. Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J. Gen. Virol. 1983, 64, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Moskophidis, D.; Battegay, M.; Bruendler, M.A.; Laine, E.; Gresser, I.; Zinkernagel, R.M. Resistance of lymphocytic choriomeningitis virus to alpha/beta interferon and to gamma interferon. J. Virol. 1994, 68, 1951–1955. [Google Scholar] [PubMed]
- Zinkernagel, R.M.; Doherty, P.C. Cytotoxic thymus-derived lymphocytes in cerebrospinal fluid of mice with lymphocytic choriomeningitis. J. Exp. Med. 1973, 138, 1266–1269. [Google Scholar] [CrossRef]
- Baenziger, J.; Hengartner, H.; Zinkernagel, R.M.; Cole, G.A. Induction or prevention of immunopathological disease by cloned cytotoxic T cell lines specific for lymphocytic choriomeningitis virus. Eur. J. Immunol. 1986, 16, 387–393. [Google Scholar] [CrossRef]
- Ou, R.; Zhou, S.; Huang, L.; Moskophidis, D. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J. Virol. 2001, 75, 8407–8423. [Google Scholar] [CrossRef]
- Nansen, A.; Christensen, J.P.; Ropke, C.; Marker, O.; Scheynius, A.; Thomsen, A.R. Role of interferon-gamma in the pathogenesis of LCMV-induced meningitis: unimpaired leucocyte recruitment, but deficient macrophage activation in interferon-gamma knock-out mice. J. Neuroimmunol. 1998, 86, 202–212. [Google Scholar] [CrossRef]
- Lehmann-Grube, F.; Assmann, U.; Loliger, C.; Moskophidis, D.; Lohler, J. Mechanism of recovery from acute virus infection. I. Role of T lymphocytes in the clearance of lymphocytic choriomeningitis virus from spleens of mice. J. Immunol. 1985, 134, 608–615. [Google Scholar] [PubMed]
- Zinkernagel, R.M.; Moskophidis, D.; Kundig, T.; Oehen, S.; Pircher, H.; Hengartner, H. Effector T-cell induction and T-cell memory versus peripheral deletion of T cells. Immunol. Rev. 1993, 133, 199–223. [Google Scholar] [CrossRef] [PubMed]
- Benson, L.; Hotchin, J. Antibody formation in persistent tolerant infection with lymphocytic choriomeningitis virus. Nature 1969, 222, 1045–1047. [Google Scholar] [CrossRef] [PubMed]
- Hotchin, J.; Weigand, H. Studies of Lymphocytic Choriomeningitis in Mice I. The Relationship Between Age at Inoculation and Outcome of Infection. J. Immunol. 1961, 86, 392–400. [Google Scholar] [PubMed]
- Pfau, C.J.; Gresser, I.; Hunt, K.D. Lethal role of interferon in lymphocytic choriomeningitis virus-induced encephalitis. J. Gen. Virol. 1983, 64, 1827–1830. [Google Scholar] [CrossRef]
- Lehmann-Grube, F.; Ambrassat, J. A new method to detect lymphocytic choriomeningitis virus-specific antibody in human sera. J. Gen. Virol. 1977, 37, 85–92. [Google Scholar] [CrossRef]
- Lehmann-Grube, F.; Lohler, J.; Utermohlen, O.; Gegin, C. Antiviral immune responses of lymphocytic choriomeningitis virus-infected mice lacking CD8+ T lymphocytes because of disruption of the beta 2-microglobulin gene. J. Virol. 1993, 67, 332–339. [Google Scholar]
- Byrne, J.A.; Oldstone, M.B. Biology of cloned cytotoxic T lymphocytes specific for lymphocytic choriomeningitis virus: clearance of virus in vivo. J. Virol. 1984, 51, 682–686. [Google Scholar]
- Müller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef]
- Christensen, J.P.; Marker, O.; Thomsen, A.R. The role of CD4+ T cells in cell-mediated immunity to LCMV: Studies in MHC class I and class II deficient mice. Scand. J. Immunol. 1994, 40, 373–382. [Google Scholar] [CrossRef]
- Leist, T.P.; Cobbold, S.P.; Waldmann, H.; Aguet, M.; Zinkernagel, R.M. Functional analysis of T lymphocyte subsets in antiviral host defense. J. Immunol. 1987, 138, 2278–2281. [Google Scholar] [PubMed]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. CCS 2017, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Bergthaler, A.; Flatz, L.; Hegazy, A.N.; Johnson, S.; Horvath, E.; Löhning, M.; Pinschewer, D.D.; Zinkernagel, R.M. Viral replicative capacity is the primary determinant of lymphocytic choriomeningitis virus persistence and immunosuppression. Proc. Natl. Acad. Sci. USA 2010, 107, 21641–21646. [Google Scholar] [CrossRef]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef]
- Sevilla, N.; Kunz, S.; Holz, A.; Lewicki, H.; Homann, D.; Yamada, H.; Campbell, K.P.; de La Torre, J.C.; Oldstone, M.B. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J. Exp. Med. 2000, 192, 1249–1260. [Google Scholar] [CrossRef]
- Sevilla, N.; McGavern, D.B.; Teng, C.; Kunz, S.; Oldstone, M.B. Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J. Clin. Invest. 2004, 113, 737–745. [Google Scholar] [CrossRef]
- Ng, C.T.; Sullivan, B.M.; Oldstone, M.B.A. The role of dendritic cells in viral persistence. Curr. Opin. Virol. 2011, 1, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Sevilla, N.; Rojek, J.M.; Oldstone, M.B. Use of alternative receptors different than alpha-dystroglycan by selected isolates of lymphocytic choriomeningitis virus. Virology 2004, 325, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Smelt, S.C.; Borrow, P.; Kunz, S.; Cao, W.; Tishon, A.; Lewicki, H.; Campbell, K.P.; Oldstone, M.B. Differences in affinity of binding of lymphocytic choriomeningitis virus strains to the cellular receptor alpha-dystroglycan correlate with viral tropism and disease kinetics. J. Virol. 2001, 75, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Clingan, J.M.; Ostrow, K.; Hosiawa, K.A.; Chen, Z.J.; Matloubian, M. Differential roles for RIG-I-like receptors and nucleic acid-sensing TLR pathways in controlling a chronic viral infection. J. Immunol. 2012, 188, 4432–4440. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Perez, M.; Kunz, S. Cellular entry of lymphocytic choriomeningitis virus. J. Virol. 2008, 82, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, K.H.; Richardson-Burns, S.; Alexopoulou, L.; Tyler, K.L.; Flavell, R.A.; Oldstone, M.B. Does Toll-like receptor 3 play a biological role in virus infections? Virology 2004, 322, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Cao, W.; Liu, Y.J. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Tanaka, N.; Hata, N.; Oda, E.; Taniguchi, T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998, 425, 112–116. [Google Scholar] [CrossRef]
- Marie, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Weber, F. RIG-I-like receptors and negative-strand RNA viruses: RLRly bird catches some worms. Cytokine Growth Factor Rev. 2014, 25, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Hata, N.; Asagiri, M.; Nakaya, T.; Taniguchi, T.; Tanaka, N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998, 441, 106–110. [Google Scholar] [CrossRef]
- Yang, H.; Lin, C.H.; Ma, G.; Baffi, M.O.; Wathelet, M.G. Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. J. Biol. Chem. 2003, 278, 15495–15504. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sobrido, L.; Zuniga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Van Pesch, V.; Lanaya, H.; Renauld, J.C.; Michiels, T. Characterization of the murine alpha interferon gene family. J. Virol. 2004, 78, 8219–8228. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Piehler, J.; Thomas, C.; Garcia, K.C.; Schreiber, G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol. Rev. 2012, 250, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Sullivan, B.M.; Teijaro, J.R.; Lee, A.M.; Welch, M.; Rice, S.; Sheehan, K.C.F.; Schreiber, R.D.; Oldstone, M.B.A. Blockade of interferon Beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 2015, 17, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Y.; Kessler, D.S.; Veals, S.A.; Levy, D.E.; Darnell, J.E., Jr. ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains. Proc. Natl. Acad. Sci. USA 1990, 87, 8555–8559. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Kessler, D.S.; Pine, R.; Darnell, J.E., Jr. Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 1989, 3, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Majoros, A.; Platanitis, E.; Kernbauer-Holzl, E.; Rosebrock, F.; Muller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Suprunenko, T.; Hofer, M.J. The emerging role of interferon regulatory factor 9 in the antiviral host response and beyond. Cytokine Growth Factor Rev. 2016, 29, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, K.; Nowicka, H.; Kostyrko, K.; Antonczyk, A.; Wesoly, J.; Bluyssen, H.A. The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses. Cytokine Growth Factor Rev. 2016, 29, 71–81. [Google Scholar] [CrossRef]
- Hofer, M.J.; Li, W.; Manders, P.; Terry, R.; Lim, S.L.; King, N.J.C.; Campbell, I.L. Mice deficient in STAT1 but not STAT2 or IRF9 develop a lethal CD4+ T-cell-mediated disease following infection with lymphocytic choriomeningitis virus. J. Virol. 2012, 86, 6932–6946. [Google Scholar] [CrossRef]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Van den Broek, M.F.; Müller, U.; Huang, S.; Aguet, M.; Zinkernagel, R.M. Antiviral defense in mice lacking both alpha/beta and gamma interferon receptors. J. Virol. 1995, 69, 4792–4796. [Google Scholar] [PubMed]
- Huber, M.; Suprunenko, T.; Ashhurst, T.; Marbach, F.; Raifer, H.; Wolff, S.; Strecker, T.; Viengkhou, B.; Jung, S.R.; Obermann, H.L.; et al. IRF9 Prevents CD8(+) T Cell Exhaustion in an Extrinsic Manner during Acute Lymphocytic Choriomeningitis Virus Infection. J. Virol. 2017, 91, e01219-17. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, K.; Kemper, P.; Stalder, A.; Zhang, J.; Hobbs, M.V.; Whitton, J.L.; Campbell, I.L. Altered tissue distribution of viral replication and T cell spreading is pivotal in the protection against fatal lymphocytic choriomeningitis in mice after neutralization of IFN-alpha/beta. J. Immunol. 1994, 153, 220. [Google Scholar] [PubMed]
- Lindqvist, R.; Mundt, F.; Gilthorpe, J.D.; Wolfel, S.; Gekara, N.O.; Kroger, A.; Overby, A.K. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflammation 2016, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ma, Y.; Wang, L.; Chi, X.; Yan, R.; Wang, S.; Li, X.; Chen, X.; Shao, W.; Chen, J.L. Alpha/beta interferon receptor deficiency in mice significantly enhances susceptibility of the animals to pseudorabies virus infection. Vet. Microbiol. 2017, 203, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.; Mohamad, S.M.; Young, D.F.; Skelton, A.J.; Leahy, T.R.; Munday, D.C.; Butler, K.M.; Morfopoulou, S.; Brown, J.R.; Hubank, M.; et al. Human IFNAR2 deficiency: Lessons for antiviral immunity. Sci. Transl. Med. 2015, 7, 307ra154. [Google Scholar] [CrossRef] [PubMed]
- Ousman, S.S.; Wang, J.; Campbell, I.L. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J. Virol. 2005, 79, 7514–7527. [Google Scholar] [CrossRef] [PubMed]
- Audige, A.; Hofer, U.; Dittmer, U.; van den Broek, M.; Speck, R.F. Evaluation of the immunomodulatory and antiviral effects of the cytokine combination IFN-alpha and IL-7 in the lymphocytic choriomeningitis virus and Friend retrovirus mouse infection models. Viral Immunol. 2011, 24, 375–385. [Google Scholar] [CrossRef]
- Sullivan, B.M.; Teijaro, J.R.; de la Torre, J.C.; Oldstone, M.B.A. Early virus-host interactions dictate the course of a persistent infection. PLoS Pathog. 2015, 11, e1004588. [Google Scholar] [CrossRef]
- Snell, L.M.; Brooks, D.G. New insights into type I interferon and the immunopathogenesis of persistent viral infections. Curr. Opin. Immunol. 2015, 34, 91–98. [Google Scholar] [CrossRef]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef]
- Talpaz, M.; Mercer, J.; Hehlmann, R. The interferon-alpha revival in CML. Ann. Hematol. 2015, 94 (Suppl. 2), S195–S207. [Google Scholar] [CrossRef]
- Hehlmann, R.; Lauseker, M.; Jung-Munkwitz, S.; Leitner, A.; Muller, M.C.; Pletsch, N.; Proetel, U.; Haferlach, C.; Schlegelberger, B.; Balleisen, L.; et al. Tolerability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon-alpha in newly diagnosed chronic myeloid leukemia. J. Clin. Oncol. 2011, 29, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Borrow, P.; Martinez-Sobrido, L.; de la Torre, J.C. Inhibition of the type I interferon antiviral response during arenavirus infection. Viruses 2010, 2, 2443–2480. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Park, C.H.; Jeong, Y.H.; Kim, Y.J.; Ha, S.J. Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8(+) T-cell exhaustion. PLoS Pathog. 2013, 9, e1003478. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Kim, B.; Oh, G.T.; Kim, Y.J. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat. Immunol. 2013, 14, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.D.; Whitmire, J.K. The depletion of NK cells prevents T cell exhaustion to efficiently control disseminating virus infection. J. Immunol. 2013, 190, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.C.; Grusdat, M.; Pandyra, A.A.; Polz, R.; Huang, J.; Sharma, P.; Deenen, R.; Kohrer, K.; Rahbar, R.; Diefenbach, A.; et al. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 2014, 40, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Aichele, P.; Unsoeld, H.; Koschella, M.; Schweier, O.; Kalinke, U.; Vucikuja, S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 2006, 176, 4525–4529. [Google Scholar] [CrossRef]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Keppler, S.J.; Rosenits, K.; Koegl, T.; Vucikuja, S.; Aichele, P. Signal 3 cytokines as modulators of primary immune responses during infections: the interplay of type I IFN and IL-12 in CD8 T cell responses. PLoS One 2012, 7, e40865. [Google Scholar] [CrossRef] [PubMed]
- Cousens, L.P.; Peterson, R.; Hsu, S.; Dorner, A.; Altman, J.D.; Ahmed, R.; Biron, C.A. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J. Exp. Med. 1999, 189, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C. Innate immune responses to LCMV infections: natural killer cells and cytokines. Curr. Top. Microbiol. Immunol. 2002, 263, 7–27. [Google Scholar] [PubMed]
- Ahmed, R.; Butler, L.D.; Bhatti, L. T4+ T helper cell function in vivo: differential requirement for induction of antiviral cytotoxic T-cell and antibody responses. J. Virol. 1988, 62, 2102–2106. [Google Scholar] [PubMed]
- Matloubian, M.; Concepcion, R.J.; Ahmed, R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 1994, 68, 8056–8063. [Google Scholar]
- Havenar-Daughton, C.; Kolumam, G.A.; Murali-Krishna, K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J. Immunol. 2006, 176, 3315–3319. [Google Scholar] [CrossRef]
- Aubert, R.D.; Kamphorst, A.O.; Sarkar, S.; Vezys, V.; Ha, S.J.; Barber, D.L.; Ye, L.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc. Natl. Acad. Sci. USA 2011, 108, 21182–21187. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Madden, P.; Henning, N.; Gregory, S.; Aid, M.; Martinot, A.J.; Barouch, D.H.; Penaloza-MacMaster, P. Regulation of CD4 T cells and their effects on immunopathological inflammation following viral infection. Immunology 2017, 152, 328–343. [Google Scholar] [CrossRef]
- Srivastava, S.; Koch, M.A.; Pepper, M.; Campbell, D.J. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J. Exp. Med. 2014, 211, 961–974. [Google Scholar] [CrossRef]
- Li, W.; Hofer, M.J.; Noçon, A.L.; Manders, P.; Campbell, I.L. Interferon regulatory factor 7 (IRF7) is required for the optimal initial control but not subsequent clearance of lymphocytic choriomeningitis virus infection in mice. Virology 2013, 439, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Cerny, A.M.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Role of interferon regulatory factor 7 in T cell responses during acute lymphocytic choriomeningitis virus infection. J. Virol. 2012, 86, 11254–11265. [Google Scholar] [CrossRef] [PubMed]
- Hangartner, L.; Zinkernagel, R.M.; Hengartner, H. Antiviral antibody responses: the two extremes of a wide spectrum. Nat. Rev. Immunol. 2006, 6, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Cerny, A.; Huegin, A.W.; Sutter, S.; Bazin, H.; Hengartner, H.H.; Zinkernagel, R.M. Immunity to lymphocytic choriomeningitis virus in B cell-depleted mice: evidence for B cell and antibody-independent protection by memory T cells. Eur. J. Immunol. 1986, 16, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Bergthaler, A.; Flatz, L.; Verschoor, A.; Hegazy, A.N.; Holdener, M.; Fink, K.; Eschli, B.; Merkler, D.; Sommerstein, R.; Horvath, E.; et al. Impaired antibody response causes persistence of prototypic T cell-contained virus. PLoS Biol. 2009, 7, e1000080. [Google Scholar] [CrossRef]
- Recher, M.; Lang, K.S.; Navarini, A.; Hunziker, L.; Lang, P.A.; Fink, K.; Freigang, S.; Georgiev, P.; Hangartner, L.; Zellweger, R.; et al. Extralymphatic virus sanctuaries as a consequence of potent T-cell activation. Nat. Med. 2007, 13, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.P.; Kauffmann, S.O.; Thomsen, A.R. Deficient CD4+ T cell priming and regression of CD8+ T cell functionality in virus-infected mice lacking a normal B cell compartment. J. Immunol. 2003, 171, 4733–4741. [Google Scholar] [CrossRef] [PubMed]
- Moseman, E.A.; Wu, T.; de la Torre, J.C.; Schwartzberg, P.L.; McGavern, D.B. Type I interferon suppresses virus-specific B cell responses by modulating CD8(+) T cell differentiation. Sci. Immunol. 2016, 1, eaah3565. [Google Scholar]
- Daugan, M.; Murira, A.; Mindt, B.C.; Germain, A.; Tarrab, E.; Lapierre, P.; Fritz, J.H.; Lamarre, A. Type I Interferon Impairs Specific Antibody Responses Early during Establishment of LCMV Infection. Front. Immunol. 2016, 7, 564. [Google Scholar] [CrossRef]
- Jung, A.; Kato, H.; Kumagai, Y.; Kumar, H.; Kawai, T.; Takeuchi, O.; Akira, S. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J. Virol. 2008, 82, 196–206. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Lewis, K.L.; Firner, S.; Thiel, V.; Hugues, S.; Reith, W.; Ludewig, B.; Reizis, B. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc. Natl. Acad. Sci. USA 2012, 109, 3012–3017. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.H.; Cui, W.; Larosa, D.F.; Taylor, D.K.; Zhang, J.; Goldstein, D.R.; Wherry, E.J.; Kaech, S.M.; Turka, L.A. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute lymphocytic choriomeningitis virus infection. J. Immunol. 2008, 181, 3804–3810. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.E.; Fenger, C.; Issazadeh-Navikas, S.; Krug, A.; Liljestrom, P.; Goriely, S.; Paludan, S.R.; Finsen, B.; Christensen, J.P.; Thomsen, A.R. Differential impact of interferon regulatory factor 7 in initiation of the type I interferon response in the lymphocytic choriomeningitis virus-infected central nervous system versus the periphery. J. Virol. 2012, 86, 7384–7392. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Cervantes-Barragan, L.; Verschoor, A.; Navarini, A.A.; Recher, M.; Pellegrini, M.; Flatz, L.; Bergthaler, A.; Honda, K.; Ludewig, B.; et al. Hematopoietic cell-derived interferon controls viral replication and virus-induced disease. Blood 2009, 113, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Gil, M.P.; Wang, X.; Louten, J.; Chu, W.M.; Biron, C.A. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 2007, 204, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hofer, M.J.; Jung, S.R.; Lim, S.L.; Campbell, I.L. IRF7-dependent type I interferon production induces lethal immune-mediated disease in STAT1 knockout mice infected with lymphocytic choriomeningitis virus. J. Virol. 2014, 88, 7578–7588. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, D.R.; Folmsbee, S.S.; Muller, W.J.; Longnecker, R. The Type I Interferon Response Determines Differences in Choroid Plexus Susceptibility between Newborns and Adults in Herpes Simplex Virus Encephalitis. mBio 2016, 7, e00437-16. [Google Scholar] [CrossRef] [PubMed]
- Hahm, B.; Trifilo, M.J.; Zuniga, E.I.; Oldstone, M.B. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity 2005, 22, 247–257. [Google Scholar] [CrossRef]
- Kessler, D.S.; Veals, S.A.; Fu, X.Y.; Levy, D.E. Interferon-alpha regulates nuclear translocation and DNA-binding affinity of ISGF3, a multimeric transcriptional activator. Genes Dev. 1990, 4, 1753. [Google Scholar] [CrossRef]
- Qureshi, S.A.; Salditt-Georgieff, M.; Darnell, J.E. Tyrosine-Phosphorylated Stat1 and Stat2 Plus a 48-kDa Protein All Contact DNA in Forming Interferon-Stimulated-Gene Factor 3. Proc. Natl. Acad. Sci. USA 1995, 92, 3829–3833. [Google Scholar] [CrossRef]
- Veals, S.A.; Santa Maria, T.; Levy, D.E. Two domains of ISGF3 gamma that mediate protein-DNA and protein-protein interactions during transcription factor assembly contribute to DNA-binding specificity. Mol. Cell. Biol. 1993, 13, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Matsumoto, M.; Sato, M.; Kashiwazaki, Y.; Kimura, T.; Kitagawa, M.; Yokochi, T.; Tan, R.S.; Takasugi, T.; Kadokawa, Y.; et al. Regulation of IFN-alpha/beta genes: evidence for a dual function of the transcription factor complex ISGF3 in the production and action of IFN-alpha/beta. Genes Cells 1996, 1, 995. [Google Scholar] [CrossRef] [PubMed]
- John, J.; McKendry, R.; Pellegrini, S.; Flavell, D.; Kerr, I.M.; Stark, G.R. Isolation and characterization of a new mutant human cell line unresponsive to alpha and beta interferons. Mol. Cell. Biol. 1991, 11, 4189–4195. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Kadokawa, Y.; Harada, H.; Matsumoto, M.; Sato, M.; Kashiwazaki, Y.; Tarutani, M.; Tan, R.S.; Takasugi, T.; Matsuyama, T.; et al. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells 1996, 1, 115–124. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Origin | Replication and Pathogenicity 1 |
|---|---|---|
| Armstrong 53b | Originally isolated from an infected patient in 1993 [14] and obtained from a triple plaque purified clone and two passages in BHK-21 (baby hamster kidney) cells [15]. | Slowly replicating. Peripheral infection causes acute infection; virus clearance within 2 weeks from wild-type (WT) mice [3,16]. Intracranial infection results in lethal lymphocytic choriomeningitis (LCM) [17,18]. |
| Clone-13 | 13th isolate of Armstrong that differed from the parental strain in that it persisted in mice [3] | Faster replicating than LCMV-Arm 53b. Peripheral infection with high dose virus via intravenous route causes persistence, whereas low doses are cleared [3,16]. |
| Traub | Isolated from persistently infected mouse [4] | Faster replicating. Peripheral infection results in chronic infection that is cleared within 2–4 months [16,19]. Intracranial infection causes LCM [20]. |
| WE 2 | Isolated from an infected patient (WE) with meningo-encephalitis in in 1935 [5] | Slowly replicating. Peripheral infection is cleared within 2 weeks [16,19,21,22]. Mice survive intracranial infection ([23] as reviewed in [22]) |
| Aggressive | Isolated from an LCMV-WE [UBC] carrier mouse [7] Note, UBC is a derivative of WE [24] | Slowly replicating. Peripheral infection is cleared within 2 weeks [16]. Intracranial infection with low doses causes LCM, however high dose infected mice survive [25] |
| Docile | Isolated from an LCMV-WE [UBC] carrier mouse [7] Note, UBC is a derivative of WE [24] | More quickly replicating than parent strain. Peripheral infection results in persistent infection [16,19,22]. Intracranial infection with high dose results in lifelong persistence, but low dose infected mice survive [25] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suprunenko, T.; Hofer, M.J. Complexities of Type I Interferon Biology: Lessons from LCMV. Viruses 2019, 11, 172. https://doi.org/10.3390/v11020172
Suprunenko T, Hofer MJ. Complexities of Type I Interferon Biology: Lessons from LCMV. Viruses. 2019; 11(2):172. https://doi.org/10.3390/v11020172
Chicago/Turabian StyleSuprunenko, Tamara, and Markus J. Hofer. 2019. "Complexities of Type I Interferon Biology: Lessons from LCMV" Viruses 11, no. 2: 172. https://doi.org/10.3390/v11020172
APA StyleSuprunenko, T., & Hofer, M. J. (2019). Complexities of Type I Interferon Biology: Lessons from LCMV. Viruses, 11(2), 172. https://doi.org/10.3390/v11020172