Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
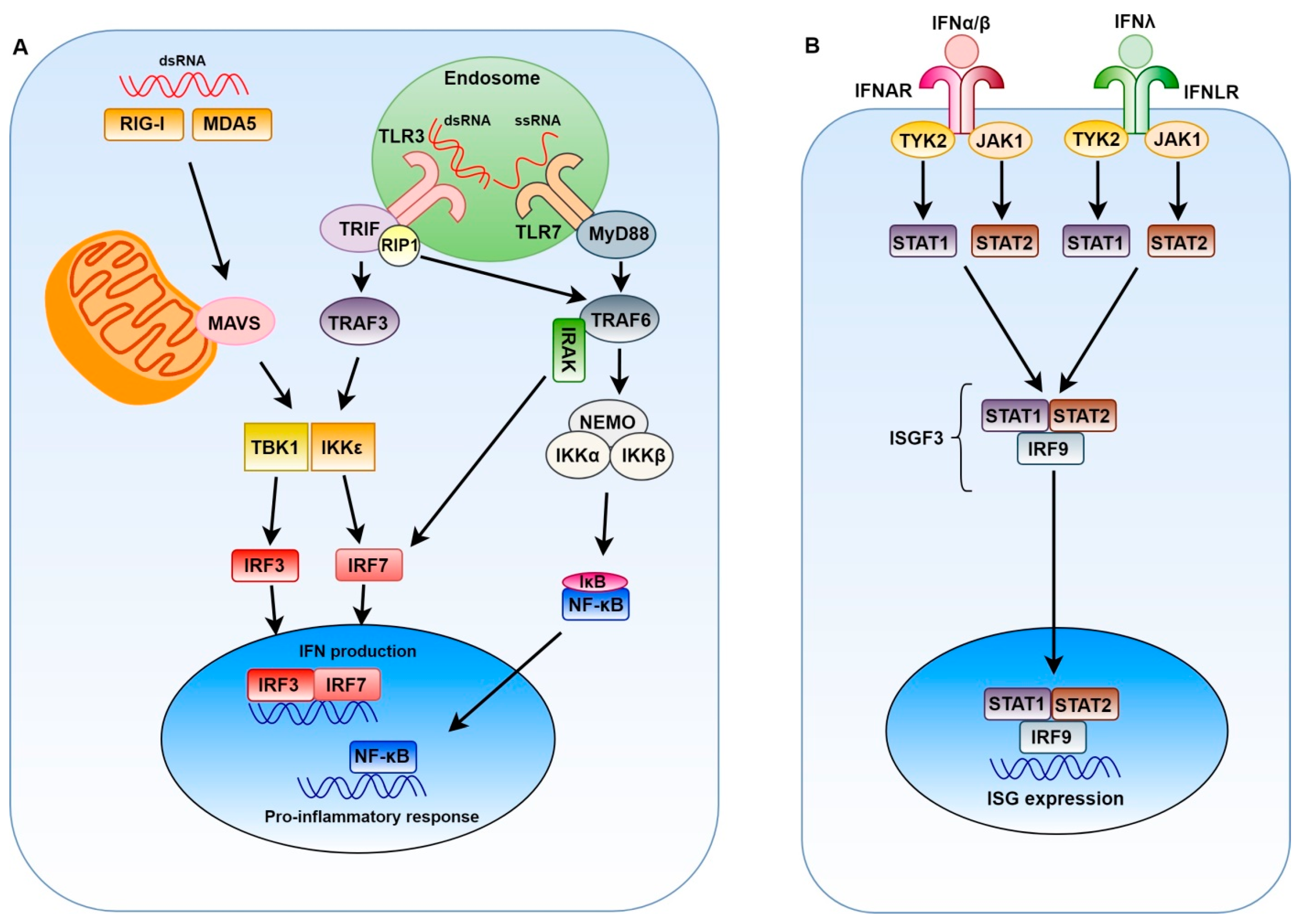
2. Innate Immune Response to Positive-Sense Single-Stranded RNA Viruses
3. Coronaviruses
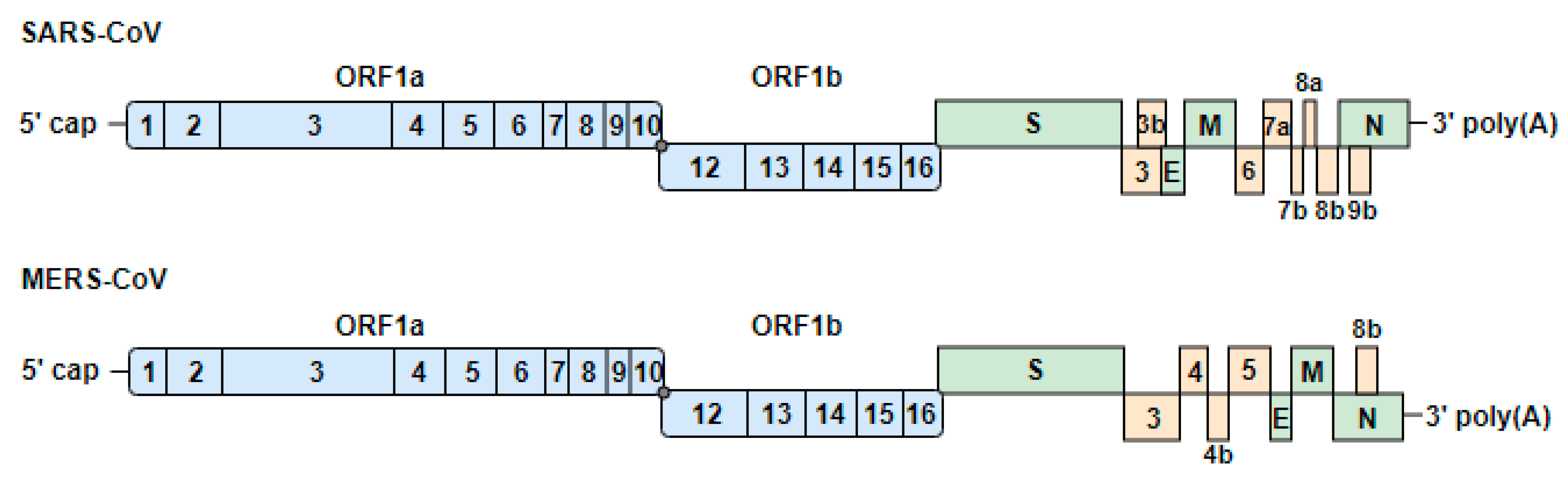
3.1. Genome Organization
3.2. The Role of Interferons in Coronavirus Pathogenesis
3.3. Innate Immune Evasion and Coronavirus Pathogenesis
4. Alphaviruses
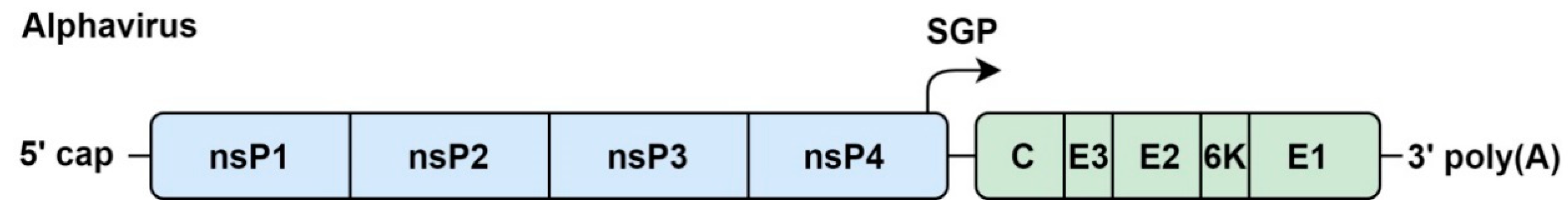
4.1. Genome Organization
4.2. The Role of Interferons in Alphavirus Pathogenesis
4.3. Innate Immune Evasion and Alphavirus Pathogenesis
5. Flaviviruses
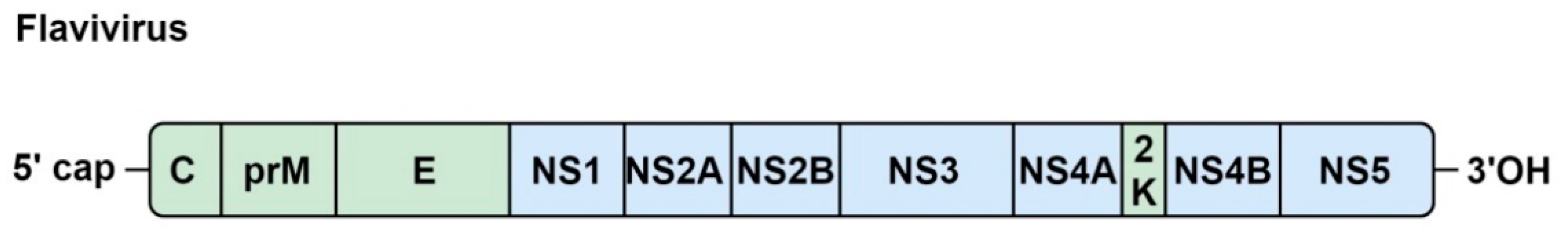
5.1. Genome Organization
5.2. The Role of Interferons in Flavivirus Pathogenesis
5.3. Innate Immune Evasion and Flavivirus Pathogenesis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brechot, C.; Bryant, J.; Endtz, H.; Garry, R.F.; Griffin, D.E.; Lewin, S.R.; Mercer, N.; Osterhaus, A.; Picot, V.; Vahlne, A.; et al. 2018 International Meeting of the Global Virus Network. Antiviral Res. 2019, 163, 140–148. [Google Scholar] [CrossRef]
- WHO. Middle East respiratory syndrome coronavirus (MERS-CoV). Available online: https://www.who.int/news-room/fact-sheets/detail/middle-east-respiratory-syndrome-coronavirus-(MERS-CoV) (accessed on 16 September 2019).
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Hilleman, M.R. Strategies and mechanisms for host and pathogen survival in acute and persistent viral infections. Proc. Natl. Acad. Sci. USA 2004, 101, 14560–14566. [Google Scholar] [CrossRef]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host. Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Harak, C.; Lohmann, V. Ultrastructure of the replication sites of positive-strand RNA viruses. Virology 2015, 479, 418–433. [Google Scholar] [CrossRef]
- Overby, A.K.; Popov, V.L.; Niedrig, M.; Weber, F. Tick-borne encephalitis virus delays interferon induction and hides its double-stranded RNA in intracellular membrane vesicles. J. Virol. 2010, 84, 8470–8483. [Google Scholar] [CrossRef]
- Chen, S.; Wu, Z.; Wang, M.; Cheng, A. Innate Immune Evasion Mediated by Flaviviridae Non-Structural Proteins. Viruses 2017, 9. [Google Scholar] [CrossRef]
- Uno, N.; Ross, T.M. Dengue virus and the host innate immune response. Emerg. Microbes Infect. 2018, 7, 167. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; Vazquez, C.; Horner, S.M. Hepatitis C Virus. Strategies to Evade Antiviral Responses. Future Virol. 2014, 9, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Kindler, E.; Thiel, V.; Weber, F. Interaction of SARS and MERS Coronaviruses with the Antiviral Interferon Response. Adv. Virus Res. 2016, 96, 219–243. [Google Scholar] [CrossRef] [PubMed]
- Pardy, R.D.; Valbon, S.F.; Richer, M.J. Running interference: Interplay between Zika virus and the host interferon response. Cytokine 2019, 119, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; Gong, M.J.; Zhao, F.R.; Shao, J.J.; Xie, Y.L.; Zhang, Y.G.; Chang, H.Y. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef]
- Onoguchi, K.; Yoneyama, M.; Takemura, A.; Akira, S.; Taniguchi, T.; Namiki, H.; Fujita, T. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 2007, 282, 7576–7581. [Google Scholar] [CrossRef]
- Egli, A.; Santer, D.M.; O’Shea, D.; Tyrrell, D.L.; Houghton, M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg. Microbes Infect. 2014, 3, e51. [Google Scholar] [CrossRef]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Durbin, J.E. Contribution of type III interferons to antiviral immunity: Location, location, location. J. Biol. Chem. 2017, 292, 7295–7303. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marié, I.J.; Durbin, J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 2011, 1, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.T.; Gale, M.; Loo, Y.M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef] [PubMed]
- Belgnaoui, S.M.; Paz, S.; Hiscott, J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 2011, 23, 564–572. [Google Scholar] [CrossRef]
- Uzé, G.; Monneron, D. IL-28 and IL-29: Newcomers to the interferon family. Biochimie 2007, 89, 729–734. [Google Scholar] [CrossRef]
- Jilg, N.; Lin, W.; Hong, J.; Schaefer, E.A.; Wolski, D.; Meixong, J.; Goto, K.; Brisac, C.; Chusri, P.; Fusco, D.N.; et al. Kinetic differences in the induction of interferon stimulated genes by interferon-α and interleukin 28B are altered by infection with hepatitis C virus. Hepatology 2014, 59, 1250–1261. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Invest. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- WHO. Issues consensus document on the epidemiology of SARS. Wkly. Epidemiol. Rec. 2003, 78, 373–375. [Google Scholar]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.; Guan, Y.; Rozanov, M.; Spaan, W.J.; Gorbalenya, A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- Liu, D.X.; Fung, T.S.; Chong, K.K.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res. 2014, 109, 97–109. [Google Scholar] [CrossRef]
- Haagmans, B.L.; Kuiken, T.; Martina, B.E.; Fouchier, R.A.; Rimmelzwaan, G.F.; van Amerongen, G.; van Riel, D.; de Jong, T.; Itamura, S.; Chan, K.H.; et al. Pegylated interferon-alpha protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat. Med. 2004, 10, 290–293. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef]
- Falzarano, D.; de Wit, E.; Rasmussen, A.L.; Feldmann, F.; Okumura, A.; Scott, D.P.; Brining, D.; Bushmaker, T.; Martellaro, C.; Baseler, L.; et al. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat. Med. 2013, 19, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Yao, Y.; Yeung, M.L.; Deng, W.; Bao, L.; Jia, L.; Li, F.; Xiao, C.; Gao, H.; Yu, P.; et al. Treatment With Lopinavir/Ritonavir or Interferon-β1b Improves Outcome of MERS-CoV Infection in a Nonhuman Primate Model of Common Marmoset. J. Infect. Dis. 2015, 212, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Kumaki, Y.; Ennis, J.; Rahbar, R.; Turner, J.D.; Wandersee, M.K.; Smith, A.J.; Bailey, K.W.; Vest, Z.G.; Madsen, J.R.; Li, J.K.; et al. Single-dose intranasal administration with mDEF201 (adenovirus vectored mouse interferon-α) confers protection from mortality in a lethal SARS-CoV BALB/c mouse model. Antiviral Res. 2011, 89, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Loutfy, M.R.; Blatt, L.M.; Siminovitch, K.A.; Ward, S.; Wolff, B.; Lho, H.; Pham, D.H.; Deif, H.; LaMere, E.A.; Chang, M.; et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: A preliminary study. JAMA 2003, 290, 3222–3228. [Google Scholar] [CrossRef] [PubMed]
- Omrani, A.S.; Saad, M.M.; Baig, K.; Bahloul, A.; Abdul-Matin, M.; Alaidaroos, A.Y.; Almakhlafi, G.A.; Albarrak, M.M.; Memish, Z.A.; Albarrak, A.M. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: A retrospective cohort study. Lancet Infect. Dis. 2014, 14, 1090–1095. [Google Scholar] [CrossRef]
- Shalhoub, S.; Farahat, F.; Al-Jiffri, A.; Simhairi, R.; Shamma, O.; Siddiqi, N.; Mushtaq, A. IFN-α2a or IFN-β1a in combination with ribavirin to treat Middle East respiratory syndrome coronavirus pneumonia: A retrospective study. J. Antimicrob. Chemother. 2015, 70, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Al-Tawfiq, J.A.; Momattin, H.; Dib, J.; Memish, Z.A. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: An observational study. Int. J. Infect. Dis. 2014, 20, 42–46. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Alothman, A.; Balkhy, H.H.; Al-Dawood, A.; AlJohani, S.; Al Harbi, S.; Kojan, S.; Al Jeraisy, M.; Deeb, A.M.; Assiri, A.M.; et al. Treatment of Middle East Respiratory Syndrome with a combination of lopinavir-ritonavir and interferon-β1b (MIRACLE trial): Study protocol for a randomized controlled trial. Trials 2018, 19, 81. [Google Scholar] [CrossRef]
- Smits, S.L.; de Lang, A.; van den Brand, J.M.; Leijten, L.M.; van IJcken, W.F.; Eijkemans, M.J.; van Amerongen, G.; Kuiken, T.; Andeweg, A.C.; Osterhaus, A.D.; et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010, 6, e1000756. [Google Scholar] [CrossRef]
- Yu, S.Y.; Hu, Y.W.; Liu, X.Y.; Xiong, W.; Zhou, Z.T.; Yuan, Z.H. Gene expression profiles in peripheral blood mononuclear cells of SARS patients. World J. Gastroenterol. 2005, 11, 5037–5043. [Google Scholar] [CrossRef]
- Reghunathan, R.; Jayapal, M.; Hsu, L.Y.; Chng, H.H.; Tai, D.; Leung, B.P.; Melendez, A.J. Expression profile of immune response genes in patients with Severe Acute Respiratory Syndrome. BMC Immunol. 2005, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Faure, E.; Poissy, J.; Goffard, A.; Fournier, C.; Kipnis, E.; Titecat, M.; Bortolotti, P.; Martinez, L.; Dubucquoi, S.; Dessein, R.; et al. Distinct immune response in two MERS-CoV-infected patients: Can we go from bench to bedside? PLoS ONE 2014, 9, e88716. [Google Scholar] [CrossRef] [PubMed]
- Hogan, R.J.; Gao, G.; Rowe, T.; Bell, P.; Flieder, D.; Paragas, J.; Kobinger, G.P.; Wivel, N.A.; Crystal, R.G.; Boyer, J.; et al. Resolution of primary severe acute respiratory syndrome-associated coronavirus infection requires Stat1. J. Virol. 2004, 78, 11416–11421. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.B.; Chen, J.; Morrison, T.E.; Whitmore, A.; Funkhouser, W.; Ward, J.M.; Lamirande, E.W.; Roberts, A.; Heise, M.; Subbarao, K.; et al. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog. 2010, 6, e1000849. [Google Scholar] [CrossRef] [PubMed]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G.; et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef] [PubMed]
- Mahlakõiv, T.; Ritz, D.; Mordstein, M.; DeDiego, M.L.; Enjuanes, L.; Müller, M.A.; Drosten, C.; Staeheli, P. Combined action of type I and type III interferon restricts initial replication of severe acute respiratory syndrome coronavirus in the lung but fails to inhibit systemic virus spread. J. Gen. Virol. 2012, 93, 2601–2605. [Google Scholar] [CrossRef]
- Totura, A.L.; Whitmore, A.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, e00638-15. [Google Scholar] [CrossRef]
- Sheahan, T.; Morrison, T.E.; Funkhouser, W.; Uematsu, S.; Akira, S.; Baric, R.S.; Heise, M.T. MyD88 is required for protection from lethal infection with a mouse-adapted SARS-CoV. PLoS Pathog. 2008, 4, e1000240. [Google Scholar] [CrossRef]
- Zhao, J.; Li, K.; Wohlford-Lenane, C.; Agnihothram, S.S.; Fett, C.; Gale, M.J.; Baric, R.S.; Enjuanes, L.; Gallagher, T.; McCray, P.B.; et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 4970–4975. [Google Scholar] [CrossRef]
- Nicholls, J.M.; Poon, L.L.; Lee, K.C.; Ng, W.F.; Lai, S.T.; Leung, C.Y.; Chu, C.M.; Hui, P.K.; Mak, K.L.; Lim, W.; et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet 2003, 361, 1773–1778. [Google Scholar] [CrossRef]
- Lo, A.W.; Tang, N.L.; To, K.F. How the SARS coronavirus causes disease: Host or organism? J. Pathol. 2006, 208, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Lam, C.W.; Wu, A.K.; Ip, W.K.; Lee, N.L.; Chan, I.H.; Lit, L.C.; Hui, D.S.; Chan, M.H.; Chung, S.S.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Chien, J.Y.; Hsueh, P.R.; Cheng, W.C.; Yu, C.J.; Yang, P.C. Temporal changes in cytokine/chemokine profiles and pulmonary involvement in severe acute respiratory syndrome. Respirology 2006, 11, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.J.; Su, I.J.; Theron, M.; Wu, Y.C.; Lai, S.K.; Liu, C.C.; Lei, H.Y. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 2005, 75, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.J.; Ran, L.; Xu, L.; Danesh, A.; Bermejo-Martin, J.F.; Cameron, C.M.; Muller, M.P.; Gold, W.L.; Richardson, S.E.; Poutanen, S.M.; et al. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 2007, 81, 8692–8706. [Google Scholar] [CrossRef] [PubMed]
- Min, C.K.; Cheon, S.; Ha, N.Y.; Sohn, K.M.; Kim, Y.; Aigerim, A.; Shin, H.M.; Choi, J.Y.; Inn, K.S.; Kim, J.H.; et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci. Rep. 2016, 6, 25359. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Choe, P.G.; Park, W.B.; Oh, H.S.; Kim, E.J.; Nam, E.Y.; Na, S.H.; Kim, M.; Song, K.H.; Bang, J.H.; et al. Clinical Progression and Cytokine Profiles of Middle East Respiratory Syndrome Coronavirus Infection. J. Korean Med. Sci. 2016, 31, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.; Deming, D.; Paddock, C.D.; Cheng, A.; Yount, B.; Vogel, L.; Herman, B.D.; Sheahan, T.; Heise, M.; Genrich, G.L.; et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007, 3, e5. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef]
- De Wilde, A.H.; Raj, V.S.; Oudshoorn, D.; Bestebroer, T.M.; van Nieuwkoop, S.; Limpens, R.W.; Posthuma, C.C.; van der Meer, Y.; Bárcena, M.; Haagmans, B.L.; et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-α treatment. J. Gen. Virol. 2013, 94, 1749–1760. [Google Scholar] [CrossRef]
- Zielecki, F.; Weber, M.; Eickmann, M.; Spiegelberg, L.; Zaki, A.M.; Matrosovich, M.; Becker, S.; Weber, F. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J. Virol. 2013, 87, 5300–5304. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef]
- Zhao, J.; Falcón, A.; Zhou, H.; Netland, J.; Enjuanes, L.; Pérez Breña, P.; Perlman, S. Severe acute respiratory syndrome coronavirus protein 6 is required for optimal replication. J. Virol. 2009, 83, 2368–2373. [Google Scholar] [CrossRef] [PubMed]
- Dediego, M.L.; Pewe, L.; Alvarez, E.; Rejas, M.T.; Perlman, S.; Enjuanes, L. Pathogenicity of severe acute respiratory coronavirus deletion mutants in hACE-2 transgenic mice. Virology 2008, 376, 379–389. [Google Scholar] [CrossRef]
- Yount, B.; Roberts, R.S.; Sims, A.C.; Deming, D.; Frieman, M.B.; Sparks, J.; Denison, M.R.; Davis, N.; Baric, R.S. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J. Virol. 2005, 79, 14909–14922. [Google Scholar] [CrossRef]
- Schaecher, S.R.; Stabenow, J.; Oberle, C.; Schriewer, J.; Buller, R.M.; Sagartz, J.E.; Pekosz, A. An immunosuppressed Syrian golden hamster model for SARS-CoV infection. Virology 2008, 380, 312–321. [Google Scholar] [CrossRef]
- Menachery, V.D.; Mitchell, H.D.; Cockrell, A.S.; Gralinski, L.E.; Yount, B.L.; Graham, R.L.; McAnarney, E.T.; Douglas, M.G.; Scobey, T.; Beall, A.; et al. MERS-CoV Accessory ORFs Play Key Role for Infection and Pathogenesis. MBio 2017, 8. [Google Scholar] [CrossRef]
- Cockrell, A.S.; Yount, B.L.; Scobey, T.; Jensen, K.; Douglas, M.; Beall, A.; Tang, X.C.; Marasco, W.A.; Heise, M.T.; Baric, R.S. A mouse model for MERS coronavirus-induced acute respiratory distress syndrome. Nat. Microbiol. 2016, 2, 16226. [Google Scholar] [CrossRef]
- Thornbrough, J.M.; Jha, B.K.; Yount, B.; Goldstein, S.A.; Li, Y.; Elliott, R.; Sims, A.C.; Baric, R.S.; Silverman, R.H.; Weiss, S.R. Middle East Respiratory Syndrome Coronavirus NS4b Protein Inhibits Host RNase L Activation. MBio 2016, 7, e00258. [Google Scholar] [CrossRef]
- Frieman, M.; Yount, B.; Heise, M.; Kopecky-Bromberg, S.A.; Palese, P.; Baric, R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007, 81, 9812–9824. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Channappanavar, R.; Jankevicius, G.; Fett, C.; Zhao, J.; Athmer, J.; Meyerholz, D.K.; Ahel, I.; Perlman, S. The Conserved Coronavirus Macrodomain Promotes Virulence and Suppresses the Innate Immune Response during Severe Acute Respiratory Syndrome Coronavirus Infection. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Mielech, A.M.; Chen, Y.; Mesecar, A.D.; Baker, S.C. Nidovirus papain-like proteases: Multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 2014, 194, 184–190. [Google Scholar] [CrossRef]
- Knaap, R.C.M.; Fernández-Delgado, R.; Dalebout, T.J.; Oreshkova, N.; Bredenbeek, P.J.; Enjuanes, L.; Sola, I.; Snijder, E.J.; Kikkert, M. The deubiquitinating activity of Middle East respiratory syndrome coronavirus papain-like protease delays the innate immune response and enhances virulence in a mouse model. bioRxiv 2019. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L.; Josset, L.; Gralinski, L.E.; Scobey, T.; Agnihothram, S.; Katze, M.G.; Baric, R.S. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2’-o-methyltransferase activity. J. Virol. 2014, 88, 4251–4264. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Gralinski, L.E.; Mitchell, H.D.; Dinnon, K.H.; Leist, S.R.; Yount, B.L.; Graham, R.L.; McAnarney, E.T.; Stratton, K.G.; Cockrell, A.S.; et al. Middle East Respiratory Syndrome Coronavirus Nonstructural Protein 16 Is Necessary for Interferon Resistance and Viral Pathogenesis. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Morales, L.; Oliveros, J.C.; Fernandez-Delgado, R.; tenOever, B.R.; Enjuanes, L.; Sola, I. SARS-CoV-Encoded Small RNAs Contribute to Infection-Associated Lung Pathology. Cell Host Microbe 2017, 21, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.V.D.; Trinta, K.S. Chikungunya virus: Clinical aspects and treatment–A Review. Mem. Inst. Oswaldo Cruz 2017, 112, 523–531. [Google Scholar] [CrossRef]
- Galán-Huerta, K.A.; Rivas-Estilla, A.M.; Fernández-Salas, I.; Farfan-Ale, J.A.; Ramos-Jiménez, J. Chikungunya virus: A general overview. Medicina Universitaria 2015, 17, 175–183. [Google Scholar] [CrossRef]
- Leung, J.Y.; Ng, M.M.; Chu, J.J. Replication of alphaviruses: A review on the entry process of alphaviruses into cells. Adv. Virol. 2011, 2011, 249640. [Google Scholar] [CrossRef]
- Schilte, C.; Couderc, T.; Chretien, F.; Sourisseau, M.; Gangneux, N.; Guivel-Benhassine, F.; Kraxner, A.; Tschopp, J.; Higgs, S.; Michault, A.; et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 2010, 207, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.; Anraku, I.; Le, T.T.; Larcher, T.; Major, L.; Roques, P.; Schroder, W.A.; Higgs, S.; Suhrbier, A. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 2010, 84, 8021–8032. [Google Scholar] [CrossRef] [PubMed]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Invest. 2010, 120, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Her, Z.; Ong, E.K.; Chen, J.M.; Dimatatac, F.; Kwek, D.J.; Barkham, T.; Yang, H.; Rénia, L.; Leo, Y.S.; et al. Persistent arthralgia induced by Chikungunya virus infection is associated with interleukin-6 and granulocyte macrophage colony-stimulating factor. J. Infect. Dis. 2011, 203, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Chow, A.; Sun, Y.J.; Kwek, D.J.; Lim, P.L.; Dimatatac, F.; Ng, L.C.; Ooi, E.E.; Choo, K.H.; Her, Z.; et al. IL-1beta, IL-6, and RANTES as biomarkers of Chikungunya severity. PLoS ONE 2009, 4, e4261. [Google Scholar] [CrossRef]
- Teng, T.S.; Kam, Y.W.; Lee, B.; Hapuarachchi, H.C.; Wimal, A.; Ng, L.C.; Ng, L.F. A Systematic Meta-analysis of Immune Signatures in Patients with Acute Chikungunya Virus Infection. J. Infect. Dis. 2015, 211, 1925–1935. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, I.; Vomaske, J.; Totonchy, T.; Kreklywich, C.N.; Haberthur, K.; Springgay, L.; Brien, J.D.; Diamond, M.S.; Defilippis, V.R.; Streblow, D.N. Chikungunya virus infection results in higher and persistent viral replication in aged rhesus macaques due to defects in anti-viral immunity. PLoS Negl. Trop. Dis. 2013, 7, e2343. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.P. Immunopathology of Chikungunya Virus Infection: Lessons Learned from Patients and Animal Models. Annu. Rev. Virol. 2017, 4, 413–427. [Google Scholar] [CrossRef]
- Her, Z.; Teng, T.S.; Tan, J.J.; Teo, T.H.; Kam, Y.W.; Lum, F.M.; Lee, W.W.; Gabriel, C.; Melchiotti, R.; Andiappan, A.K.; et al. Loss of TLR3 aggravates CHIKV replication and pathology due to an altered virus-specific neutralizing antibody response. EMBO Mol. Med. 2015, 7, 24–41. [Google Scholar] [CrossRef]
- Rudd, P.A.; Wilson, J.; Gardner, J.; Larcher, T.; Babarit, C.; Le, T.T.; Anraku, I.; Kumagai, Y.; Loo, Y.M.; Gale, M.; et al. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. J. Virol. 2012, 86, 9888–9898. [Google Scholar] [CrossRef]
- Gardner, C.L.; Burke, C.W.; Higgs, S.T.; Klimstra, W.B.; Ryman, K.D. Interferon-alpha/beta deficiency greatly exacerbates arthritogenic disease in mice infected with wild-type chikungunya virus but not with the cell culture-adapted live-attenuated 181/25 vaccine candidate. Virology 2012, 425, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Couderc, T.; Chrétien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.S.; Foo, S.S.; Simamarta, D.; Lum, F.M.; Teo, T.H.; Lulla, A.; Yeo, N.K.; Koh, E.G.; Chow, A.; Leo, Y.S.; et al. Viperin restricts chikungunya virus replication and pathology. J. Clin. Invest. 2012, 122, 4447–4460. [Google Scholar] [CrossRef]
- Poddar, S.; Hyde, J.L.; Gorman, M.J.; Farzan, M.; Diamond, M.S. The Interferon-Stimulated Gene IFITM3 Restricts Infection and Pathogenesis of Arthritogenic and Encephalitic Alphaviruses. J. Virol. 2016, 90, 8780–8794. [Google Scholar] [CrossRef] [PubMed]
- Clavarino, G.; Cláudio, N.; Couderc, T.; Dalet, A.; Judith, D.; Camosseto, V.; Schmidt, E.K.; Wenger, T.; Lecuit, M.; Gatti, E.; et al. Induction of GADD34 is necessary for dsRNA-dependent interferon-β production and participates in the control of Chikungunya virus infection. PLoS Pathog. 2012, 8, e1002708. [Google Scholar] [CrossRef]
- Werneke, S.W.; Schilte, C.; Rohatgi, A.; Monte, K.J.; Michault, A.; Arenzana-Seisdedos, F.; Vanlandingham, D.L.; Higgs, S.; Fontanet, A.; Albert, M.L.; et al. ISG15 is critical in the control of Chikungunya virus infection independent of UbE1L mediated conjugation. PLoS Pathog. 2011, 7, e1002322. [Google Scholar] [CrossRef]
- Hawman, D.W.; Stoermer, K.A.; Montgomery, S.A.; Pal, P.; Oko, L.; Diamond, M.S.; Morrison, T.E. Chronic joint disease caused by persistent Chikungunya virus infection is controlled by the adaptive immune response. J. Virol. 2013, 87, 13878–13888. [Google Scholar] [CrossRef]
- Fros, J.J.; Pijlman, G.P. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 2016, 8. [Google Scholar] [CrossRef]
- White, L.K.; Sali, T.; Alvarado, D.; Gatti, E.; Pierre, P.; Streblow, D.; Defilippis, V.R. Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff. J. Virol. 2011, 85, 606–620. [Google Scholar] [CrossRef]
- Frolova, E.I.; Fayzulin, R.Z.; Cook, S.H.; Griffin, D.E.; Rice, C.M.; Frolov, I. Roles of nonstructural protein nsP2 and Alpha/Beta interferons in determining the outcome of Sindbis virus infection. J. Virol. 2002, 76, 11254–11264. [Google Scholar] [CrossRef]
- Chan, Y.H.; Teo, T.H.; Utt, A.; Tan, J.J.; Amrun, S.N.; Abu Bakar, F.; Yee, W.X.; Becht, E.; Lee, C.Y.P.; Lee, B.; et al. Mutating chikungunya virus non-structural protein produces potent live-attenuated vaccine candidate. EMBO Mol. Med. 2019, 11, e10092. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, M.D.; Mazzon, M.; Jacobs, M.; Amara, A. Pathogenesis of flavivirus infections: Using and abusing the host cell. Cell Host Microbe 2009, 5, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.S.; Rasotgi, V.; Jain, S.; Gupta, V. Discovery of fifth serotype of dengue virus (DENV-5): A new public health dilemma in dengue control. Med. J. Armed Forces India 2015, 71, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef]
- Paixão, E.S.; Teixeira, M.G.; Rodrigues, L.C. Zika, chikungunya and dengue: The causes and threats of new and re-emerging arboviral diseases. BMJ Glob. Health 2018, 3, e000530. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Abdolmohammadi, K.; Fatahi, Y.; Dalili, H.; Rasoolinejad, M.; Rezaei, F.; Salehi-Vaziri, M.; Shafiei-Jandaghi, N.Z.; Gooshki, E.S.; Zaim, M.; et al. Zika Virus Infection, Basic and Clinical Aspects: A Review Article. Iran J. Public Health 2019, 48, 20–31. [Google Scholar] [CrossRef]
- Da Silva, M.H.M.; Moises, R.N.C.; Alves, B.E.B.; Pereira, H.W.B.; de Paiva, A.A.P.; Morais, I.C.; Nascimento, Y.M.; Monteiro, J.D.; de Souto, J.T.; Nascimento, M.S.L.; et al. Innate immune response in patients with acute Zika virus infection. Med. Microbiol. Immunol. 2019. [Google Scholar] [CrossRef]
- Lum, F.M.; Lye, D.C.B.; Tan, J.J.L.; Lee, B.; Chia, P.Y.; Chua, T.K.; Amrun, S.N.; Kam, Y.W.; Yee, W.X.; Ling, W.P.; et al. Longitudinal Study of Cellular and Systemic Cytokine Signatures to Define the Dynamics of a Balanced Immune Environment During Disease Manifestation in Zika Virus-Infected Patients. J. Infect. Dis. 2018, 218, 814–824. [Google Scholar] [CrossRef]
- Kam, Y.W.; Leite, J.A.; Lum, F.M.; Tan, J.J.L.; Lee, B.; Judice, C.C.; Teixeira, D.A.T.; Andreata-Santos, R.; Vinolo, M.A.; Angerami, R.; et al. Specific Biomarkers Associated with Neurological Complications and Congenital Central Nervous System Abnormalities From Zika Virus-Infected Patients in Brazil. J. Infect. Dis. 2017, 216, 172–181. [Google Scholar] [CrossRef]
- Her, Z.; Kam, Y.W.; Gan, V.C.; Lee, B.; Thein, T.L.; Tan, J.J.; Lee, L.K.; Fink, K.; Lye, D.C.; Rénia, L.; et al. Severity of Plasma Leakage Is Associated With High Levels of Interferon γ-Inducible Protein 10, Hepatocyte Growth Factor, Matrix Metalloproteinase 2 (MMP-2), and MMP-9 During Dengue Virus Infection. J. Infect. Dis. 2017, 215, 42–51. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, X.; Hong, W.; Qiu, S.; Wang, J.; Yu, L.; Zeng, Y.; Tan, X.; Zhang, F. Slow resolution of inflammation in severe adult dengue patients. BMC Infect. Dis. 2016, 16, 291. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Jain, A.; Garg, R.K.; Kumar, R.; Agrawal, O.P.; Lakshmana Rao, P.V. Serum levels of IL-8, IFNγ, IL-10, and TGF β and their gene expression levels in severe and non-severe cases of dengue virus infection. Arch. Virol. 2015, 160, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Malavige, G.N.; Gomes, L.; Alles, L.; Chang, T.; Salimi, M.; Fernando, S.; Nanayakkara, K.D.; Jayaratne, S.; Ogg, G.S. Serum IL-10 as a marker of severe dengue infection. BMC Infect. Dis. 2013, 13, 341. [Google Scholar] [CrossRef] [PubMed]
- Butthep, P.; Chunhakan, S.; Yoksan, S.; Tangnararatchakit, K.; Chuansumrit, A. Alteration of cytokines and chemokines during febrile episodes associated with endothelial cell damage and plasma leakage in dengue hemorrhagic fever. Pediatr. Infect. Dis. J. 2012, 31, e232–e238. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Tesh, R.B.; Azar, S.R.; Muruato, A.E.; Hanley, K.A.; Auguste, A.J.; Langsjoen, R.M.; Paessler, S.; Vasilakis, N.; Weaver, S.C. Characterization of a Novel Murine Model to Study Zika Virus. Am. J. Trop. Med. Hyg. 2016, 94, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Dowall, S.D.; Graham, V.A.; Rayner, E.; Atkinson, B.; Hall, G.; Watson, R.J.; Bosworth, A.; Bonney, L.C.; Kitchen, S.; Hewson, R. A Susceptible Mouse Model for Zika Virus Infection. PLoS Negl. Trop. Dis. 2016, 10, e0004658. [Google Scholar] [CrossRef] [PubMed]
- Aliota, M.T.; Caine, E.A.; Walker, E.C.; Larkin, K.E.; Camacho, E.; Osorio, J.E. Characterization of Lethal Zika Virus Infection in AG129 Mice. PLoS Negl. Trop. Dis. 2016, 10, e0004682. [Google Scholar] [CrossRef]
- Julander, J.G.; Siddharthan, V. Small-Animal Models of Zika Virus. J. Infect. Dis. 2017, 216, S919–S927. [Google Scholar] [CrossRef]
- Shresta, S.; Kyle, J.L.; Snider, H.M.; Basavapatna, M.; Beatty, P.R.; Harris, E. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas T- and B-cell-dependent immunity are less critical. J. Virol. 2004, 78, 2701–2710. [Google Scholar] [CrossRef]
- Johnson, A.J.; Roehrig, J.T. New mouse model for dengue virus vaccine testing. J. Virol. 1999, 73, 783–786. [Google Scholar]
- Shresta, S.; Sharar, K.L.; Prigozhin, D.M.; Beatty, P.R.; Harris, E. Murine model for dengue virus-induced lethal disease with increased vascular permeability. J. Virol. 2006, 80, 10208–10217. [Google Scholar] [CrossRef] [PubMed]
- Orozco, S.; Schmid, M.A.; Parameswaran, P.; Lachica, R.; Henn, M.R.; Beatty, R.; Harris, E. Characterization of a model of lethal dengue virus 2 infection in C57BL/6 mice deficient in the alpha/beta interferon receptor. J. Gen. Virol. 2012, 93, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Hollidge, B.; Daye, S.; Zeng, X.; Blancett, C.; Kuszpit, K.; Bocan, T.; Koehler, J.W.; Coyne, S.; Minogue, T.; et al. Neuropathogenesis of Zika Virus in a Highly Susceptible Immunocompetent Mouse Model after Antibody Blockade of Type I Interferon. PLoS Negl. Trop. Dis. 2017, 11, e0005296. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Woodruff, E.M.; Trapecar, M.; Fontaine, K.A.; Ezaki, A.; Borbet, T.C.; Ott, M.; Sanjabi, S. Dampened antiviral immunity to intravaginal exposure to RNA viral pathogens allows enhanced viral replication. J. Exp. Med. 2016, 213, 2913–2929. [Google Scholar] [CrossRef]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef]
- Christofferson, R.C.; McCracken, M.K.; Johnson, A.M.; Chisenhall, D.M.; Mores, C.N. Development of a transmission model for dengue virus. Virol. J. 2013, 10, 127. [Google Scholar] [CrossRef]
- Piret, J.; Carbonneau, J.; Rhéaume, C.; Baz, M.; Boivin, G. Predominant role of IPS-1 over TRIF adaptor proteins in early innate immune response against Zika virus in mice. J. Gen. Virol. 2018, 99, 209–218. [Google Scholar] [CrossRef]
- Perry, S.T.; Prestwood, T.R.; Lada, S.M.; Benedict, C.A.; Shresta, S. Cardif-mediated signaling controls the initial innate response to dengue virus in vivo. J. Virol. 2009, 83, 8276–8281. [Google Scholar] [CrossRef]
- Chen, S.T.; Lin, Y.L.; Huang, M.T.; Wu, M.F.; Cheng, S.C.; Lei, H.Y.; Lee, C.K.; Chiou, T.W.; Wong, C.H.; Hsieh, S.L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature 2008, 453, 672–676. [Google Scholar] [CrossRef]
- Sariol, C.A.; Martínez, M.I.; Rivera, F.; Rodríguez, I.V.; Pantoja, P.; Abel, K.; Arana, T.; Giavedoni, L.; Hodara, V.; White, L.J.; et al. Decreased dengue replication and an increased anti-viral humoral response with the use of combined Toll-like receptor 3 and 7/8 agonists in macaques. PLoS ONE 2011, 6, e19323. [Google Scholar] [CrossRef]
- Weisblum, Y.; Oiknine-Djian, E.; Vorontsov, O.M.; Haimov-Kochman, R.; Zakay-Rones, Z.; Meir, K.; Shveiky, D.; Elgavish, S.; Nevo, Y.; Roseman, M.; et al. Zika Virus Infects Early- and Midgestation Human Maternal Decidual Tissues, Inducing Distinct Innate Tissue Responses in the Maternal-Fetal Interface. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liang, Y.; Yi, P.; Xu, L.; Hawkins, H.K.; Rossi, S.L.; Soong, L.; Cai, J.; Menon, R.; Sun, J. Outcomes of Congenital Zika Disease Depend on Timing of Infection and Maternal-Fetal Interferon Action. Cell Rep. 2017, 21, 1588–1599. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Yockey, L.J.; Jurado, K.A.; Arora, N.; Millet, A.; Rakib, T.; Milano, K.M.; Hastings, A.K.; Fikrig, E.; Kong, Y.; Horvath, T.L.; et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Corry, J.; Arora, N.; Good, C.A.; Sadovsky, Y.; Coyne, C.B. Organotypic models of type III interferon-mediated protection from Zika virus infections at the maternal-fetal interface. Proc. Natl. Acad. Sci. USA 2017, 114, 9433–9438. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Miner, J.J.; Cao, B.; Arora, N.; Smith, A.M.; Kovacs, A.; Mysorekar, I.U.; Coyne, C.B.; Diamond, M.S. Gestational Stage and IFN-λ Signaling Regulate ZIKV Infection in Utero. Cell Host Microbe 2017, 22, 366–376. [Google Scholar] [CrossRef]
- Beatty, P.R.; Puerta-Guardo, H.; Killingbeck, S.S.; Glasner, D.R.; Hopkins, K.; Harris, E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 2015, 7, 304ra141. [Google Scholar] [CrossRef]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; García-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef]
- Ashour, J.; Morrison, J.; Laurent-Rolle, M.; Belicha-Villanueva, A.; Plumlee, C.R.; Bernal-Rubio, D.; Williams, K.L.; Harris, E.; Fernandez-Sesma, A.; Schindler, C.; et al. Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe 2010, 8, 410–421. [Google Scholar] [CrossRef]
- Ding, Q.; Gaska, J.M.; Douam, F.; Wei, L.; Kim, D.; Balev, M.; Heller, B.; Ploss, A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. USA 2018, 115, E6310–E6318. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelemans, T.; Kikkert, M. Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses 2019, 11, 961. https://doi.org/10.3390/v11100961
Nelemans T, Kikkert M. Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses. 2019; 11(10):961. https://doi.org/10.3390/v11100961
Chicago/Turabian StyleNelemans, Tessa, and Marjolein Kikkert. 2019. "Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections" Viruses 11, no. 10: 961. https://doi.org/10.3390/v11100961
APA StyleNelemans, T., & Kikkert, M. (2019). Viral Innate Immune Evasion and the Pathogenesis of Emerging RNA Virus Infections. Viruses, 11(10), 961. https://doi.org/10.3390/v11100961