1. Introduction
HIV-1 spreads between T cells primarily through two modes of transmission: the release of cell-free virus particles followed by their uptake by (more or less distantly located) cells expressing the viral receptor/co-receptor, and the cell-to-cell transmission of particles to an adjacent cell via the virological synapse (VS), i.e., when infected and uninfected cells transiently align. The formation of the HIV-1 VS is initiated by the viral envelope glycoprotein (Env) on the surface of productively infected cells binding to its receptor, CD4, on target T cells [
1] and is followed by the polarization of Gag at the cell–cell contact site [
1,
2]. Virus particles are then released in high concentrations towards the target cell [
3], facilitating efficient infection while also possibly shielding virus particles from some neutralizing antibodies ([
4] and recently reviewed in [
5]). Indeed, as demonstrated in a recent study using physiologically relevant cell culture systems [
6], it is possible that virus that is not released in close proximity to a target cell is rapidly inactivated, emphasizing the importance of VS-mediated transmission. However, given that Env is fusogenic at neutral pH, it would seem likely at first that VS-mediated contacts should frequently result in cell–cell fusion, thus forming a multinucleated infected cell (syncytium). While we now know that small, T cell-based syncytia arise early in HIV-1 infection and can spread the virus by cell–cell contact [
7,
8,
9,
10,
11,
12], the majority of infected T cells observed in lymphoid tissue are mononucleated, documenting that most HIV-1 VSs ultimately result in complete cell separation and the generation of a new, productively infected cell. This is likely due to tight regulation at the VS that acts to prevent excessive syncytium formation (reviewed in [
13,
14]).
Multiple independent studies have identified viral and host functions which, together, prevent excessive HIV-1-induced cell–cell fusion at the VS. Firstly, Env is rapidly downregulated from the surface of infected cells in the absence of Gag [
15,
16]. Secondly, upon Gag multimerization at the plasma membrane, Env is trapped by immature Gag through Env’s cytoplasmic tail and maintained in a poorly fusogenic state [
17]. This trapping by Gag ends only after Env’s incorporation into virus particles when Gag precursor gets cleaved, i.e., upon maturation [
18,
19,
20,
21]. The residual fusion activity of Gag-trapped Env on infected cells has been shown to be inhibited by several host membrane proteins that accumulate at the producer cell side of the VS, including tetraspanins and phosphorylated ezrin (p-ezrin) [
22,
23,
24]. Tetraspanins inhibit HIV-1-induced cell–cell fusion at a post-hemifusion stage [
23], while ezrin is implicated in F-actin organization and the recruitment of the tetraspanin CD81 to the VS [
24]. It remains unclear how and whether these protein functions are coordinated, though based on other cell–cell fusion regulation paradigms (discussed below), additional host proteins are likely required to mediate the efficient inhibition of HIV-1-induced fusion by tetraspanins and ezrin.
EWI-F (CD9P-1/FPRP) is an immunoglobulin superfamily (IgSF) member and partner of tetraspanins CD9 and CD81 [
25]. EWI-F was shown to be a potent inhibitor of cell–cell fusion in myoblasts, where EWI-F knockout resulted in more frequent fusion than CD9/CD81 double knockout [
26]. However, EWI-F is poorly expressed in T cells [
27], the primary host cell type for HIV-1. A related protein, EWI-2 (IGSF8/PGRL) [
28,
29], which also associates with tetraspanins and is expressed in T cells [
25,
27], has been documented to play a role in hepatitis C virus (HCV) entry [
30,
31] and T cell immunological synapse (IS) formation [
32]. The latter study also suggested that EWI-2 has a yet undetermined involvement in HIV-1 particle production [
32]. Furthermore, both EWI-F and EWI-2 interact with ezrin to organize the cytoskeleton in concert with tetraspanins [
27]. EWI-2 thus lies at the nexus of tetraspanins, ezrin, and the actin cytoskeleton (which can also inhibit cell–cell fusion) [
33].
2. Materials and Methods
2.1. Cell Lines and Cell Culture
The following cells were obtained through the NIH AIDS Reagent Program (Germantown, MD, USA), Division of AIDS, NIAID, NIH: HeLa cells from Dr. Richard Axel [
34], TZM-bl cells from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc. [
35,
36,
37,
38,
39], CEM.NKR CCR5+Luc+ (CEM-luc) cells from Dr. John Moore and Dr. Catherine Spenlehauer [
40,
41], CEM-T4 cells from Dr. J.P. Jacobs [
42], and CEM-SS cells from Dr. Peter L. Nara [
34,
43,
44].
HEK 293T, HeLa, and TZM-bl cells were maintained in Dulbecco’s Modification of Eagle’s Medium (DMEM) (Corning, Corning, NY, USA, Cat. #10-017-CV) containing 10% fetal bovine serum (FBS; Corning, Corning, NY, USA, Cat. #35-010-CV) and antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin; Invitrogen, Carlsbad, CA, USA). CEM-luc cells were maintained in RPMI 1640 medium (Corning, Corning, NY, USA, Cat. #10-104-CV) supplemented with 10% FBS and 0.8 mg/mL geneticin sulfate (G418). CEM2n, a kind gift from R. Harris [
45], and CEM-SS cells were maintained in RPMI medium supplemented with 10% FBS and antibiotics.
Human primary blood mononuclear cells (PBMCs) were isolated as buffy coats from the whole blood of healthy donors by Ficoll density centrifugation. CD4+ T cells were enriched from PBMCs by negative selection using the MACS CD4+ T Cell Isolation Kit (Miltenyi Biotec, Auburn, CA, USA, Cat. #130-096-533) or the EasySep Human CD4+ T Cell Isolation Kit (STEMCELL Technologies, Vancouver, BC, Canada, Cat. #17952) according to manufacturer’s instructions. Primary CD4+ T cells were activated in RPMI containing 10% FBS, 50 units/mL IL-2, antibiotics, and 5 µg/mL phytohemagglutinin. After 48 h of activation, cells were washed and subsequently maintained and expanded in the same medium but without phytohemagglutinin. Cells were used for infections at 4–7 days post isolation.
2.2. Antibodies
Mouse monoclonal antibody (mAb) to EWI-2 (8A12) was a kind gift from Dr. Eric Rubinstein [
25]. Mouse mAb to HIV-1 p24 (AG3.0) was obtained through the NIH AIDS Reagent Program (Germantown, MD, USA), Division of AIDS, NIAID, NIH, from Dr. Jonathan Allan [
46]. Rabbit antiserum to HIV-1 p6 was a kind gift from David E. Ott. Rabbit polyclonal antibody (pAb) to HIV-1 p24 was obtained from Advanced Biotechnologies (Eldersburg, MD, USA, Cat. #13-203-000). Secondary antibodies were as follows: Alexa Fluor 488-conjugated donkey pAb to mouse IgG (#A21202), Alexa Fluor 488-conjugated donkey pAb to rabbit IgG (Cat. #A21206), Alexa Fluor 594-conjugated donkey pAb to mouse IgG (Cat. #R37115), Alexa Fluor 594-conjugated donkey pAb to rabbit IgG (Cat. #A21207), Alexa Fluor 647-conjugated donkey pAb to mouse IgG (Cat. #A31571), and Alexa Fluor 647-conjugated goat pAb to mouse IgG (Cat. #A21235), all from Invitrogen (Carlsbad, CA, USA). Zenon labeling of primary antibodies with either Alexa Fluor 488 or Alexa Fluor 594 was carried out using Zenon Labeling Kits according to the manufacturer’s instructions (Molecular Probes, Eugene, OR, USA, Cat. #Z25002 and #Z25007).
2.3. Plasmids and Virus Strains
Respectively, pcDNA3, pCDNA3.1, and pCMV SPORT6 (Invitrogen, Carlsbad, CA, USA) were vectors for EWI-2, CD81, and L6 overexpression (EWI-2 was a kind gift from Dr. Eric Rubinstein; Université Paris-Sud, Villejuif, France). Proviral plasmids pNL4-3 and pNL4-3 ΔEnv (KFS) were kind gifts from Dr. Eric Freed (National Cancer Institute, Frederick, MD, USA) [
47]. NL4-3-derived fluorescent protein-tagged proviral plasmids pNL-sfGI, pNL-sfGI ΔEnv, pNL-CI, and pNL-CI ΔEnv [
10] were kind gifts from Dr. Benjamin Chen (Mount Sinai School of Medicine, New York, NY, USA). Vesicular stomatitis virus glycoprotein (VSV-G) was used to pseudotype viral stocks produced in HEK 293T cells. The lentiviral vector FG12 [
48], previously modified to include a puromycin resistance cassette [
24], was further modified to remove the GFP reporter cassette by digestion with AfeI and PshAI and subsequent blunt-end relegation.
2.4. Virus Stocks and Infections
VSV-G-pseudotyped virus stocks of NL4-3, NL4-3 ∆Env, NL-sfGI, NL-CI, and NL-CI ∆Env were produced in HEK 293T cells transfected with the proviral plasmid and pVSV-G (at 17:3 ratio) using calcium phosphate precipitation. For shRNA encoding lentiviruses, shEWI-2 and shScramble, stocks were produced in HEK 293T cells transfected with FG12-shRNA vector, ΔR8.2 packaging vector, and pVSV-G (at a ratio of 3:7:1. Supernatants were harvested 2 days after transfection, cleared by centrifugation at 2000 rcf for 10 min, filtered, and stored at −80 °C.
To infect CEM2n cells by spinoculation, two million cells were incubated with RPMI/10% FBS containing 90 µL of virus stock (resulting in ~3% of the cells being infected) or medium alone (for uninfected controls), for 20 min at 37 °C, followed by centrifugation at 1200 rcf for 2 h at 37 °C. Cell pellets were allowed to recover at 37 °C for 15 min, centrifuged at 300 rcf for 2 min, and resuspended in fresh RPMI/10% FBS. Cells were incubated at 37 °C, the medium was refreshed 2 days post infection, and the cells were used 1 day later for all subsequent experiments.
To infect primary CD4+ T cells, 1 or 2 million cells were incubated in RPMI/10% FBS/IL-2 containing 200 or 400 µL of virus, respectively, and spinoculated as described above. Cells were resuspended in fresh RPMI/10% FBS/PS/IL-2 and incubated at 37 °C/5% CO2. Cells were used 2-3 days post infection for all subsequent experiments.
To infect CEM-SS cells by shaking, one or two million cells suspended in CO2-independent medium (Gibco, Grand Island, NY, USA, Cat #18045088) supplemented with 10% FBS were mixed with VSV-G-pseudotyped virus stocks and shaken at 220 rpm for 2 h at 37 °C. Cells were then washed and plated in fresh RPMI/10% FBS, and used for experiments as described. For CEM-SS infection by spinoculation, the procedure was performed as described above with some modifications; one or two million cells were incubated in RPMI/10% FBS containing 40–50 µL (analyzing surface expression and post-synapse enrichment, respectively) of virus stock or medium alone (for uninfected controls). Following spinoculation, cells were incubated at 37 °C for 2 days before being used for subsequent experiments.
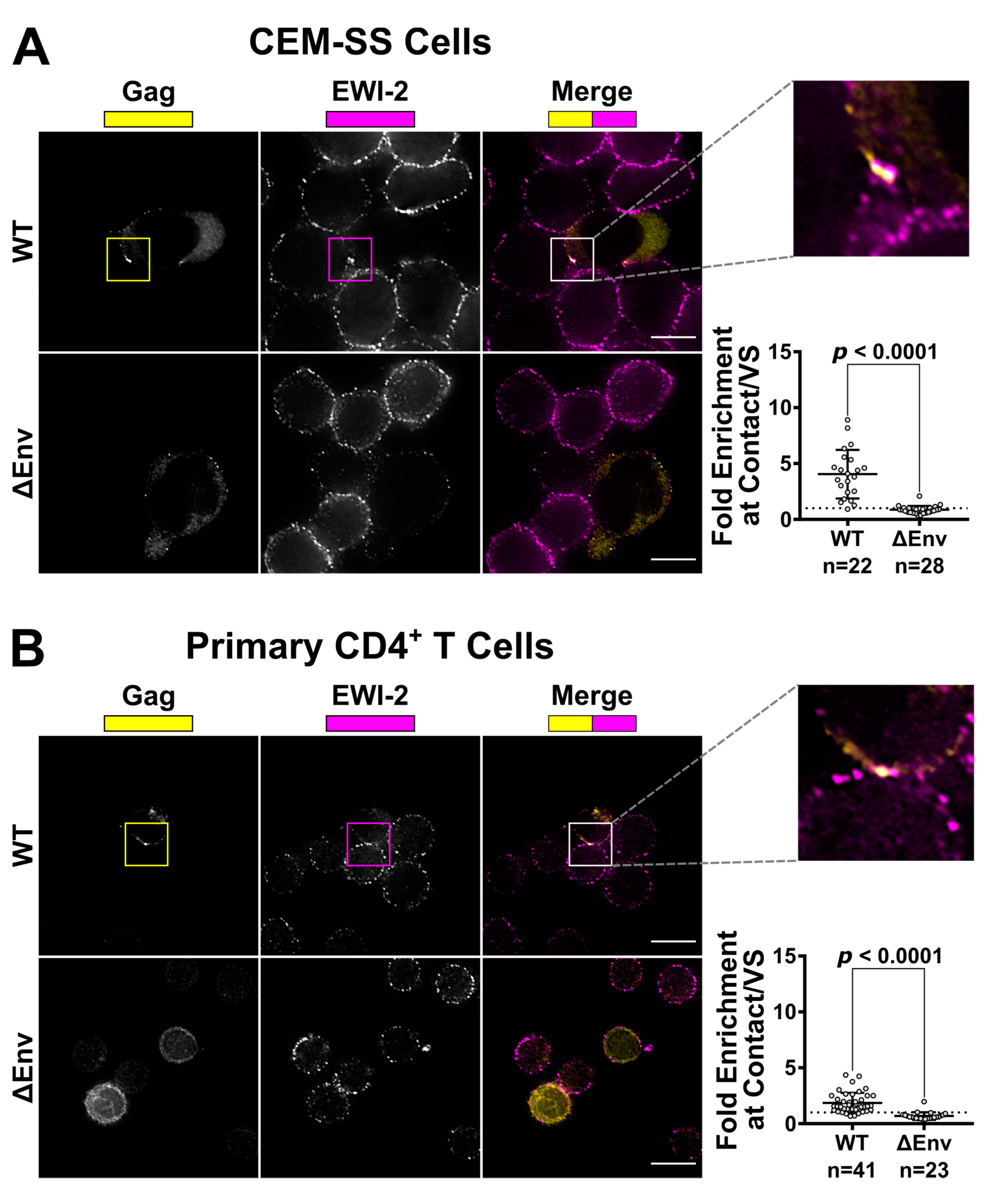
2.5. Imaging and Quantification of EWI-2 Accumulation at the VS
CEM-SS and primary CD4+ T cells were infected by shaking or spinoculation, respectively, with VSV-G-pseudotyped WT or ΔEnv virus then treated as follows: For CEM-SS cells, two days post infection, uninfected CEM-SS target cells were labeled with CMAC (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions, mixed with infected cells at a 1:1 or 1:2 ratio (infected:target), seeded onto the microwell of a 35 mm glass-bottom dish (MatTek Corporation, Ashland, MA, USA, Cat. #P35G-1.5-14-C) coated with poly-L-Lysine (Trevigen, Gaithersburg, MD, USA), and incubated at 37° C for 3 to 4.5 h. Cells were then chilled on ice and surface-labeled with 1:200 mouse anti-EWI-2 mAb in RPMI/10% FBS for 45 min at 4 °C. Surface-labeled cells were fixed with 4% PFA in PBS at 4 °C for 10 min, and blocked and permeabilized overnight with 1% BSA and 0.2% Triton X-100 in PBS (block/perm buffer). All CEM-SS conditions were labeled with Alexa Fluor 647-conjugated anti-mouse secondary pAb in block/perm buffer at 1:500 dilution. Cells were subsequently stained with Alexa Fluor 594 Zenon-labeled anti-p24 AG3.0 mouse mAb and fixed again with 4% PFA in PBS. Cells were kept in PBS for imaging.
For primary cells, uninfected cells were mixed with infected cells at a 1:1 ratio (infected:target), seeded onto 8-well glass-bottom plates (CellVis, Mountain View, CA, USA, Cat. #C8-1.5H-N) coated with 1:10 poly-L-Lysine in double-distilled water (ddH2O), and incubated for 2 to 2.5 h at 37 °C. Cells were surface-labeled for EWI-2 and fixed as above, then blocked and permeabilized with block/perm buffer for 10 min. Cells were then labeled with a mixture of rabbit anti-p24 and anti-p6 antibodies, each at 1:1000 dilution, in PBS with 1% BSA (block) for 45 min. Subsequently, cells were labeled with Alexa Fluor-conjugated secondary pAbs as indicated. Cells were kept in PBS for imaging.
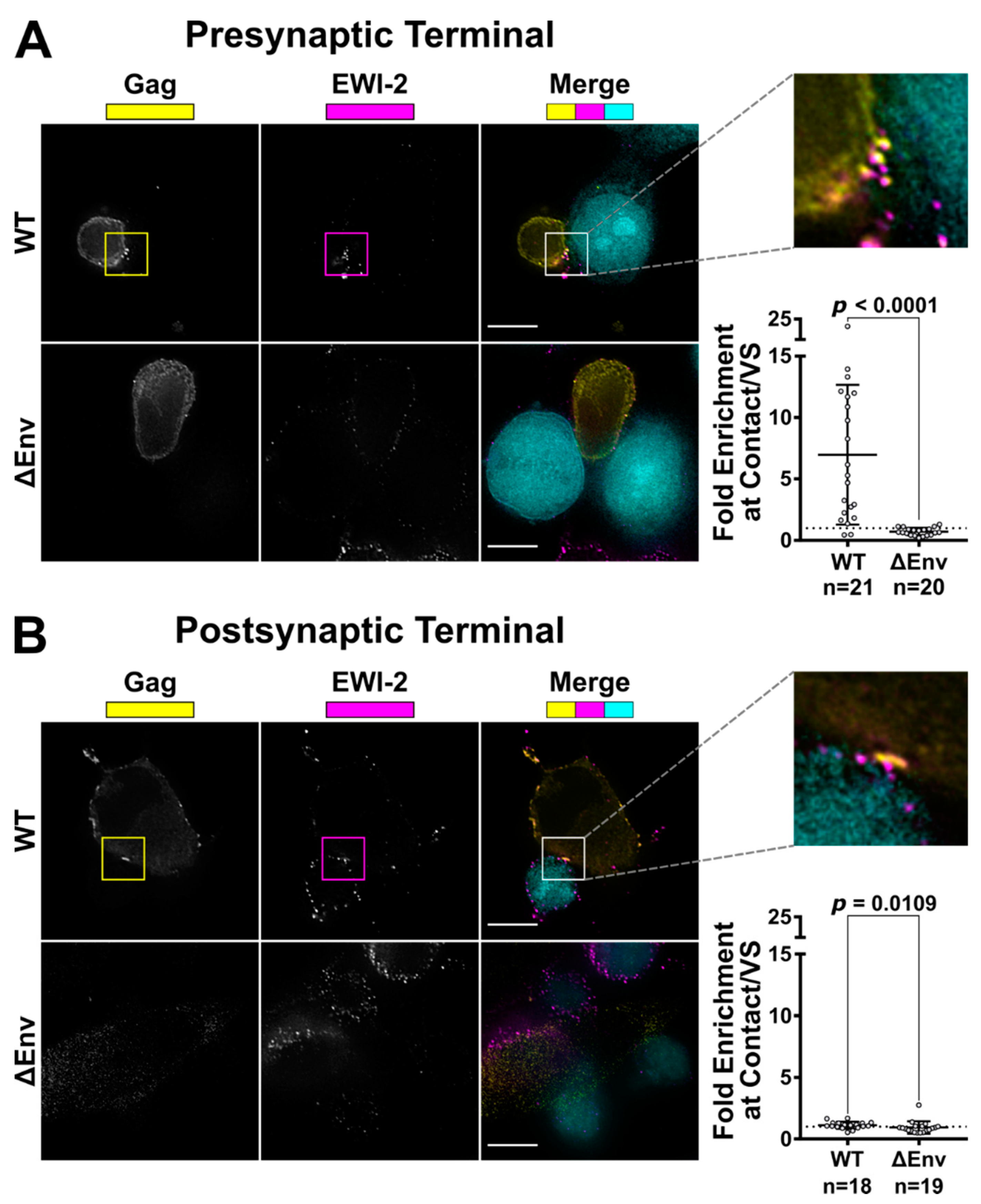
To visualize the only producer cell-associated EWI-2 at the VS, 10,000 target TZM-bl cells (which have nearly-undetectable levels of EWI-2) were seeded onto 8-well glass-bottom plates coated with 1:10 poly-L-Lysine in ddH2O. The next day, those TZM-bl cells were labeled with CMAC at 1:250 dilution in serum-free DMEM, and then co-cultured with 150,000 CEM-SS cells (either uninfected or infected with NL-CI or NL-CI ∆Env 2 days prior as described above) per well for 2.5 h at 37 °C in RPMI/10% FBS. The cells were then surface-labeled with 1:200 mouse anti-EWI-2 mAb in RPMI/10% FBS on ice for 45 min. Cells were subsequently fixed with 4% PFA in PBS and permeabilized with block/perm for 10 min. After permeabilization, the cells were labeled using a mixture of rabbit anti-p24 and anti-p6 antibodies, each at 1:1000 dilution, in block for 45 min. Cells were subsequently labeled using Alexa Fluor-conjugated secondary pAbs (anti-mouse-Alexa Fluor 647 and anti-rabbit-Alexa Fluor 488) each at 1:500 in block for 45 min. Cells were kept in PBS for imaging.
To visualize only target cell-associated EWI-2 at the VS, HeLa producer cells (which have nearly-undetectable levels of EWI-2) were plated (10,000 cells per well) in 8-well glass-bottom plates coated with 1:10 poly-L-Lysine in ddH2O. Twenty-four hours later, cells were transfected with NL-sfGI, NL-sfGI ∆Env, or empty vector, using FuGENE6 transfection reagent at a ratio of 3:1 (FuGENE6:DNA) according to manufacturer’s instructions (Promega, Madison, WI, USA, Cat. #E2691). Twenty-four hours post-transfection, 100,000–150,000 uninfected CEM-SS cells (labeled with CMAC at a 1:250 dilution in serum-free RPMI) were added to form VSs with provirus-transfected HeLa cells. After 2-2.5 h of coculture, cells were surface-labeled with 1:200 mouse anti-EWI-2 mAb in RPMI/10% FBS for 45 min at 4 °C. Surface-labeled cells were fixed with 4% PFA in PBS at 4 °C for 10 min, and then incubated with block/perm for 10 min, before labeling with a mixture of rabbit anti-p24 and anti-p6 antibodies, each at 1:1000 dilution, in block for 45 min. Subsequently, cells were labeled with secondary pAbs (anti-mouse-Alexa Fluor 647 and anti-rabbit-Alexa Fluor 594), each at 1:500 in block. Cells were kept in PBS for imaging.
Images were acquired on a DeltaVision epifluorescence microscope (GE/Applied Precision, Issaquah, WA, USA) with an Olympus IX-70 base using an Olympus 60× PlanApo 1.42 NA objective and equipped with a CoolSNAP HQ CCD camera (Photometrics, Tucson, AZ, USA). Images were imported into Fiji Version 2.0.0-rc-69/1.52p [
49] for analysis following deconvolution and cropping using Softworx software (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). The VS was identified using the Gag channel and the level of EWI-2 accumulation was determined by measuring its signal intensity at the VS. For ΔEnv controls, cell–cell contacts were identified using the differential interference contrast (DIC) channel and treated analogous to a VS. The EWI-2-associated signal intensity at non-contact sites was determined by manually outlining the surface of the cell, excluding any regions that were in contact with an adjacent cell, and calculating the mean EWI-2 intensity within the selected area. To determine the level of enrichment at the VS (or cell–cell contact for ΔEnv controls), an “unbiased” approach was applied to account for the EWI-2 signal contributed by both the target and producer cell at each VS/contact. Enrichment was calculated as the EWI-2 signal intensity at the VS/contact divided by the sum of the EWI-2 signal at non-contact sites of the producer and target cell in that particular VS/contact.
2.6. Proteomic Analysis of EWI-2 Levels in HIV-1 Infected Cells
To identify HIV-1-dependent changes in the abundance of total EWI-2, we re-analysed data from two previous studies [
50,
51]. In brief, primary human CD4
+ T cells were infected with pNL4-3-∆Env-Nef-P2A-SBP-∆LNGFR (HIV-AFMACS) at MOI≤0.5, enriched by antibody-free magnetic cell sorting (AFMACS) [
52] and analysed 48 h after infection [
51]. CEM-T4 T cells were infected with pNL4-3-∆Env-EGFP at MOI=1.5 and analysed 48 h after infection [
50]. TMT-labeled tryptic peptides from whole cell lysates were subjected to off-line high pH reversed-phase (HpRP)-HPLC fractionation and analysed using an Orbitrap Fusion Tribrid mass spectrometer (ThermoFisher Scientific, Waltham, MA, USA) coupled to a Dionex UltiMate 3000 UHPLC (Thermo Scientific, Waltham, MA, USA). Details of sample processing and data analysis have been previously described [
50,
51] and proteomic data from primary human CD4
+ T cells are available from the ProteomeX-change Consortium using dataset identifier PXD012263 (
http://proteomecentral.proteomexchange.org).
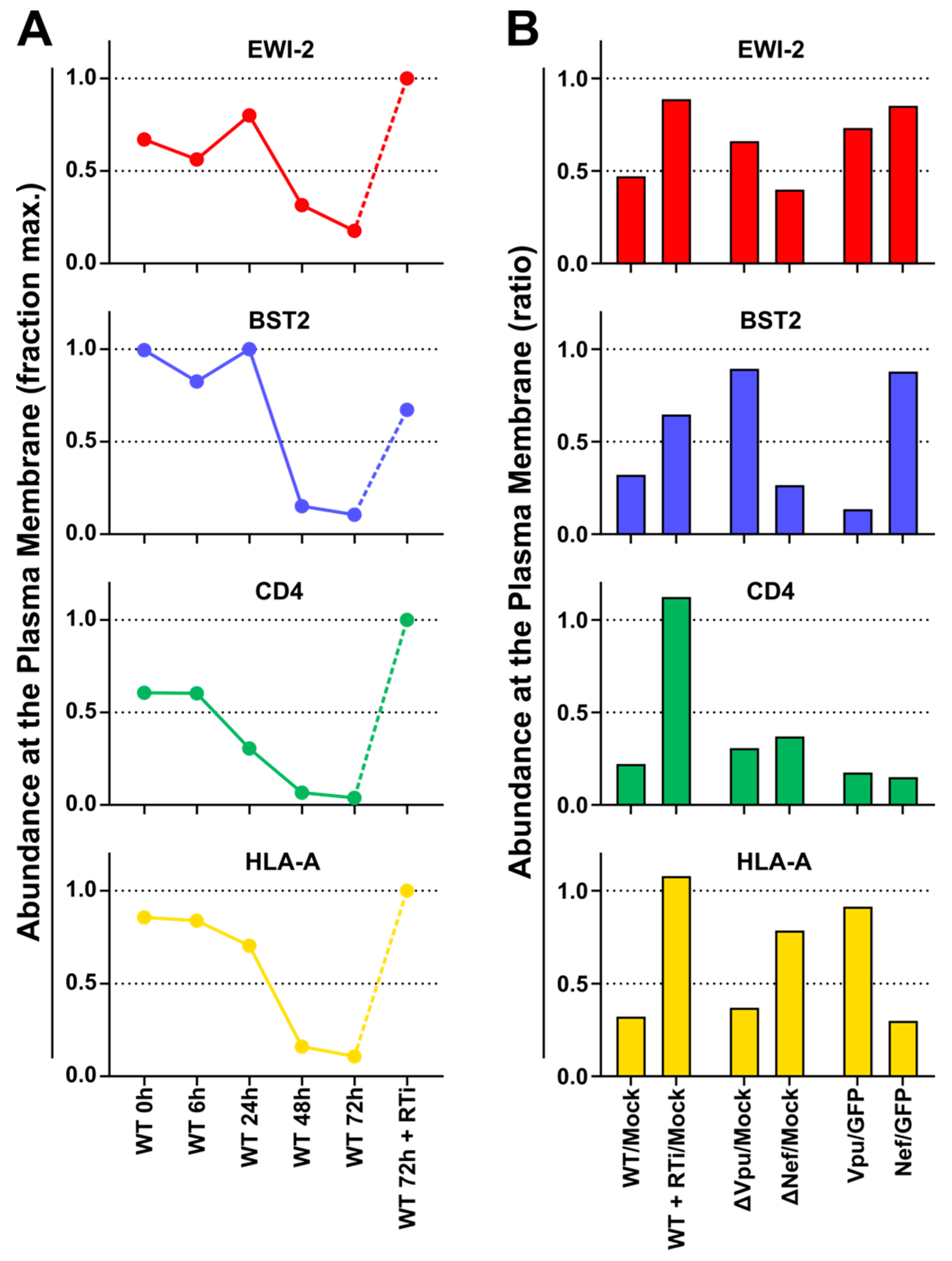
To characterise HIV-1-dependent changes in the abundance of plasma membrane EWI-2, we re-analysed data from a previous study [
53]. In brief, for the TMT-based time course experiment, CEM-T4 T cells were infected with pNL4-3-∆Env-EGFP at MOI=10 and analysed at the indicated time points after infection. For the SILAC-based single time point experiments, cells were pre-labeled with light, medium or heavy lysine and arginine and either infected with WT or Vpu-/Nef-deficient pNL4-3-∆Env-EGFP at MOI=10 and analysed 72 h after infection, or transduced with GFP or Vpu/Nef and selected with puromycin. Sialylated cell surface glycoproteins were enriched by selective aminooxy-biotinylation followed by immunoaffinity purification using streptavidin-conjugated beads (plasma membrane profiling). Tryptic peptides were labeled with TMT reagents (time course experiment only), subjected to off-line High pH Reversed-Phase (HpRP)-HPLC fractionation and analysed using an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific, Waltham, MA, USA) coupled to a Dionex UltiMate 3000 UHPLC (Thermo Scientific, Waltham, MA, USA). Details of sample processing and data analysis have been previously described [
53] and time course proteomic data are available from the ProteomeX-change Consortium using dataset identifier PXD002934 (
http://proteomecentral.proteomexchange.org).
2.7. Determining Surface Levels of EWI-2 by Microscopy
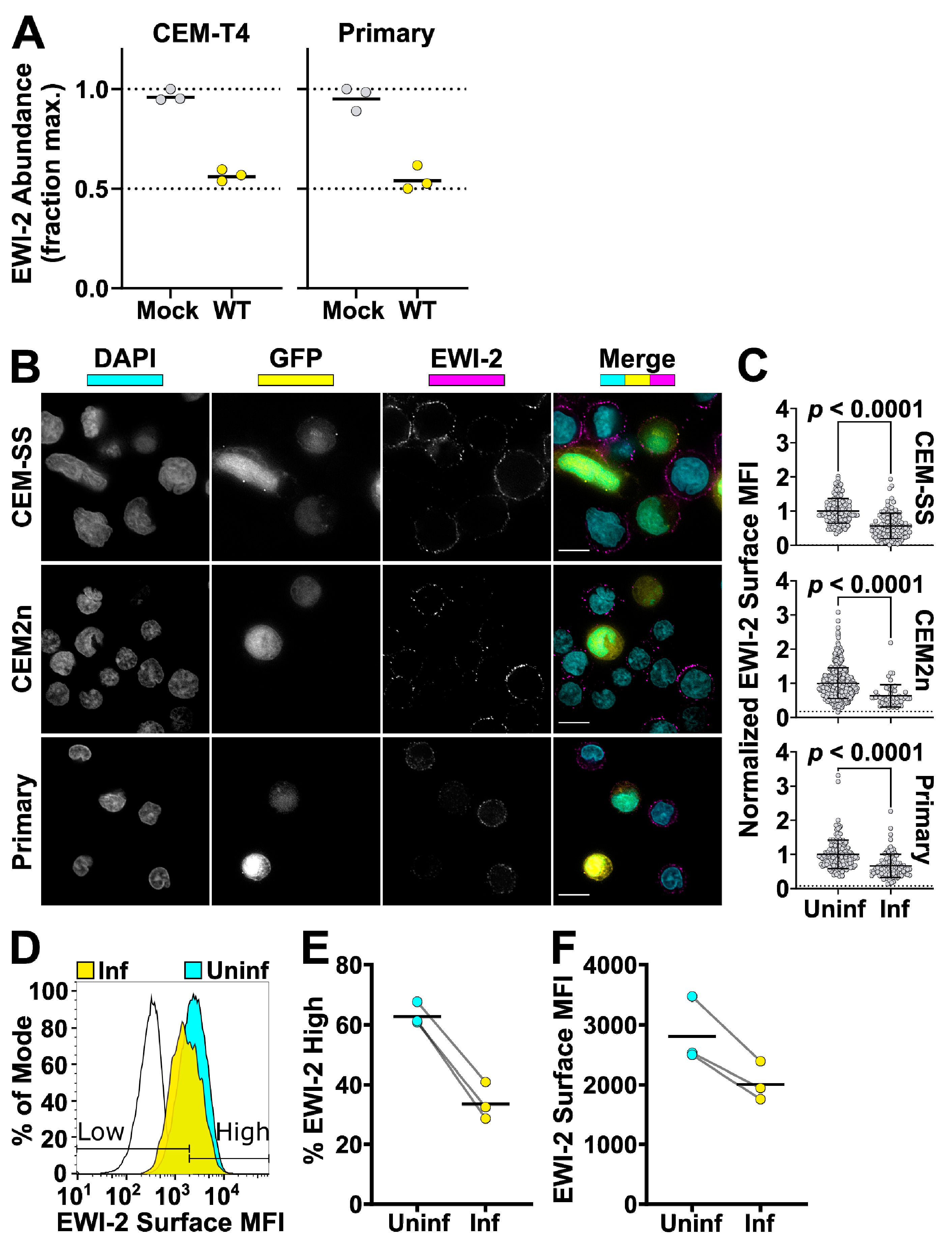
To compare EWI-2 surface expression between infected and uninfected cells, CEM-SS, CEM2n cells, and primary CD4+ T cells were infected with VSV-G-pseudotyped NL-sfGI as described above. Two to three days post infection, 3 × 105 infected cells were plated onto each well of 8-well glass-bottom plates coated with 1:10 poly-L-Lysine in ddH2O. Two additional wells were used for uninfected controls. After 2 h of incubation at 37 °C, the medium was replaced with ice cold RPMI/10% FBS containing mouse anti-EWI-2 mAb at 1:200 dilution for surface labeling, and incubated for 45 min at 4 °C. Following the primary antibody incubation, cells were washed with RPMI/10% FBS and fixed with 4% PFA in PBS for 10 min at 4 °C, blocked and permeabilized with PBS containing 1% BSA and 100 µg/mL digitonin for 10 min, and incubated with the indicated secondary antibody in block for 45 min at room temperature. Cells were washed with block and imaged in PBS. At least 50 fields containing infected cells were selected for each biological replicate and imaged, deconvolved, and cropped using the DeltaVision microscope and Softworx software described above. After deconvolution, Fiji was used to manually select the cell surface at the midline of each cell and the mean intensity of EWI-2-associated signal was quantified and subsequently subtracted by the mean intensity of an area that did not contain cells. Cell–cell contact sites were excluded from the quantification. Background subtracted intensity values of all cells were normalized to the average surface associated intensity of the entire uninfected cell population, internal controls contained in the same wells as infected cells, contained within respective biological replicates. This normalization allowed for the direct comparison of surface expression trends between biological replicates that accounts for potential variation in protein labeling efficiency between replicates. The virus-associated fluorescent reporter channel was used to segregate measurements into uninfected and infected. The data shown in Figure 3B are pooled from 2–3 independent biological replicates, each consisting of two technical replicates, all of which were sampled randomly until a minimum of 50 infected cells were quantified.
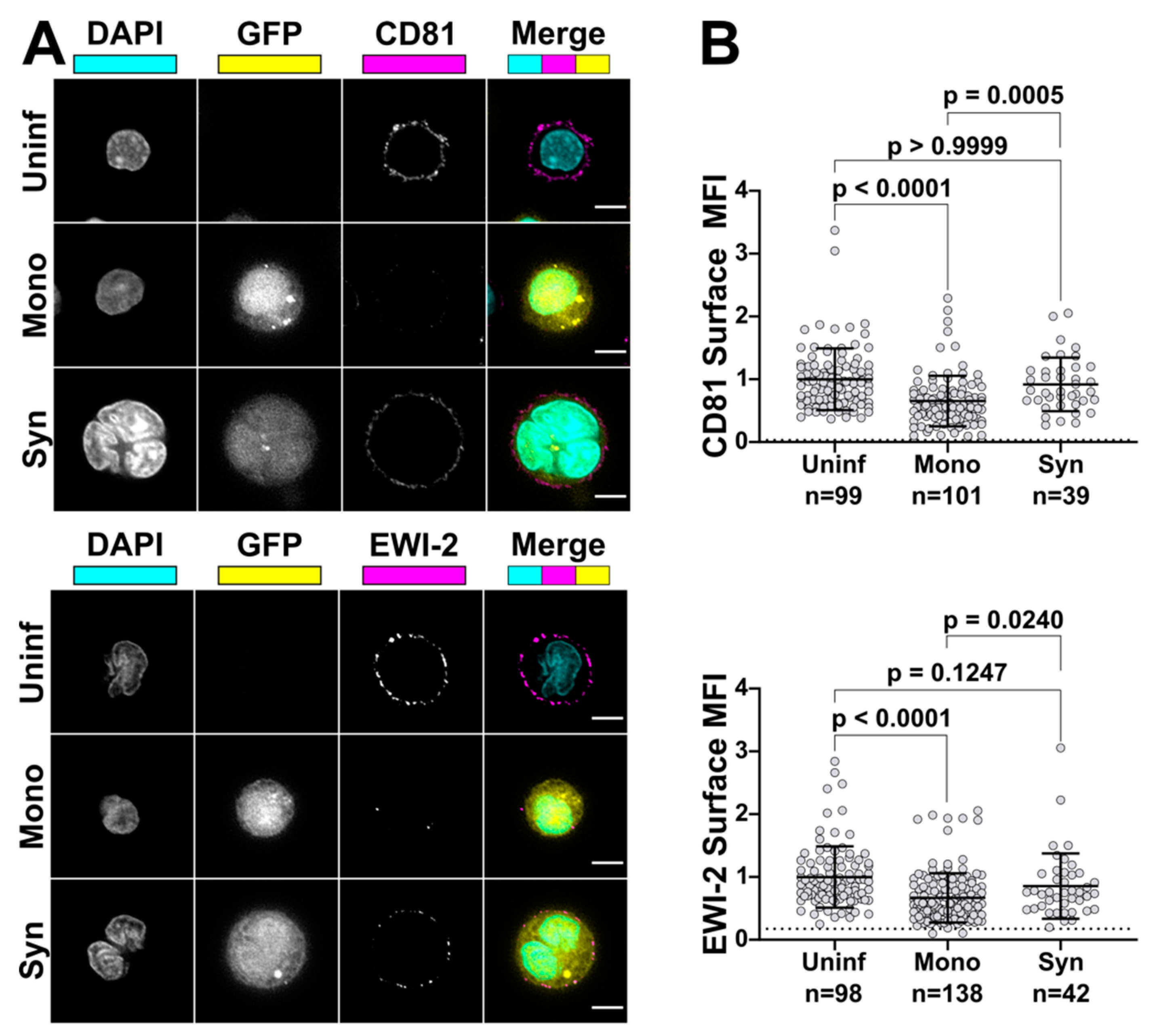
To compare EWI-2 surface expression levels between mononucleated infected cells and HIV-1-induced syncytia, primary CD4+ T cells were infected with VSV-G-pseudotyped virus as described above. Three days post infection, 3 × 105 infected cells were plated onto each well of 8-well glass-bottom plates coated with 1:10 poly-L-Lysine in ddH2O alongside two wells of uninfected cells as controls. Cells were incubated at 37 °C for 2 h and surface labeled as described above using either mouse anti-EWI-2 or mouse anti-CD81 mAb at 1:200 or 1:100, respectively. Samples were fixed, permeabilized, and labeled with appropriate AlexaFluor conjugated antibodies and DAPI as described above. Cells were imaged in PBS and at least 50 fields containing 10–20 cells each and containing at least some infected cells with multinucleated appearance (determined by DAPI and GFP signal) were selected for each biological replicate and imaged, deconvolved, and cropped as described above. Fiji was then used to analyze the surface expression of each protein of interest as described above. The virus-associated fluorescent reporter channel (GFP) was used to segregate measurements into infected and uninfected populations, and nuclear staining (DAPI) was used to further segregate infected cells into mononucleated and multinucleated infected cells. The EWI-2/CD81 channel was not viewed at all during imaging and field selection, or throughout image processing. The data shown in Figure 6 are pooled from 2–3 biological replicates, with two technical replicates each, all of which were sampled randomly until a minimum of 15 syncytia per biological replicate were quantified.
2.8. Determining Surface EWI-2 Signal on Infected Cells by Flow Cytometry
CEM2n cells infected as described above were harvested after three days and incubated in cold PBS with 5 mM EDTA for 15 min (3.0 × 105 cells/tube). Cells were pelleted at 400 rcf for 7 min at 4 °C and resuspended in cold RMPI/10% FBS containing mouse anti-EWI-2 mAb at 1:200 dilution. After a 45 min incubation at 4 °C, cells were washed with cold RPMI/10% FBS and resuspended in ice cold PBS with 5 mM EDTA. To fix, an equal volume of PBS with 8% PFA was added and samples were incubated on ice for 10 min. Cells were washed and stained with Alexa Fluor 594-conjugated secondary antibody at 1:500 in block for 45 min at room temperature, before being washed, resuspended in PBS, and analyzed using a BD LSRII flow cytometer. Data were analyzed using FlowJo V10 (Becton, Dickinson & Company, Franklin Lakes, NJ, USA). Samples were gated for infected and uninfected populations by GFP expression. EWI-2high and EWI-2low gates were set based in part on controls lacking primary antibody, and in part by adjusting the gates to reflect the number of uninfected EWI-2high cells as measured by microscopy. The data shown are the collection of three independent biological replicates, each consisting of two technical replicates.
2.9. HeLa-based HIV-1-Induced Cell–Cell Fusion Assay
First, 50,000 HeLa cells were plated in each well of a 24-well plate and, the next day, transfected (using FuGENE6; see
Section 2.5) in duplicate with 100 ng of either pNL-sfGI or pNL-sfGI ΔEnv along with 500 ng total expression vector carrying CD81 or EWI-2. L6, a tetraspanin-like protein that does not inhibit cell–cell fusion, was co-transfected instead of CD81 or EWI-2 as a positive control for maximum fusion activity, For dose response assays, 125, 250, or 500 ng of either EWI-2 or CD81 plasmid was ”stuffed” with L6 expression plasmid to maintain 500 ng of total protein expression plasmid in each condition. No cytotoxicity was observed upon transfection for any of the experimental conditions. Then, 24 h post-transfection, producer HeLa cells were co-cultured with 10
6 TZM-bl target cells (which, upon producer-target cell fusion, express firefly luciferase under control of the HIV-1 LTR) per well for 3 h before unattached target cells were washed off and the medium was refreshed. 14-18 h later, cells were lysed for at least 30 min on ice using 1% Triton X-100, 2 mM EDTA, 50 mM Tris-HCl, 200 mM NaCl, with 1% protease inhibitor cocktail. Lysates were precleared by centrifugation at 20,000 rcf for 5 min at 4 °C and stored at –80 °C until use for luciferase activity assays. Note that the timepoints used here ensure that there is not enough time for the development of any luciferase signal resulting from productive infection of target TZM-bl cells through virus transmission and that only cell–cell fusion contributes to the luciferase activity measured.
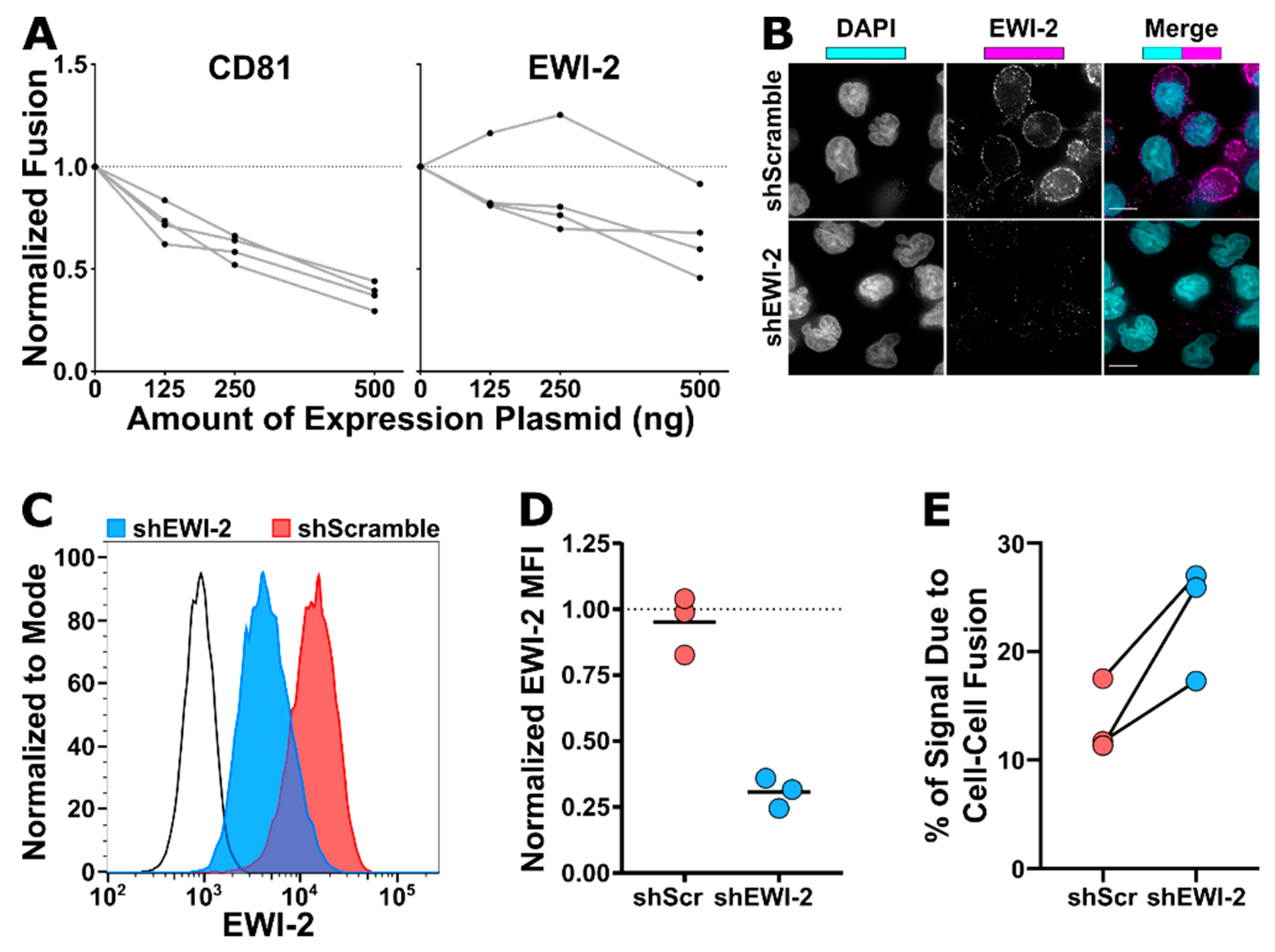
Each lysate was incubated with an equal volume of firefly luciferase reagent (Promega, Madison, WI, USA, Cat. #E1500) for 1 min in a 96-well white-walled plate (ThermoFisher Scientific, Waltham, MA, USA, Cat. #7571) before collecting luminescence signal intensity on a microplate reader (BioTek Synergy 2, BioTek, Winooski, VT, USA). Background luminescence was determined using a lysis buffer blank and subtracted from all experimental samples. Luminescence intensity was used as a quantitative measurement of relative HeLa-TZM syncytium formation against the non-fusogenic (therefore incapable of forming syncytia) ΔEnv control by dividing each value by the Δenv value (which effectively corresponds to any leaky expression of luciferase in TZM-bl cells as no cell–cell fusion occurs at all in this condition). To then determine relative fusion activity of cells transfected with EWI-2 and CD81, those values were normalized to the L6 condition. Normalized fusion is therefore the fold difference of cell–cell fusion activity taking place when cells were co-transfected with the indicated amount of either CD81 or EWI-2 plasmid, compared to the activity taking place when cells were co-transfected with L6. The data shown are the collection of four independent biological replicates.
2.10. Establishment of EWI-2 Knockdown CEM-SS Cells
The shRNA-encoding sequences targeting either EWI-2 (modified from previously described EWI-2-targeting siRNA [
27] or a scrambled control, were introduced to the lentiviral vector FG12 (as described in 2.3) using oligos containing shRNA sequences, a loop sequence, and an AgeI site, flanked by BbsI and XhoI restriction site overhangs, as previously described [
24], (EWI-2 sense, 5′- ACCGGGGCTTCGAAAACGGTGATCTTCAAGAGAGATCACCGTTTTCGAAGCCCTTTTTTACCGGTC-3′, and anti-sense, 5′- TCGAGACCGGTAAAAAAGGGCTTCGAAAACGGTGATCTCTCTTGAAGATCACCGTTTTCGAAGCCC-3′; scramble sense, 5′- ACCGGGCAGATGCGTCCAGTTAGATTCAAGAGATCTAACTGGACGCATCTGCCTTTTTTACCGGTC-3′, and anti-sense, 5′- TCGAGACCGGTAAAAAAGGCAGATGCGTCCAGTTAGATCTCTTGAATCTAACTGGACGCATCTGCC-3′). A PolII promoter was first obtained by ligating the oligo with PBS-hU6 digested with BbsI and XhoI restriction endonucleases (New England BioLabs, Ipswich, MA, USA). The PolII-shRNA constructs were obtained by digesting the resulting PBS-hU6 vector with XbaI and XhoI, and the insert was subsequently ligated into the FG12 vector digested with the same enzymes.
VSV-G pseudotyped FG12-shRNA lentiviruses were used to transduce CEM-SS cells by spinoculating one million cells with 500 µL of lentiviral supernatant (either shEWI-2 or shScramble). Cells were incubated at 37 °C for 2 days in RPMI/10% FBS and positively transduced cells were then selected for puromycin resistance by supplementing the media with 0.5 µg/mL of puromycin for 8 days. Subsequently, shEWI-2 and shScramble CEM-SS cells were maintained in RPMI/10% FBS/0.25 µg/mL puromycin.
EWI-2 knockdown was analyzed by flow cytometry and microscopy. For flow cytometry analysis, 3.0 × 10 5 shScramble and shEWI-2 cells, alongside parental CEM-SS controls, were pelleted at 400 rcf for 7 min, resuspended in 1:1000 Live/Dead Fixable Near-IR stain (Invitrogen, Carlsbad, CA, USA, Cat. #L10119) in PBS for 30–45 min, washed with RPMI/10% FBS and fixed for 10 min in 4% PFA in PBS by resuspending the cells in PBS and then adding an equal volume of 8% PFA in PBS. Fixed samples were washed with 1 mL of PBS, blocked and permeabilized in 100 µL of block/perm buffer for 10 min, and washed with PBS containing 1% BSA. EWI-2 was labeled using mAb 8A12 diluted 1:200 in block for 45 min, washed with block, and stained with Alexa Fluor 488-conjugated secondary antibody in block for 45 min. Cells were then washed and resuspended in PBS for flow cytometry analysis using a BD LSRII flow cytometer. Data were analyzed using FlowJo V10. Samples were gated for live cells, and EWI-2 expression was measured by the mean fluorescence intensity of EWI-2 signal in the live cell population and normalized to the parental control expression within each biological replicate. The data are the result of three independent biological replicates with two technical replicates each. For microscopy, 2.5 × 105 shScramble and shEWI-2 cells, alongside parental CEM-SS controls, were plated on 8-well glass bottom plates coated with 1:10 poly-L-lysine in ddH2O. After 2 h at 37 °C, cells were fixed for 10 min using 4% PFA in PBS, washed, and incubated with block/perm for 10 min. Cells were washed with block and incubated with 1:200 mAb 8A12 for 45 min, washed, and stained with 1:500 Alexa Fluor 647-conjugated secondary antibody and 1:2500 DAPI in block for 45 min. Cells were washed with block and imaged in PBS using a 60× objective as described above. Images were deconvolved and cropped by DeltaVision microscope and Softworx software described above and imported into Fiji for analysis.
2.11. CEM-luc-based HIV-1-Induced Cell–Cell Fusion Assay
Two million shScramble or shEWI-2 cells were spinoculated as described above with 1.7 or 2 µL of VSV-G pseudotyped NL4-3, alongside parental CEM-SS cells spinoculated with 25 µL of VSV-G pseudotyped NL4-3 ΔEnv to achieve an infection rate of ~30% for each condition. Cells were incubated at 37 °C for 2 days and then co-cultured with uninfected CEM-luc cells in RPMI/10% FBS containing the following drug treatments; 1:1000 DMSO for vehicle control, 1 µM Efavirenz (EFV) (NIH AIDS Reagent Program, Cat. #4624) to inhibit transmission, or 1 µM EFV with 0.5 µM HIV-1 IIIB C34 peptide (C34) (NIH AIDS Reagent Program, Cat. #9824) to inhibit both transmission and cell–cell fusion. After 24 h, the co-culture medium was refreshed, and all conditions were incubated at 37 °C in RPMI/10% FBS containing 1 µM EFV and 0.5 µM C34. 24 h later, cells were pelleted at 1000 rcf for 5 min at 4 °C and resuspended in luciferase reporter lysis buffer (Promega, Cat. #E4530) with 1% protease inhibitor cocktail (Millipore Sigma, Darmstadt, Germany, Cat. #P8340) to lyse on ice for 15 min. Lysates were cleared by centrifugation at 20,000 rcf for 5 min at 4 °C and stored at –80 °C until use for luciferase activity assays.
In parallel, infected cells were prepared for flow cytometry analysis alongside uninfected controls, to determine the infection rate across each condition at the start of the co culture with uninfected CEM-luc cells. Cells were pelleted and resuspended in 1:1000 Live/Dead Fixable Near-IR stain in PBS as described above, washed and resuspended in PBS. An equal volume of 8% PFA in PBS was added to fix the cells in a final concentration of 4% PFA in PBS for 10 min. Cells were washed and resuspended in block/perm, incubated for 10 min, washed with block, and resuspended for an overnight incubation in 1:100 AG3.0 in block. Cells were washed and stained with 1:500 Alexa Fluor 488-conjugated secondary antibody for 45 min followed by a wash with block. Cells were resuspended in PBS and analyzed by flow cytometry using a BD LSRII flow cytometer. Data was analyzed using FlowJo V10. Live cells were gated using the Live/Dead signal, and the percentage of infected cells in the live population was determined by gating on the AG3.0 associated signal.
Each lysate was incubated with an equal volume of firefly luciferase reagent for 1 min in a 96-well white-walled plate before collecting luminescence signal intensity on a microplate reader as described above (2.9). Background luminescence was determined using a lysis buffer blank and subtracted from all experimental samples. Relative luminescence units (RLUs) were normalized based on the infection level of each cell type determined by flow cytometry analysis, and the average RLU value from the ΔEnv infected, DMSO treated condition was subtracted from all conditions. All samples treated with both EFV and C34 had RLU values below that of the ΔEnv DMSO condition (data not shown), validating the efficacy of the inhibitors for complete inhibition of transmission to target CEM-luc cells. To determine the proportion of luciferase expression due to cell–cell fusion, the average RLU value from the EFV-treated condition (syncytium formation-dependent signal) was divided by that of the DMSO-treated (signal from both transmission and syncytium formation) and multiplied by 100. Data represent the percentage of luciferase signal due to syncytium formation between infected shScramble or shEWI-2 cells and uninfected CEM-luc cells from three independent biological replicates each consisting of 1–2 technical replicates.
2.12. Statistical Analysis
All statistical analyses were carried out in GraphPad Prism 8 (GraphPad Software, San Diego, CA) as indicated in Figure legends.
4. Discussion
The transient alignment of infected (producer) and uninfected (target) cells allows for efficient transmission of virus particles. However, because of the presence of viral Env and CD4/co-receptor at the surface of producer and target cell, respectively, rather than separating after particle transfer, these cells could also easily fuse with each other, thus forming a syncytium. This study now identifies EWI-2 as a host protein that contributes to the maintenance of viral homeostasis through fusion inhibition.
Our investigations were partially prompted by two recent reports. In one of those studies, Rubinstein and colleagues documented a role for EWI-F, a close relative of EWI-2, in myoblast fusion regulation [
26]. EWI-F was shown to act as fusion repressor in cooperation with the tetraspanins CD9 and CD81. With the other study, Yáñez-Mó and colleagues [
32] showed the presence of EWI-2 at sites of contact between uninfected T cells and T cells stably expressing HIV-1 Env. In separate experiments, HIV-1-infected EWI-2 knockdown cells were also shown to have somewhat increased virus production and the authors mentioned (as data not shown) that this was accompanied by augmented syncytium formation, indicating that EWI-2 could be involved in the regulation of HIV-1-induced membrane fusion. Importantly, however, the study did not address the question of whether the reported increase in syncytium formation was (potentially) caused by the action of EWI-2 in producer or target cells, nor did it provide a dissection of where EWI-2 accumulates (producer and/or target cells). The authors did speculate that EWI-2, together with α-actinin, might be active in target cells, there possibly contributing to α-actinin’s actin bundling activity, thus ultimately inhibiting virus entry/fusion. They also explicitly stated, however, that even if their speculation about where α-actinin acts during virus replication should eventually be confirmed (with subsequent studies), they cannot exclude an involvement of the partner protein EWI-2 in the “subsequent steps of the viral life cycle”. Our study now reveals that EWI-2 indeed acts during the late phase of the HIV-1 replication cycle: It accumulates on the producer cell side of the VS (
Figure 1 and
Figure 2). Surprisingly, unlike tetraspanins, which have fusion-inhibitory roles at both sides of the VS (and thus are present at both the viral pre- and post-synapse [
22,
62]), EWI-2 accumulates (and inhibits fusion) only at the presynaptic terminal of the VS. This leads us to speculate whether EWI-2 accumulation at the presynaptic terminal might contribute to unique intracellular signaling events in HIV-1-infected cells [
32,
63], such as tuning T cell receptor function.
Paralleling what we previously documented for tetraspanins [
22], we found that fusion with uninfected target cells was inhibited by EWI-2, and we established that it does so in a dose-dependent manner (
Figure 5). Also analogous to our findings about tetraspanins [
54,
56], we demonstrate that while EWI-2 accumulates at the virological presynapse, overall this protein is downregulated in infected cells (
Figure 3). Our proteomic analysis (
Figure 4) now shows that EWI-2 depletion from the infected cell surface, as is also the case for tetraspanins [
56,
57], is primarily mediated by Vpu (
Figure 4). Since EWI-2 is a known interactor of tetraspanins CD81 and CD9, it is possible that EWI-2 downregulation by Vpu (with or without Nef) is “direct” (e.g., the canonical Vpu “targets” BST2 and CD4, as well as SNAT1 [
53]) or “indirect,” possibly through its association with tetraspanins. Note, this is also true of CD81/other tetraspanins, which may likewise be “direct” or “indirect” targets (e.g., by their association with EWI-2). Our data do not distinguish these possibilities, and further mechanistic studies would be required to delineate the detailed mechanism of Vpu-mediated depletion. It should also be noted that in Table S1 of [
64], EWI-2 depletion in CEM-T4 cells is (somewhat) dependent on the expression of Vpr. The effect size is modest and likely “indirect,” and does not contradict the Vpu and Nef data shown here. It does, however, suggest that the mechanism of EWI-2 depletion in HIV-1 infected T cells may be complex.
Overall, the combination of these two features (enrichment during assembly and transmission at the VS, and regulation by HIV-1 accessory proteins in infected cells), together with the fusion-preventing functions, strongly suggests that a particular host factor plays an important role in virus replication.
We expect that EWI-2 also inhibits the fusion of virus particles to target cells, as tetraspanins do [
54,
56,
59], and we are currently testing that hypothesis (within the context of an extensive follow-up analysis aimed at dissecting the molecular determinants responsible for EWI-2′s fusion-inhibitory functions). It seems likely that tetraspanins and EWI-2 are not only tolerated but indeed enriched at virus budding sites because the benefit of cell–cell fusion inhibition at the VS is balanced against any negative effect of a reduction in virus infectivity. This is demonstrated by the fact that, in a native (unmanipulated) context, it is simultaneously true that (A) HIV-1-infected T cells routinely exhibit enrichment of these fusion inhibitors at virus release sites, (B) that cell–cell fusion is relatively infrequent, and (C) that HIV-1 spreads efficiently in those cell cultures.
As mentioned, while fusion inhibition operates at many levels and is orchestrated by HIV-1 proteins during infection, syncytia do nevertheless form, including
in vivo [
7,
8,
9] and when using a transmitted/founder (T/F) R5-tropic Env or even full-length replication-competent T/F virus [
10,
12]. However, these syncytia seem to remain small, at 4 or fewer nuclei and the vast majority having only two nuclei [
9]. Very large syncytia (dozens to thousands of nuclei) are only induced by HIV-1 infection of certain T cell lines, especially Sup-T1 cells [
65], or
in vivo but only with the involvement of macrophage or dendritic cells [
66,
67,
68]. It is therefore possible that T cell–T cell fusion is inhibited not only when a mononucleated infected cell encounters a target cell, but also when a syncytium encounters a target cell. An alternative explanation is that syncytia may be less viable as they grow larger, though some evidence contradicts that [
69]. Here, we present evidence that host fusion-inhibitory proteins EWI-2 and CD81 are present at higher levels on the surface of small T cell syncytia when compared to mononucleated infected cells in the same culture. Because we find that the fusion-inhibitory capacity of EWI-2 and CD81 is also dose-dependent, it would therefore be expected that a higher “dose” of EWI-2 and/or CD81 in syncytia would make them less likely to undergo cell–cell fusion a second (or third) time. We are currently formally testing this hypothesis, and also investigating the surface levels on syncytia of other host proteins normally downregulated upon HIV-1 infection. Without implicating any particular fusion-inhibitory protein, we have in the past found evidence that indeed fusion-inhibitory factors may also be acting at syncytium-target cell VSs [
9]: in Movie S7 of that report, we showed an example of a small syncytium containing two nuclei undergoing cell–cell fusion and acquiring a third nucleus. Subsequently, that syncytium encountered uninfected target cells and transferred virus particles to them through close contact but did not undergo further cell–cell fusion and instead fully separated from them despite exhibiting the ability to fuse only hours earlier. We can now speculate that, as a result of the cell–cell fusion event we captured at the beginning of that sequence, this syncytium likely acquired a dose of EWI-2 and/or CD81, which subsequently allowed the syncytium to mediate cell-to-cell virus transfer at the VS without further cell–cell fusion.
Finally, repressing HIV-1 Env-induced cell–cell fusion not only allows for a continued increase in the number of infected cells (as that number doubles each time producer and target cells separate after virus transmission), but keeping Env’s fusion activity at bay may also be beneficial for the virus for other reasons. For instance, we and others have recently shown that lowering Env’s fusion activity also allows HIV-1 to overcome a restriction factor (APOBEC3G; [
60]), and even antiviral drugs [
70]. Further, large syncytia, that could form if Env-induced cell–cell fusion is uncontrolled, are likely prone to be attacked by innate immune cells. It is therefore critical that HIV-1 recruits fusion-inhibitory host factors such as EWI-2 to the VS to prevent excess cell–cell fusion and keep T cell syncytia small when they do form.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





