Nuclear Transit and HIV LTR Binding of NF-κB Subunits Held by IκB Proteins: Implications for HIV-1 Activation


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Transfection
2.2. Nuclear and Cytoplasmic Extract Preparation
2.3. Immunoblotting
2.4. Chromatin Immunoprecipitation Assay (ChIP)
2.5. HIV-1 RNA Quantitation
2.6. Image Quantitation
2.7. Statistical Analyses
3. Results
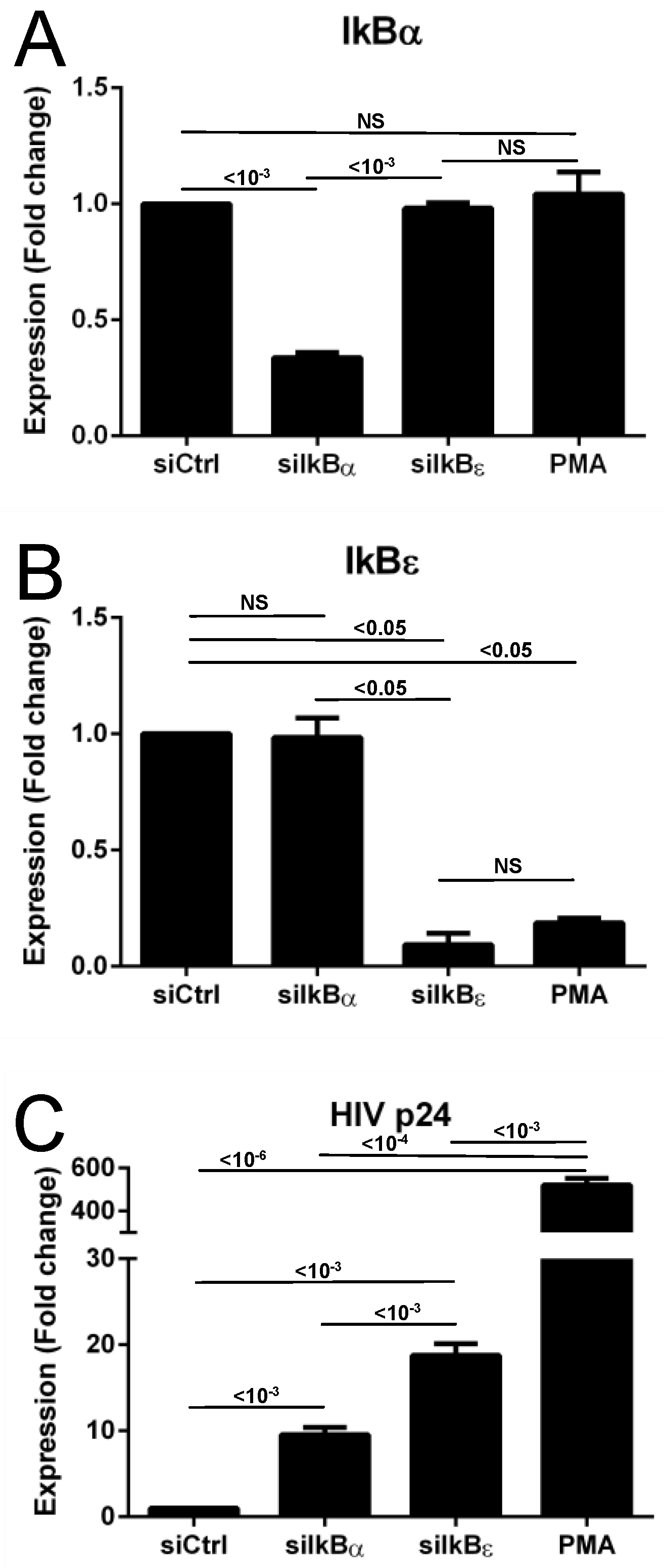
3.1. siRNA Transfection and HIV Transcription Activation
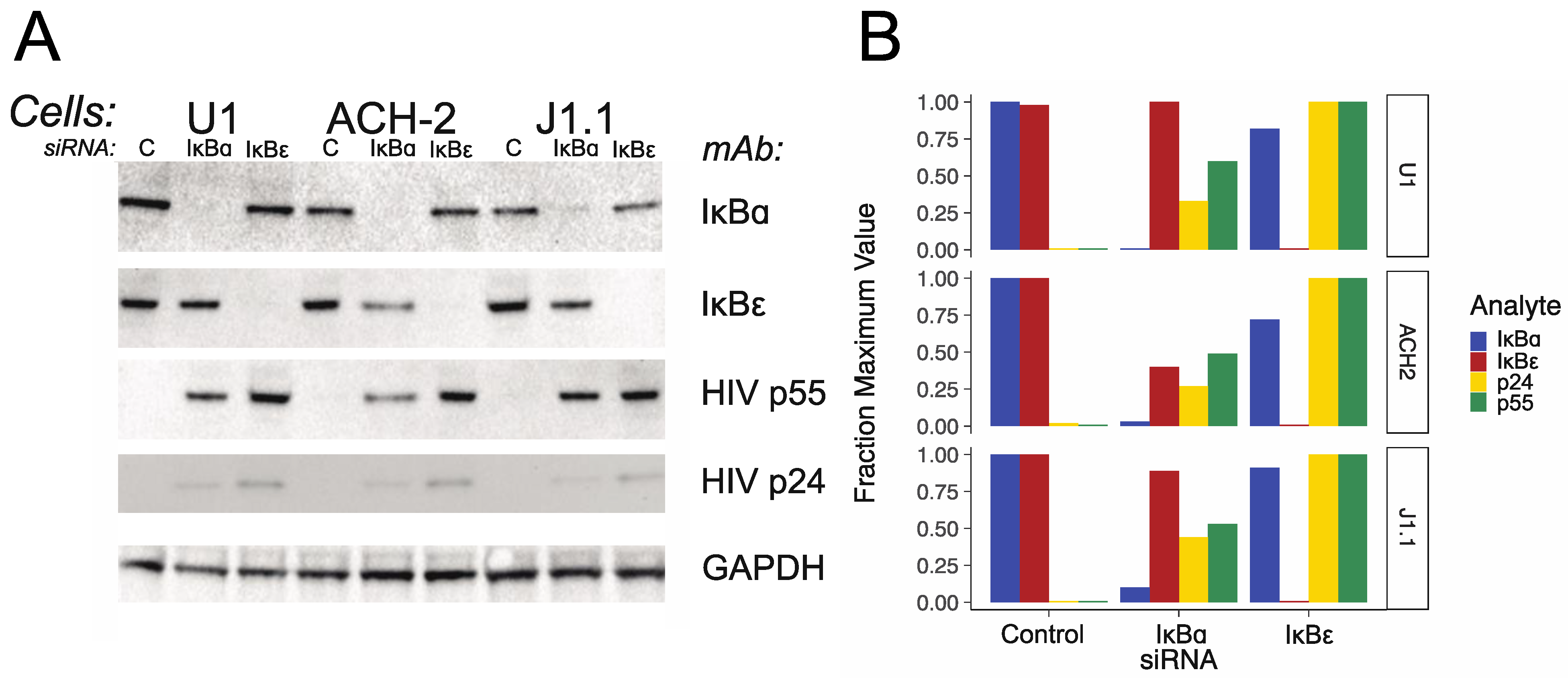
3.2. IκBα and IκBε siRNA Transfection and IκBα and IκBε and HIV Protein Expression
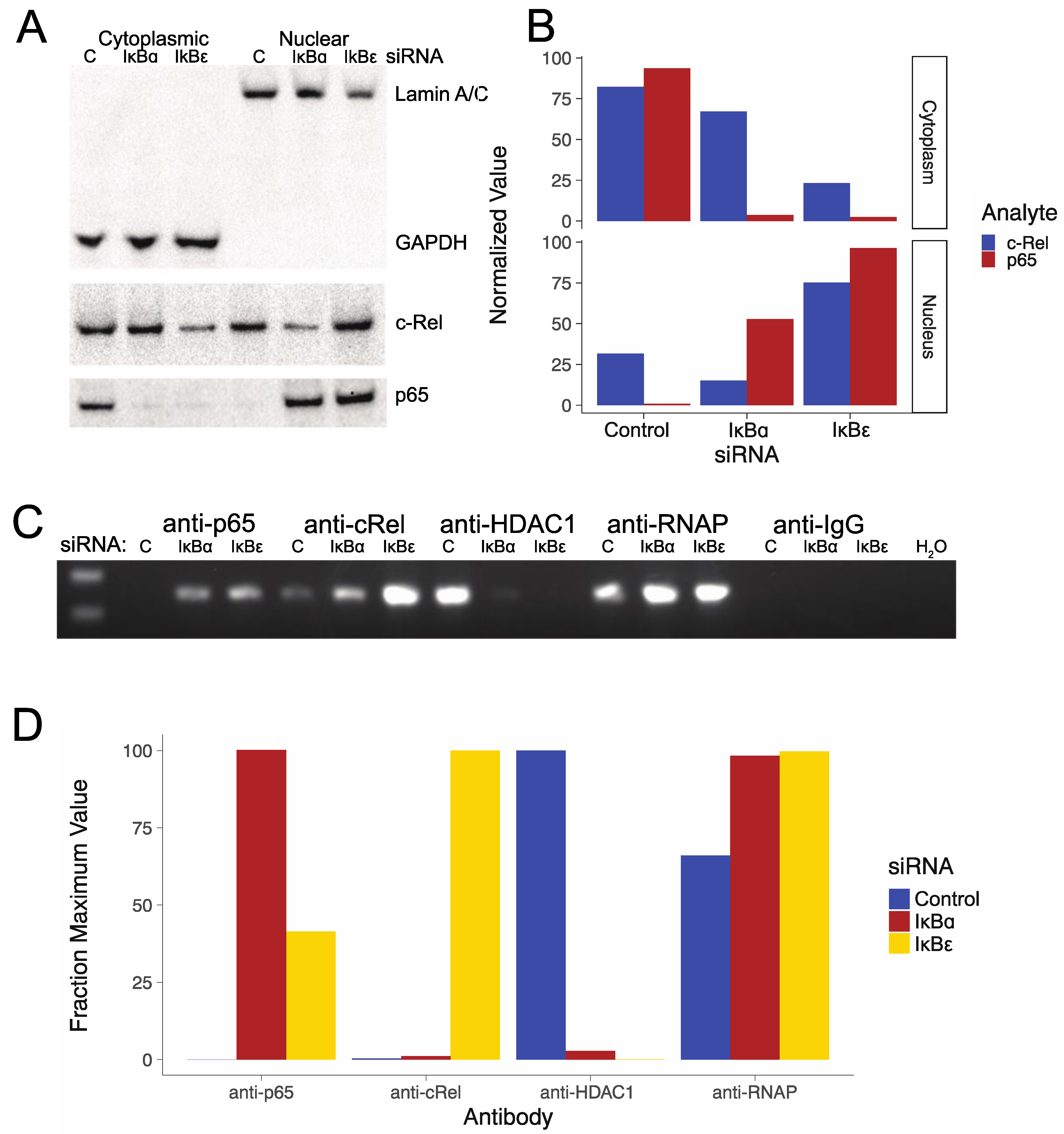
3.3. Nuclear Translocation of Activating NF-κB Subunits Associated with Transfection of IκBα and IκBε siRNAs
3.4. HIV LTR NF-κB Chromatin Immunoprecipitation After Transfection of IκBα and IκBε siRNAs
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Sengupta, S.; Siliciano, R.F. Targeting the latent reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gonzalez, S.; Colomer-Lluch, M.; Serra-Moreno, R. Barriers for hiv cure: The latent reservoir. AIDS Res. Hum. Retrovir. 2018, 34, 739–759. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.M.; Margolis, D.M. Hiv persistence on antiretroviral therapy and barriers to a cure. Adv. Exp. Med. Biol. 2018, 1075, 165–185. [Google Scholar] [PubMed]
- Pitman, M.C.; Lau, J.S.Y.; McMahon, J.H.; Lewin, S.R. Barriers and strategies to achieve a cure for hiv. Lancet HIV 2018, 5, e317–e328. [Google Scholar] [CrossRef]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. Novel latency reversal agents for HIV-1 cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. Hiv latency in isolated patient cd4+ T cells may be due to blocks in hiv transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef]
- Singh, A.; Weinberger, L.S. Stochastic gene expression as a molecular switch for viral latency. Curr. Opin. Microbiol. 2009, 12, 460–466. [Google Scholar] [CrossRef][Green Version]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Laughlin, M.; Zeichner, S.; Kolson, D.; Alwine, J.; Seshamma, T.; Pomerantz, R.; Gonzalez-Scaran, F. Sodium butryate treatment of cells latently infected with hiv-1 results in the expression of unspliced viral rna. Virology 1993, 196, 496–505. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in hiv-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef]
- Elliott, J.H.; Wightman, F.; Solomon, A.; Ghneim, K.; Ahlers, J.; Cameron, M.J.; Smith, M.Z.; Spelman, T.; McMahon, J.; Velayudham, P.; et al. Activation of hiv transcription with short-course vorinostat in hiv-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014, 10, e1004473. [Google Scholar] [CrossRef] [PubMed]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed]
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; Van Driessche, B.; Corazza, F.; Gatot, J.S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8, 117–138. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. Bet inhibitors rvx-208 and pfi-1 reactivate HIV-1 from latency. Sci. Rep. 2017, 7, 16646. [Google Scholar] [CrossRef]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Prins, J.M.; Jurriaans, S.; van Praag, R.M.; Blaak, H.; van Rij, R.; Schellekens, P.T.; ten Berge, I.J.; Yong, S.L.; Fox, C.H.; Roos, M.T.; et al. Immuno-activation with anti-CD3 and recombinant human Il-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 1999, 13, 2405–2410. [Google Scholar] [CrossRef]
- McKernan, L.N.; Momjian, D.; Kulkosky, J. Protein kinase c: One pathway towards the eradication of latent HIV-1 reservoirs. Adv. Virol. 2012, 2012, 805347. [Google Scholar] [CrossRef]
- Jiang, G.; Dandekar, S. Targeting NF-kappaB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retrovir. 2015, 31, 4–12. [Google Scholar] [CrossRef]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef]
- Martin, A.R.; Pollack, R.A.; Capoferri, A.; Ambinder, R.F.; Durand, C.M.; Siliciano, R.F. Rapamycin-mediated mtor inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J. Clin. Investig. 2017, 127, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Spivak, A.M.; Larragoite, E.T.; Coletti, M.L.; Macedo, A.B.; Martins, L.J.; Bosque, A.; Planelles, V. Janus kinase inhibition suppresses PKC-induced cytokine release without affecting HIV-1 latency reversal ex vivo. Retrovirology 2016, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Lafeuillade, A.; Poggi, C.; Chadapaud, S.; Hittinger, G.; Chouraqui, M.; Pisapia, M.; Delbeke, E. Pilot study of a combination of highly active antiretroviral therapy and cytokines to induce HIV-1 remission. J. Acquir. Immune Defic. Syndr. 2001, 26, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Stellbrink, H.J.; Hufert, F.T.; Tenner-Racz, K.; Lauer, J.; Schneider, C.; Albrecht, H.; Racz, P.; van Lunzen, J. Kinetics of productive and latent hiv infection in lymphatic tissue and peripheral blood during triple-drug combination therapy with or without additional interleukin-2. Antivir. Ther. 1998, 3, 209–214. [Google Scholar] [PubMed]
- Dybul, M.; Hidalgo, B.; Chun, T.W.; Belson, M.; Migueles, S.A.; Justement, J.S.; Herpin, B.; Perry, C.; Hallahan, C.W.; Davey, R.T.; et al. Pilot study of the effects of intermittent interleukin-2 on human immunodeficiency virus (HIV)-specific immune responses in patients treated during recently acquired hiv infection. J. Infect. Dis. 2002, 185, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Engel, D.; Mizell, S.B.; Hallahan, C.W.; Fischette, M.; Park, S.; Davey, R.T., Jr.; Dybul, M.; Kovacs, J.A.; Metcalf, J.A.; et al. Effect of interleukin-2 on the pool of latently infected, resting cd4+ T cells in hiv-1-infected patients receiving highly active anti-retroviral therapy. Nat. Med. 1999, 5, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Fenard, D.; Bisgrove, D.; Verdin, E.; Greene, W.C. Prostratin antagonizes HIV latency by activating nf-kappab. J. Biol. Chem. 2004, 279, 42008–42017. [Google Scholar] [CrossRef]
- Korin, Y.D.; Brooks, D.G.; Brown, S.; Korotzer, A.; Zack, J.A. Effects of prostratin on T-cell activation and human immunodeficiency virus latency. J. Virol. 2002, 76, 8118–8123. [Google Scholar] [CrossRef]
- Kulkosky, J.; Sullivan, J.; Xu, Y.; Souder, E.; Hamer, D.H.; Pomerantz, R.J. Expression of latent haart-persistent HIV type 1 induced by novel cellular activating agents. AIDS Res. Hum. Retrovir. 2004, 20, 497–505. [Google Scholar] [CrossRef]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the aids retrovirus promoter by the cellular transcription factor, SP1. Science 1986, 232, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Edwards, N.L.; Duckett, C.S.; Agranoff, A.B.; Schmid, R.M.; Nabel, G.J. A cooperative interaction between NF-kappa B and SP1 is required for HIV-1 enhancer activation. EMBO J. 1993, 12, 3551–3558. [Google Scholar] [CrossRef] [PubMed]
- El Kharroubi, A.; Piras, G.; Zensen, R.; Martin, M.A. Transcriptional activation of the integrated chromatin-associated human immunodeficiency virus type 1 promoter. Mol. Cell. Biol. 1998, 18, 2535–2544. [Google Scholar] [CrossRef] [PubMed]
- Berkhout, B.; Jeang, K.T. Functional roles for the TATA promoter and enhancers in basal and Tat-induced expression of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 1992, 66, 139–149. [Google Scholar]
- Kaufman, J.D.; Valandra, G.; Roderiquez, G.; Bushar, G.; Giri, C.; Norcross, M.A. Phorbol ester enhances human immunodeficiency virus-promoted gene expression and acts on a repeated 10-base-pair functional enhancer element. Mol. Cell. Biol. 1987, 7, 3759–3766. [Google Scholar] [CrossRef]
- Zeichner, S.L.; Kim, J.Y.; Alwine, J.C. Linker-scanning mutational analysis of the transcriptional activity of the human immunodeficiency virus type 1 long terminal repeat. J. Virol. 1991, 65, 2436–2444. [Google Scholar]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The t cell activation factor NF-ATC positively regulates HIV-1 replication and gene expression in t cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef]
- Tong-Starksen, S.E.; Luciw, P.A.; Peterlin, B.M. Human immunodeficiency virus long terminal repeat responds to T-cell activation signals. Proc. Natl. Acad. Sci. USA 1987, 84, 6845–6849. [Google Scholar] [CrossRef]
- Zeichner, S.L.; Hirka, G.; Andrews, P.W.; Alwine, J.C. Differentiation-dependent human immunodeficiency virus long terminal repeat regulatory elements active in human teratocarcinoma cells. J. Virol. 1992, 66, 2268–2273. [Google Scholar]
- Marshall, N.F.; Price, D.H. Purification of p-tefb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar] [CrossRef]
- Marshall, N.F.; Peng, J.; Xie, Z.; Price, D.H. Control of rna polymerase ii elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 1996, 271, 27176–27183. [Google Scholar] [CrossRef]
- Yang, X.; Herrmann, C.H.; Rice, A.P. The human immunodeficiency virus tat proteins specifically associate with tak in vivo and require the carboxyl-terminal domain of rna polymerase ii for function. J. Virol. 1996, 70, 4576–4584. [Google Scholar]
- Gold, M.O.; Yang, X.; Herrmann, C.H.; Rice, A.P. Pitalre, the catalytic subunit of tak, is required for human immunodeficiency virus tat transactivation in vivo. J. Virol. 1998, 72, 4448–4453. [Google Scholar]
- West, M.J.; Lowe, A.D.; Karn, J. Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J. Virol. 2001, 75, 8524–8537. [Google Scholar] [CrossRef]
- Barboric, M.; Nissen, R.M.; Kanazawa, S.; Jabrane-Ferrat, N.; Peterlin, B.M. Nf-kappab binds p-tefb to stimulate transcriptional elongation by rna polymerase II. Mol. Cell 2001, 8, 327–337. [Google Scholar] [CrossRef]
- Andersen, J.L.; DeHart, J.L.; Zimmerman, E.S.; Ardon, O.; Kim, B.; Jacquot, G.; Benichou, S.; Planelles, V. Hiv-1 vpr-induced apoptosis is cell cycle dependent and requires bax but not ant. PLoS Pathog. 2006, 2, e127. [Google Scholar] [CrossRef]
- Basseres, D.S.; Baldwin, A.S. Nuclear factor-kappab and inhibitor of kappab kinase pathways in oncogenic initiation and progression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef]
- Ghosh, S.; Hayden, M.S. New regulators of nf-kappab in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-kappaB p50 promotes hiv latency through hdac recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef]
- Williams, S.A.; Greene, W.C. Regulation of HIV-1 latency by T-cell activation. Cytokine 2007, 39, 63–74. [Google Scholar] [CrossRef]
- Geeraert, L.; Kraus, G.; Pomerantz, R.J. Hide-and-seek: The challenge of viral persistence in HIV-1 infection. Annu. Rev. Med. 2008, 59, 487–501. [Google Scholar] [CrossRef]
- Hoffmann, A.; Leung, T.H.; Baltimore, D. Genetic analysis of NF-kappaB/rel transcription factors defines functional specificities. EMBO J. 2003, 22, 5530–5539. [Google Scholar] [CrossRef]
- Natoli, G.; Saccani, S.; Bosisio, D.; Marazzi, I. Interactions of NF-kappaB with chromatin: The art of being at the right place at the right time. Nat. Immunol. 2005, 6, 439–445. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Carmody, R.J.; Ruan, Q.; Palmer, S.; Hilliard, B.; Chen, Y.H. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science 2007, 317, 675–678. [Google Scholar] [CrossRef]
- Motoyama, M.; Yamazaki, S.; Eto-Kimura, A.; Takeshige, K.; Muta, T. Positive and negative regulation of nuclear factor-kappab-mediated transcription by ikappab-zeta, an inducible nuclear protein. J. Biol. Chem. 2005, 280, 7444–7451. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamazaki, S.; Uematsu, S.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Kuwata, H.; Takeuchi, O.; Takeshige, K.; et al. Regulation of toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein ikappabzeta. Nature 2004, 430, 218–222. [Google Scholar] [CrossRef]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the complexities of the nf-kappab signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef]
- Pasparakis, M.; Luedde, T.; Schmidt-Supprian, M. Dissection of the nf-kappab signalling cascade in transgenic and knockout mice. Cell Death Differ. 2006, 13, 861–872. [Google Scholar] [CrossRef]
- Klement, J.F.; Rice, N.R.; Car, B.D.; Abbondanzo, S.J.; Powers, G.D.; Bhatt, P.H.; Chen, C.H.; Rosen, C.A.; Stewart, C.L. Ikappabalpha deficiency results in a sustained nf-kappab response and severe widespread dermatitis in mice. Mol. Cell. Biol. 1996, 16, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.E.; Phillips, R.J.; Erdjument-Bromage, H.; Tempst, P.; Ghosh, S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell 1995, 80, 573–582. [Google Scholar] [CrossRef]
- Griffin, B.D.; Moynagh, P.N. In vivo binding of nf-kappab to the ikappabbeta promoter is insufficient for transcriptional activation. Biochem. J. 2006, 400, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, S.T.; Epinat, J.C.; Rice, N.R.; Israel, A. I kappa b epsilon, a novel member of the I kappa B family, controls rela and crel nf-kappa b activity. EMBO J. 1997, 16, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Nabel, G.J. A new member of the i kappab protein family, i kappab epsilon, inhibits rela (p65)-mediated nf-kappab transcription. Mol. Cell. Biol. 1997, 17, 6184–6190. [Google Scholar] [CrossRef]
- Memet, S.; Laouini, D.; Epinat, J.C.; Whiteside, S.T.; Goudeau, B.; Philpott, D.; Kayal, S.; Sansonetti, P.J.; Berche, P.; Kanellopoulos, J.; et al. Ikappabepsilon-deficient mice: Reduction of one T cell precursor subspecies and enhanced ig isotype switching and cytokine synthesis. J. Immunol. 1999, 163, 5994–6005. [Google Scholar]
- Fernandez, G.; Zaikos, T.D.; Khan, S.Z.; Jacobi, A.M.; Behlke, M.A.; Zeichner, S.L. Targeting ikappab proteins for HIV latency activation: The role of individual ikappab and nf-kappab proteins. J. Virol. 2013, 87, 3966–3978. [Google Scholar] [CrossRef]
- Folks, T.M.; Justement, J.; Kinter, A.; Dinarello, C.A.; Fauci, A.S. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 1987, 238, 800–802. [Google Scholar] [CrossRef]
- Perez, V.L.; Rowe, T.; Justement, J.S.; Butera, S.T.; June, C.H.; Folks, T.M. An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J. Immunol. 1991, 147, 3145–3148. [Google Scholar]
- Clouse, K.A.; Powell, D.; Washington, I.; Poli, G.; Strebel, K.; Farrar, W.; Barstad, P.; Kovacs, J.; Fauci, A.S.; Folks, T.M. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J. Immunol. 1989, 142, 431–438. [Google Scholar]
- Folks, T.M.; Clouse, K.A.; Justement, J.; Rabson, A.; Duh, E.; Kehrl, J.H.; Fauci, A.S. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc. Natl. Acad. Sci. USA 1989, 86, 2365–2368. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, G.; Zaikos, T.D.; Khan, S.Z.; Jacobi, A.; Behlke, M.; Zeichner, S.L. Targeting iκb proteins for hiv latency activation: The role of individual iκb and nf-κb proteins. J. Virol. 2019, in press. [Google Scholar]
- Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. Introduction to nf-kappab: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Emiliani, S.; Fischle, W.; Ott, M.; Van Lint, C.; Amella, C.A.; Verdin, E. Mutations in the tat gene are responsible for human immunodeficiency virus type 1 postintegration latency in the u1 cell line. J. Virol. 1998, 72, 1666–1670. [Google Scholar] [PubMed]
- Emiliani, S.; Van Lint, C.; Fischle, W.; Paras, P., Jr.; Ott, M.; Brady, J.; Verdin, E. A point mutation in the hiv-1 tat responsive element is associated with postintegration latency. Proc. Natl. Acad. Sci. USA 1996, 93, 6377–6381. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Gerondakis, S. The c-rel transcription factor in development and disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef]
- Schwartz, C.; Bouchat, S.; Marban, C.; Gautier, V.; Van Lint, C.; Rohr, O.; Le Douce, V. On the way to find a cure: Purging latent HIV-1 reservoirs. Biochem. Pharmacol. 2017, 146, 10–22. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.Z.; Gasperino, S.; Zeichner, S.L. Nuclear Transit and HIV LTR Binding of NF-κB Subunits Held by IκB Proteins: Implications for HIV-1 Activation. Viruses 2019, 11, 1162. https://doi.org/10.3390/v11121162
Khan SZ, Gasperino S, Zeichner SL. Nuclear Transit and HIV LTR Binding of NF-κB Subunits Held by IκB Proteins: Implications for HIV-1 Activation. Viruses. 2019; 11(12):1162. https://doi.org/10.3390/v11121162
Chicago/Turabian StyleKhan, Sohrab Z., Sofia Gasperino, and Steven L. Zeichner. 2019. "Nuclear Transit and HIV LTR Binding of NF-κB Subunits Held by IκB Proteins: Implications for HIV-1 Activation" Viruses 11, no. 12: 1162. https://doi.org/10.3390/v11121162
APA StyleKhan, S. Z., Gasperino, S., & Zeichner, S. L. (2019). Nuclear Transit and HIV LTR Binding of NF-κB Subunits Held by IκB Proteins: Implications for HIV-1 Activation. Viruses, 11(12), 1162. https://doi.org/10.3390/v11121162