Look Who’s Talking: T-Even Phage Lysis Inhibition, the Granddaddy of Virus-Virus Intercellular Communication Research

Abstract
1. Introduction
1.1. Communication
1.1.1. ‘Signals’
1.1.2. Coercion
1.1.3. Cues
2. Overview of Lysis Inhibition
3. History of Lysis Inhibition
3.1. Initial Observations of Lysis Inhibition
3.2. Virus-Virus Intercellular Communication
4. Lysis Inhibition Laboratory Phenotypes
4.1. Plaques
4.1.1. Plaque Treatment with Chloroform Vapor
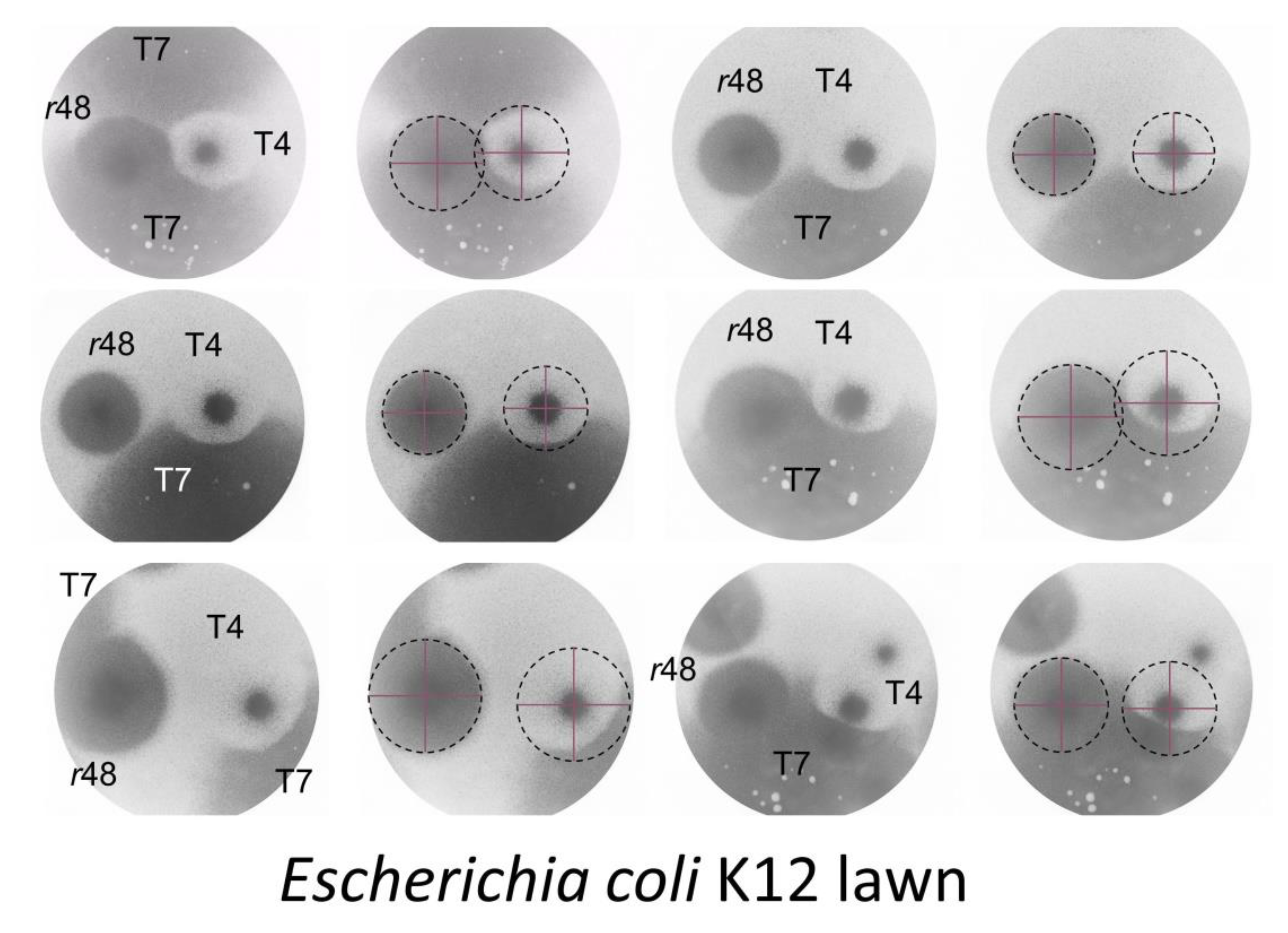
4.1.2. Colliding T4 Plaques
4.1.3. T7 Plaques Colliding with T4 Plaques
4.1.4. Wild-Type T4 Plaques, Conclusions
4.2. Broth-Growth Aspects
5. Mechanisms
5.1. Overview of Key Players
5.2. Inhibition of T-Holin
5.2.1. Protein T
5.2.2. Protein RI
5.2.3. Protein RIII
5.2.4. Genes rI.-1 and rI.1
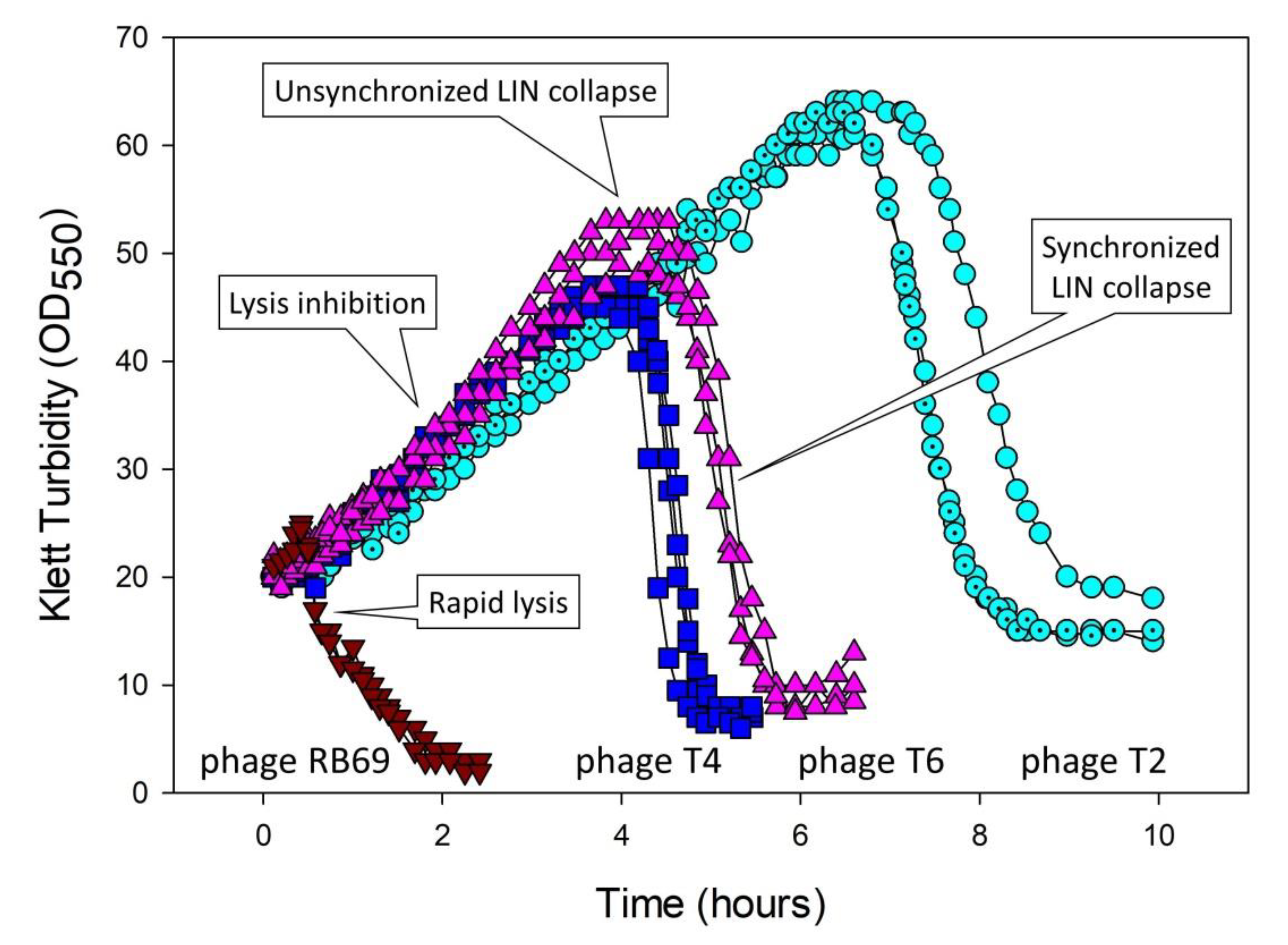
5.3. Lysis-Inhibition Collapse and Its Synchronization
5.3.1. Four Mechanisms Potentially Leading to Lysis-Inhibition Collapse
- Lysis from within (LI). RI- and RIII-mediated inhibition of T-holin ceases and normal LI therefore commences. Perhaps secondary adsorption-associated RI stabilization in the periplasm can only last so long [34] and is not otherwise replenishable over the long term during LIN.
- Membrane deterioration (MD). Eventually the plasma membrane of phage T4-infected bacteria becomes unstable, resulting in metabolic poisoning of the LINed bacterium and thereby a triggering of T-hole formation, i.e., “nonspecific deterioration of the membrane” [20] (‘membrane deterioration’; MD). LI thus commences. I consider this mechanism to be possible but nevertheless somewhat hypothetical (see also Section 5.3.2).
- Lysis from without (LO). In the course of continued secondary adsorption, cell walls become sufficiently degraded that LO commences. This results in plasma membrane disruption, metabolic poisoning of the phage-infected bacterium, and thereby LI as well.
- Secondary traumatization (ST). In the course of continued secondary adsorption, plasma membranes become sufficiently degraded as to trigger T-hole formation, thus, as above, with LI commencing. I consider this mechanism also to be somewhat hypothetical, though perhaps less hypothetical than mechanism 2.
5.3.2. Initiation of LIN Collapse: Ruling out Mechanism 2?
5.3.3. Timing of Initiation of Lysis-Inhibition Collapse versus Its Synchronization
5.4. The Phage Imm and Sp Proteins
5.5. Lysis Inhibition and Unsynchronized as Well as Synchronized Lysis-Inhibition Collapse
- LIN induction is associated with secondary adsorption as it transduces a signal which allows periplasmic RI protein to interfere with or continue to interfere with T-hole formation.
- This secondary adsorption may result in some degree of cell envelope damage but the degree of damage is reduced due to the actions of the Imm and Sp proteins.
- Lysis of a fraction of individual LINed bacteria, especially near the point of initiation of LIN collapse (unsynchronized LIN collapse) and perhaps as caused by erosions of R-protein mediated inhibition to T-hole formation (mechanism 1), results in a buildup of phage virions within the environment.
- The presence of these additional extracellular (free) virions results in an accumulation of secondary adsorptions of still-intact LINed phage-infected bacteria.
- At some point, rates of bacterial lysis accelerate as a direct consequence of increasing numbers of secondary adsorptions (mechanisms 3 or 4).
- Additional lysis gives rise to further buildups of secondary adsorptions of remaining intact phage-infected bacteria, providing a positive-feedback lysis, i.e., synchronized LIN collapse.
6. Evolutionary Ecology of Lysis Inhibition and Synchronized Lysis-Inhibition Collapse
6.1. Utility of Rapid Lysis
6.2. Utility of an Inducibly Longer Latent Period (Lysis Inhibition)
6.2.1. Reduced Densities of Phage-Uninfected Bacteria
6.2.2. Increased Densities of Phage-Infected Bacteria
6.3. Utility of Synchronized Lysis-Inhibition Collapse
7. Ecology of Lysis Inhibition
7.1. Ecology of Lysis Inhibition among Planktonic Bacteria
7.2. Ecology of Lysis Inhibition among Biofilm Bacteria
8. Other Virus-Associated Communication Mechanisms
8.1. High-Multiplicity Lysogeny Decisions
8.2. Arbitrium Systems
8.3. Autoinducer-Associated Prophage Induction
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, I.-N.; Smith, D.L.; Young, R. Holins: The protein clocks of bacteriophage infections. Ann. Rev. Microbiol. 2000, 54, 799–825. [Google Scholar] [CrossRef]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis-lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef]
- Silpe, J.E.; Bassler, B.L. A host-produced quorum-sensing autoinducer controls a phage lysis-lysogeny decision. Cell 2019, 176, 268–280. [Google Scholar] [CrossRef]
- Bull, J.J.; Pfennig, D.W.; Wang, I.-W. Genetic details, optimization, and phage life histories. Trends Ecol. Evol. 2004, 19, 76–82. [Google Scholar] [CrossRef]
- Bull, J.J. Optimality models of phage life history and parallels in disease evolution. J. Theor. Biol. 2006, 241, 928–938. [Google Scholar] [CrossRef]
- Abedon, S.T. Bacteriophage intraspecific cooperation and defection. In Contemporary Trends in Bacteriophage Research; Adams, H.T., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2009; pp. 191–215. [Google Scholar]
- Leggett, H.C.; Brown, S.P.; Reece, S.E. War and peace: Social interactions in infections. Philos. Trans. R. Soc. Lond B Biol. Sci. 2014, 369, 20130365. [Google Scholar] [CrossRef]
- Hershey, A.D. Mutation of bacteriophage with respect to type of plaque. Genetics 1946, 31, 620–640. [Google Scholar]
- Doermann, A.H. Lysis and lysis inhibition with Escherichia coli bacteriophage. J. Bacteriol. 1948, 55, 257–275. [Google Scholar]
- Abedon, S.T. Commentary: Communication between viruses guides lysis-lysogeny decisions. Front. Microbiol. 2017, 8, 983. [Google Scholar] [CrossRef]
- Abedon, S.T.; Duffy, S.; Turner, P.E. Bacteriophage ecology. In Encyclopedia of Microbiology; Schaecter, M., Ed.; Elsevier: Oxford, UK, 2009; pp. 42–57. [Google Scholar]
- Abedon, S.T. Communication among phages, bacteria, and soil environments. Soil Biology 2011, 23, 37–65. [Google Scholar]
- Demerec, M.; Fano, U. Bacteriophage-resistant mutants in Escherichia coli. Genetics 1945, 30, 119–136. [Google Scholar] [PubMed]
- Abedon, S.T. The murky origin of Snow White and her T-even dwarfs. Genetics 2000, 155, 481–486. [Google Scholar]
- Gromkoua, R.H. T-related bacteriophage isolated from Shigella sonnei. J. Virol. 1968, 2, 692–694. [Google Scholar]
- Schito, G.C. Dvelopment of coliphage N4: Ultrastructural studies. J. Virol. 1974, 13, 186–196. [Google Scholar] [PubMed]
- Abedon, S.T.; Hyman, P.; Thomas, C. Experimental examination of bacteriophage latent-period evolution as a response to bacterial availability. Appl. Environ. Microbiol. 2003, 69, 7499–7506. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Lysis of lysis inhibited bacteriophage T4-infected cells. J. Bacteriol. 1992, 174, 8073–8080. [Google Scholar] [CrossRef] [PubMed]
- Ellis, E.L.; Delbrück, M. The growth of bacteriophage. J. Gen. Physiol. 1939, 22, 365–384. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Bacteriophage lysis: Mechanisms and regulation. Microbiol. Rev. 1992, 56, 430–481. [Google Scholar]
- Abedon, S.T. Bacteriophage T4 resistance to lysis-inhibition collapse. Genet. Res. 1999, 74, 1–11. [Google Scholar] [CrossRef]
- Abedon, S.T. Lysis from without. Bacteriophage 2011, 1, 46–49. [Google Scholar] [CrossRef]
- Abedon, S.T. Lysis and the interaction between free phages and infected cells. In The Molecular Biology of Bacteriophage T4; Karam, J.D., Kutter, E., Carlson, K., Guttman, B., Eds.; ASM Press: Washington, DC, USA, 1994; pp. 397–405. [Google Scholar]
- Igler, C.; Abedon, S.T. Commentary: A host-produced quorum-sensing autoinducer controls a phage lysis-lysogeny decision. Front. Microbiol. 2019, 10, 1171. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D. The properties of X-ray inactivated bacteriophage I. Inactivation by direct effect. J. Bacteriol. 1950, 60, 697–718. [Google Scholar] [PubMed]
- Rutberg, B.; Rutberg, L. Role of superinfecting phage in lysis inhibition with phage T4 in Escherichia coli. J. Bacteriol. 1965, 90, 891–894. [Google Scholar] [PubMed]
- Benzer, S. Adverntures in the rII region. In Phage and the Origins of Molecular Biology (expanded edition); Cairns, J., Stent, G.S., Watson, J.D., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1966; pp. 157–165. [Google Scholar]
- Bode, W. Lysis inhibition in Escherichia coli infected with bacteriophage T4. J. Virol. 1967, 1, 948–955. [Google Scholar] [PubMed]
- Abedon, S.T. Selection for lysis inhibition in bacteriophage. J. Theor. Biol. 1990, 146, 501–511. [Google Scholar] [CrossRef]
- Paddison, P.; Abedon, S.T.; Dressman, H.K.; Gailbreath, K.; Tracy, J.; Mosser, E.; Neitzel, J.; Guttman, B.; Kutter, E. Lysis inhibition and fine-structure genetics in bacteriophage T4. Genetics 1998, 148, 1539–1550. [Google Scholar]
- Dressman, H.K.; Drake, J.W. Lysis and lysis inhibition in bacteriophage T4: rV mutations reside in the holin t gene. J. Bacteriol. 1999, 181, 4391–4396. [Google Scholar]
- Ramanculov, E.; Young, R. An ancient player unmasked: T4 rI encodes a t-specific antiholin. Mol. Microbiol. 2001, 41, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.A.; Struck, D.K.; Young, R. Periplasmic domains define holin-antiholin interactions in T4 lysis inhibition. J. Bacteriol. 2005, 187, 6631–6640. [Google Scholar] [CrossRef]
- Tran, T.A.; Struck, D.K.; Young, R. The T4 RI antiholin has an N-terminal signal anchor release domain that targets it for degradation by DegP. J. Bacteriol. 2007, 189, 7618–7625. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage population growth: Constraints, games, adaptation. In Bacteriophage Ecology; Abedon, S.T., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 64–93. [Google Scholar]
- Moussa, S.H.; Kuznetsov, V.; Tran, T.A.; Sacchettini, J.C.; Young, R. Protein determinants of phage T4 lysis inhibition. Protein Sci. 2012, 21, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Moussa, S.H.; Lawler, J.L.; Young, R. Genetic dissection of T4 lysis. J. Bacteriol. 2014, 196, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Young, R. The last r locus unveiled: T4 RIII is a cytoplasmic antiholin. J. Bacteriol. 2016, 198, 2448–2457. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.S. Observations on the relationship of symbiotic and lytic bacteriophage. J. Pathol. Bacteriol. 1951, 63, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Lieb, M. The establishment of lysogenicity in Escherichia coli. J. Bacteriol. 1953, 65, 642–651. [Google Scholar] [PubMed]
- Levine, M. Mutations in the temperate phage P22 and lysogeny in Salmonella. Virology 1957, 3, 22–41. [Google Scholar] [CrossRef]
- Fry, B.A. Conditions for the infection of Escherichia coli with lambda phage and for the establishment of lysogeny. J. Gen. Microbiol. 1959, 21, 676–684. [Google Scholar] [CrossRef]
- Six, E. Inheritance of prophage P2 in superinfection experiments. Virology 1961, 14, 220–233. [Google Scholar] [CrossRef]
- Brooks, K. Studies in the physiological genetics of some supporessor-sensitive mutants of bacteriophages lambda. Virology 1965, 26, 489–499. [Google Scholar] [CrossRef]
- Hoffman, D.B., Jr.; Rubenstein, I. Physical studies of lysogeny. I. Properties of intracellular parental bacteriophage DNA from λ-infected sensitive bacteria. J. Mol. Biol. 1968, 35, 375–399. [Google Scholar] [CrossRef]
- Kourilsky, P. Lysogenization by bacteriophage lambda. I. Multiple infection and the lysogenic response. Mol. Gen. Genet. 1973, 122, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Avlund, M.; Dodd, I.B.; Semsey, S.; Sneppen, K.; Krishna, S. Why do phage play dice? J. Virol. 2009, 83, 11416–11420. [Google Scholar] [CrossRef] [PubMed]
- Joh, R.I.; Weitz, J.S. To lyse or not to lyse: Transient-mediated stochastic fate determination in cells infected by bacteriophages. PLoS Comput. Biol. 2011, 7, e1002006. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Roy, K.; Williamson, K.E.; Srinivasiah, S.; Wommack, K.E.; Radosevich, M. Acyl-homoserine lactones can induce virus production in lysogenic bacteria: An alternative paradigm for prophage induction. Appl. Environ. Microbiol. 2009, 75, 7142–7152. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Kropinski, A.M.; Clokie, M.R. What does the talking?: Quorum sensing signalling genes discovered in a bacteriophage genome. PLoS ONE 2014, 9, e85131. [Google Scholar] [CrossRef] [PubMed]
- Diggle, S.P.; Gardner, A.; West, S.A.; Griffin, A.S. Evolutionary theory of bacterial quorum sensing: When is a signal not a signal? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2007, 362, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.K.; Kutter, E.M.; Mosig, G.; Berget, P.B. Bacteriophage T4; American Society for Microbiology: Washington, DC, USA, 1983. [Google Scholar]
- Karam, J.D. Molecular Biology of Bacteriophage T4; ASM Press: Washington, DC, USA, 1994. [Google Scholar]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Practical methods for determining phage growth parameters. Meth. Mol. Biol. 2009, 501, 175–202. [Google Scholar]
- Kropinski, A.M. Practical advice on the one-step growth curve. Methods Mol. Biol. 2018, 1681, 41–47. [Google Scholar] [PubMed]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with ‘Lytic or lysogenic’. FEMS Microbiol. Lett. 2016, 363, fnw047. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Bacteriophage secondary infection. Virol. Sin. 2015, 30, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sertic, V. Contribution à l’étude des phénomènes de variations du bacteríophage. Compt. Rend. Soc. Biol. 1929, 100, 614–616. [Google Scholar]
- Holmes, F.L.; Summers, W.C. Reconceiving the Gene: Seymour Benzer’s Adventures in Phage Genetics; Yale University Press: New Haven, CT, USA, 2006. [Google Scholar]
- Delbrück, M. Bacterial viruses or bacteriophages. Biol. Rev. 1946, 21, 30–40. [Google Scholar] [CrossRef]
- Abedon, S.T. Bottle lysate T4 stock preparation: What if your cultures won’t clear? T4 News 1992, 6. [Google Scholar]
- Freedman, M.L.; Krisch, R.E. Enlargement of Escherichia coli after bacteriophage infection I. description of phenomenon. J. Virol. 1971, 8, 87–94. [Google Scholar] [PubMed]
- Freedman, M.L.; Krisch, R.E. Enlargement of Escherichia coli after bacteriophage infection II. proposed mechanism. J. Virol. 1971, 8, 95–102. [Google Scholar]
- Hershey, A.D. Spontaneous mutations in bacterial viruses. Cold Spring Harbor Symp. Quant. Biol. 1946, 11, 67–77. [Google Scholar]
- Delbrück, M. The growth of bacteriophage and lysis of the host. J. Gen. Physiol. 1940, 23, 643–660. [Google Scholar] [CrossRef]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H.V. Phage particles do not communicate with each other or make decisions to either lyse or lysogenize their host cells. Arch. Virol. 2017, 162, 1465–1466. [Google Scholar] [CrossRef][Green Version]
- Weitz, J.S.; Li, G.; Gulbudak, H.; Cortez, M.H.; Whitaker, R.J. Viral invasion fitness across a continuum from lysis to latency. Virus Evol. 2019, 5, vez006. [Google Scholar] [CrossRef] [PubMed]
- Ptashne, M. Genetic Switch: Phage Lambda Revisited, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2004. [Google Scholar]
- Fogg, P.C.; Allison, H.E.; Saunders, J.R.; McCarthy, A.J. Bacteriophage lambda: A paradigm revisited. J. Virol. 2010, 84, 6876–6879. [Google Scholar] [CrossRef] [PubMed]
- Blasdel, B.G.; Abedon, S.T. Superinfection immunity. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 978-0-12-809633-8. [Google Scholar] [CrossRef]
- Mavrich, T.N.; Hatfull, G.F. Evolution of superinfection immunity in Cluster A mycobacteriophages. MBio 2019, 10, e00971-19. [Google Scholar] [CrossRef] [PubMed]
- Krone, S.M.; Abedon, S.T. Modeling phage plaque growth. In Bacteriophage Ecology; Abedon, S.T., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 415–438. [Google Scholar]
- Abedon, S.T. Detection of bacteriophages: Phage plaques. In Bacteriophages: Biology, Technology, Therapy; Harper, D.R., Abedon, S.T., Burrowes, B.H., McConville, M., Eds.; Springer International Publishing AG: Cham, Switzerland, 2018. [Google Scholar]
- Josslin, R. Physiological studies on the t gene defect in T4-infected Escherichia coli. Virology 1971, 44, 101–107. [Google Scholar] [CrossRef]
- Streisinger, G.; Mukai, F.; Dreyer, W.J.; Miller, B.; Horiuchi, S. Mutations affecting the lysozyme of phage T4. Cold Spring Harbor Symp. Quant. Biol. 1961, 26, 25–30. [Google Scholar] [CrossRef]
- Hershey, A.D.; Rotman, R. Genetic recombination between host range and plaque-type mutants of bacteriophage in a single bacterial culture. Genetics 1949, 34, 44–71. [Google Scholar]
- Lanni, Y.T. Lysis inhibition with a mutant of bacteriophage T5. Virology 1958, 5, 481–501. [Google Scholar] [CrossRef]
- Abedon, S.T. Bacteriophages and Biofilms: Ecology, Phage Therapy, Plaques; Nova Science Publishers: Hauppauge, NY, USA, 2011. [Google Scholar]
- Abedon, S.T.; Yin, J. Impact of spatial structure on phage population growth. In Bacteriophage Ecology; Abedon, S.T., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 94–113. [Google Scholar]
- Abedon, S.T. Bacteriophage exploitation of bacterial biofilms: Phage preference for less mature targets? FEMS Microbiol. Lett. 2016, 363, fnv246. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage "delay" towards enhancing bacterial escape from biofilms: A more comprehensive way of viewing resistance to bacteriophages. AIMS Microbiol. 2017, 3, 186–226. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage therapy dosing: The problem(s) with multiplicity of infection (MOI). Bacteriophage 2016, 6, e1220348. [Google Scholar] [CrossRef]
- Young, R.; Wang, I.-N. Phage lysis. In The Bacteriophages; Calendar, R., Abedon, S.T., Eds.; Oxford University Press: Oxford, UK, 2006; pp. 104–125. [Google Scholar]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Kongari, R.; Rajaure, M.; Cahill, J.; Rasche, E.; Mijalis, E.; Berry, J.; Young, R. Phage spanins: Diversity, topological dynamics and gene convergence. BMC Bioinformatics 2018, 19, 326. [Google Scholar] [CrossRef] [PubMed]
- Young, R.; Wang, I.-N.; Roof, W.D. Phages will out: Strategies of host cell lysis. Trends Microbiol. 2000, 8, 120–128. [Google Scholar] [CrossRef]
- Young, R. Phage lysis. In Phages: Their Role in Pathogenesis and Biotechnology; Waldor, M.K., Friedman, D.I., Adhya, S.L., Eds.; ASM Press: Washington, DC, USA, 2005; pp. 92–127. [Google Scholar]
- Cahill, J.; Young, R. Phage Lysis: Multiple Genes for Multiple Barriers. Adv. Virus Res. 2019, 103, 33–70. [Google Scholar] [PubMed]
- Young, R.; Bläsi, U. Holins: Form and function in bacteriophage lysis. FEMS Microbiol. Rev. 1995, 17, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Ramanculov, E.; Young, R. Functional analysis of the phage T4 holin in a context. Mol. Gen. Genom. 2001, 265, 345–353. [Google Scholar] [CrossRef]
- Josslin, R. The lysis mechanism of phage T4: Mutants affecting lysis. Virology 1970, 40, 719–726. [Google Scholar] [CrossRef]
- Burch, L.H.; Zhang, L.; Chao, F.G.; Xu, H.; Drake, J.W. The bacteriophage T4 rapid-lysis genes and their mutational proclivities. J. Bacteriol. 2011, 193, 3537–3545. [Google Scholar] [CrossRef] [PubMed]
- Kai, T.; Ueno, H.; Otsuka, Y.; Morimoto, W.; Yonesaki, T. Gene 61.3 of bacteriophage T4 is the spackle gene. Virology 1999, 260, 254–259. [Google Scholar] [CrossRef]
- Golec, P.; Wiczk, A.; Majchrzyk, A.; Los, J.M.; Wegrzyn, G.; Los, M. A role for accessory genes rI.-1 and rI.1 in the regulation of lysis inhibition by bacteriophage T4. Virus Genes 2010, 41, 459–468. [Google Scholar] [CrossRef]
- Mukai, F.; Streisinger, G.; Miller, B. The mechanism of lysis in phage T4-infected cells. Virology 1967, 33, 398–402. [Google Scholar] [CrossRef]
- Golec, P.; Karczewska-Golec, J.; Voigt, B.; Albrecht, D.; Schweder, T.; Hecker, M.; Wegrzyn, G.; Los, M. Proteomic profiles and kinetics of development of bacteriophage T4 and its rI and rIII mutants in slowly growing Escherichia coli. J. Gen. Virol. 2013, 94, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Ramanculov, E.; Young, R. Genetic analysis of the T4 holin: Timing and topology. Gene 2001, 265, 25–36. [Google Scholar] [CrossRef]
- Los, M.; Wegrzyn, G.; Neubauer, P. A role for bacteriophage T4 rI gene function in the control of phage development during pseudolysogeny and in slowly growing host cells. Res. Microbiol. 2003, 154, 547–552. [Google Scholar] [CrossRef]
- Asami, K.; Xing, X.H.; Tanji, Y.; Unno, H. Synchronized disruption of Escherichia coli cells by T4 phage infection. J. Ferment. Bioeng. 1997, 83, 511–516. [Google Scholar] [CrossRef]
- Cornett, J.B. Spackle and immunity functions of bacteriophage T4. J. Virol. 1974, 13, 312–321. [Google Scholar] [PubMed]
- Couse, N.L. Control of lysis of T4-infected Escherichia coli. J. Virol. 1968, 2, 198–207. [Google Scholar] [PubMed]
- Kao, S.H.; McClain, W.H. Baseplate protein of bacteriophage T4 with both structural and lytic functions. J. Virol. 1980, 34, 95–103. [Google Scholar]
- Nakagawa, H.; Arisaka, F.; Ishii, S.-I. Isolation and characterization of the bacteriophage T4 tail-associated lysozyme. J. Virol. 1985, 54, 460–466. [Google Scholar] [PubMed]
- Kao, S.H.; McClain, W.H. Roles of T4 gene 5 and gene s products in cell lysis. J. Virol. 1980, 34, 104–107. [Google Scholar] [PubMed]
- Lu, M.-J.; Stierof, Y.-D.; Henning, U. Location and unusual membrane topology of the immunity protein of the Escherichia coli phage T4. J. Virol. 1993, 67, 4905–4913. [Google Scholar] [PubMed]
- Lu, M.-J.; Henning, U. The immunity (imm) gene of Escherichia coli bacteriophage T4. J. Virol. 1989, 63, 3472–3478. [Google Scholar] [PubMed]
- Emrich, J. Lysis of T4-infected bacteria in the absence of lysozyme. Virology 1968, 35, 158–165. [Google Scholar] [CrossRef]
- Buller, C.S.; Dobbs, K. T4-coliphage infection of Escherichia coli with defective cell envelopes. Biochem. Biophys. Res. Com. 1971, 43, 658–665. [Google Scholar] [CrossRef]
- Abedon, S.T. Selection for bacteriophage latent period length by bacterial density: A theoretical examination. Microb. Ecol. 1989, 18, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-N.; Dykhuizen, D.E.; Slobodkin, L.B. The evolution of phage lysis timing. Evol. Ecol. 1996, 10, 545–558. [Google Scholar] [CrossRef]
- Abedon, S.T.; Herschler, T.D.; Stopar, D. Bacteriophage latent-period evolution as a response to resource availability. Appl. Environ. Microbiol. 2001, 67, 4233–4241. [Google Scholar] [CrossRef]
- Wang, I.-N. Lysis timing and bacteriophage fitness. Genetics 2006, 172, 17–26. [Google Scholar] [CrossRef]
- Heineman, R.H.; Bull, J.J. Testing optimality with experimental evolution: Lysis time in a bacteriophage. Evolution 2007, 61, 1695–1709. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, I.-N. Bacteriophage adsorption rate and optimal lysis time. Genetics 2008, 180, 471–482. [Google Scholar] [CrossRef]
- Bonachela, J.A.; Levin, S.A. Evolutionary comparison between viral lysis rate and latent period. J. Theor. Biol. 2014, 345, 32–42. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ratnieks, F.L.W. Reproductive harmony via mutual policing by workers in eusocial Hymenoptera. Am. Nat. 1988, 132, 217–236. [Google Scholar]
- Frank, S.A. Mutual policing and repression of competition in the evolution of cooperative groups. Nature 1995, 377, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Thomas-Abedon, C. Phage therapy pharmacology. Curr. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Thinking about microcolonies as phage targets. Bacteriophage 2012, 2, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Ecology of anti-biofilm agents II. bacteriophage exploitation and biocontrol of biofilm bacteria. Pharmaceuticals 2015, 8, 559–589. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Bacteriophage-mediated biocontrol of wound infections, and ecological exploitation of biofilms by phages. In Recent Clinical Techniques, Results, and Research in Wounds; Shiffman, M., Ed.; Springer International Publishing AG: Cham, Switzerland, 2018. [Google Scholar]
- Zeng, L.; Skinner, S.O.; Zong, C.; Sippy, J.; Feiss, M.; Golding, I. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell 2010, 141, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Blotnick, J.A.; Vargas-García, C.A.; Dennehy, J.J.; Zurakowski, R.; Singh, A. The effect of multiplicity of infection on the temperateness of a bacteriophage: Implications for viral fitness. IEEE 2017, 1641–1645. [Google Scholar]
- Gandon, S. Why be temperate: Lessons from bacteriophage λ. Trends Microbiol. 2016, 24, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Xiong, J.; Gu, Y.; Yin, K.; Wang, J.; Hu, Y.; Zhou, D.; Fu, X.; Qi, S.; Zhu, X.; et al. Structural and functional insights into the regulation of the lysis-lysogeny decision in viral communities. Nat. Microbiol. 2018, 3, 1285–1294. [Google Scholar] [CrossRef]
- Gallego Del, S.F.; Penades, J.R.; Marina, A. Deciphering the molecular mechanism underpinning phage arbitrium communication systems. Mol. Cell 2019, 74, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Hynes, A.P.; Moineau, S. Phagebook: The social network. Mol. Cell 2017, 65, 963–964. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harms, A.; Diard, M. Crowd controlled-host quorum sensing drives phage decision. Cell Host. Microbe 2019, 25, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.L. Phages tune in to host cell quorum sensing. Cell 2019, 176, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Stewart, F.M.; Levin, B.R. The population biology of bacterial viruses: Why be temperate. Theor. Pop. Biol. 1984, 26, 93–117. [Google Scholar] [CrossRef]
- Abedon, S.T.; LeJeune, J.T. Why bacteriophage encode exotoxins and other virulence factors. Evol. Bioinform. Online 2005, 1, 97–110. [Google Scholar] [CrossRef]
- Brown, S.P.; Le Chat, L.; De Paepe, M.; Taddei, F. Ecology of microbial invasions: Amplification allows virus carriers to invade more rapidly when rare. Curr. Biol. 2006, 16, 2048–2052. [Google Scholar] [CrossRef]
- Gama, J.A.; Reis, A.M.; Domingues, I.; Mendes-Soares, H.; Matos, A.M.; Dionisio, F. Temperate bacterial viruses as double-edged swords in bacterial warfare. PLoS ONE 2013, 8, e59043. [Google Scholar] [CrossRef]
- Paul, J.H. Prophages in marine bacteria: Dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef]
- Humbelin, M.; Suri, B.; Rao, D.N.; Hornby, D.P.; Eberle, H.; Pripfl, T.; Kenel, S.; Bickle, T.A. Type III DNA restriction and modification systems EcoP1 and EcoP15. Nucleotide sequence of the EcoP1 operon, the EcoP15 mod gene and some EcoP1 mod mutants. J. Mol. Biol. 1988, 200, 23–29. [Google Scholar] [CrossRef]
- Friedman, D.I.; Mozola, C.C.; Beeri, K.; Ko, C.C.; Reynolds, J.L. Activation of a prophage-encoded tyrosine kinase by a heterologous infecting phage results in a self-inflicted abortive infection. Mol. Microbiol. 2011, 82, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.M.; Wetzel, K.S.; Dedrick, R.M.; Montgomery, M.T.; Garlena, R.A.; Jacobs-Sera, D.; Hatfull, G.F. More evidence of collusion: A new prophage-mediated viral defense system encoded by mycobacteriophage Sbash. MBio 2019, 10, e00196-19. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.T.; Guerrero Bustamante, C.A.; Dedrick, R.M.; Jacobs-Sera, D.; Hatfull, G.F. Yet more evidence of collusion: A new viral defense system encoded by Gordonia phage CarolAnn. MBio 2019, 10, e02417-18. [Google Scholar] [CrossRef] [PubMed]
- Espeland, E.M.; Lipp, E.K.; Huq, A.; Colwell, R.R. Polylysogeny and prophage induction by secondary infection in Vibrio cholerae. Environ. Microbiol. 2004, 6, 760–763. [Google Scholar] [CrossRef]
- Lemire, S.; Figueroa-Bossi, N.; Bossi, L. Bacteriophage crosstalk: Coordination of prophage induction by trans-acting antirepressors. PLoS Genet. 2011, 7, e1002149. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, J.D.; Moineau, S. Homologous recombination between a lactococcal bacteriophage and the chromosome of its host strain. Virology 2000, 270, 65–75. [Google Scholar] [CrossRef][Green Version]
- Brüssow, H.; Desiere, F. Evolution of tailed phages-insights from comparative phage genomics. In The Bacteriophages; Calendar, R., Abedon, S.T., Eds.; Oxford University Press: Oxford, UK, 2006; pp. 26–36. [Google Scholar]
- Burrowes, B.H.; Molineux, I.J.; Fralick, J.A. Directed in vitro evolution of therapeutic bacteriophages: The Appelmans protocol. Viruses 2019, 11, 241. [Google Scholar] [CrossRef] [PubMed]
- Dehò, G.; Ghisotti, D. The satellite phage P4. In The Bacteriophages, 2nd ed.; Calendar, R., Abedon, S.T., Eds.; Oxford University Press: Oxford, UK, 2006; pp. 391–408. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Stands for… |
|---|---|
| 5 (5) | Gene product 5 (phage T4 protein and associated gene) |
| AAPI | Autoinducer-Associated Prophage Induction |
| AS | Arbitrium System |
| E (e) | Endolysin (phage T4 protein and associated gene) |
| HMLD | High-Multiplicity Lysogeny Decision |
| Imm (imm) | Immunity (phage T4 protein and associated gene) |
| LI | Lysis from withIn |
| LIN | Lysis INhibition |
| LINed | Lysis INhibited |
| LIN collapse | Lysis-INhibition collapse |
| LO | Lysis from withOut |
| MD | Membrane Deterioration |
| R (r) | Rapid lysis |
| SA | Secondary Adsorption |
| Sp (sp) | Spackle (phage T4 protein and associated gene) |
| ST | Secondary Traumatization |
| T (t) | Tithonus (phage T4 protein and associated gene) |
| Term or Abbreviation | Meaning | Overview or Discussion |
|---|---|---|
| 5 (5) | Gene product 5; 5 is the underlying phage T4 gene | Protein making up the phage T4 virion tail tube tip, which is a lysis from without (LO), cell-wall digesting lysozyme |
| Arbitrium System (AS) | Phage-encoded, autoinducer-mediated, lysis delay | As achieved by temperate phages, resulting in lysogenic rather than lytic cycles; an example of phage-associated intercellularly mediated communication |
| Autoinducer | Quorum-sensing signaling molecule | Generally, a bacterium-produced molecule but also as encoded by certain temperate phages, re: arbitrium systems (ASs) |
| Autoinducer-associated prophage induction (AAPI) | Quorum-sensing autoinducer-mediated lysis acceleration that is associated with prophage induction | As has been found in association with V. cholerae; an example of phage-associated intercellularly mediated communication |
| Coinfection | Infection of a cell by more than one phage | A consequence of simultaneous or secondary infection; generally, lysis inhibition (LIN) is not explicitly a coinfection-associated phenomenon |
| E (e) | Endolysin | The phage T4 lysis from within (LI), cell-wall digesting lysozyme protein, as encoded by gene e |
| Free phage (free virion) | A post-release mature phage virion, i.e., as not still found within its parental phage infection | Though phage virions can be fully mature prior to release, it is only free phages which represent a bacterium adsorption-capable phage state |
| Focus of infection | Localized, potentially plaque-like region of phage population growth found in association with a bacterial biofilm | The phage potential to discover new biofilms to exploit likely is function of the number of virions produced, and then disseminated, per individual focus of infection |
| High-multiplicity lysogeny decisions (HMLDs) | Coinfection-associated lysis delay by a temperate phage infection | Lysis delay is achieved with HMLDs by biasing lytic-lysogeny decisions towards lysogeny; an example of phage-associated intercellularly mediated communication |
| Homoimmune | Possessing the same temperate phage immunity type | Superinfection immunity is imposed upon homoimmune secondarily infecting phages; note that neither homoimmunity nor superinfection immunity are associated with phage T4 gene imm |
| Imm (imm) | Immunity | Phage T4 protein, as encoded by gene imm, that is associated with phage expression of superinfection exclusion, resistance to lysis from without (LO; though is a lesser component of resistance to LO than protein Sp), resistance to secondary traumatization (ST), and also resistance to a premature lysis-inhibition (LIN) collapse |
| Induction (prophage) | Conversion of a latent (lysogenic) infection into a productive infection | Canonically, i.e., as with phage lambda, this prophage induction is associated with bacterial-host DNA damage and resulting SOS response |
| Lysis acceleration | Occurrence of sooner phage-induced bacterial lysis | As associated with (i) temperate-phage display of lytic rather than lysogenic cycles, (ii) premature termination of lytic cycles (re: premature LIN collapse or synchronized LIN collapse), or (iii) prophage induction during lysogenic cycles |
| Lysis delay | Later lysis; longer phage infection (latent) period, including as achieved by lysogenic cycles | As associated with (i) delayed termination of lytic cycles such as seen with lysis inhibition (LIN) or unsynchronized LIN collapse, (ii) decisions to display lysogenic cycles during lytic-lysogeny decisions, or (iii) ongoing display of lysogenic cycles rather than prophage induction |
| Lysis from within (LI) | Phage-induced bacterial lysis occurring at the end of phage lytic infections as stimulated intracellularly | With T4 phages, LI is associated, at a minimum, with genes e (endolysin) and t (holin); contrast LI with lysis from without (LO); see also unsynchronized lysis-inhibition (LIN) collapse; LI is a possible mechanism (mechanism 1) underlying at least certain aspects of lysis-inhibition (LIN) collapse |
| Lysis from without (LO) | Phage-induced bacterial lysis that is dependent especially on multiple phage adsorptions, thus as stimulated extracellularly | LO technically is not dependent on phage infection of a bacterium; contrast lysis from within (LI); LO is a possible mechanism (mechanism 3) underlying at least certain aspects of lysis-inhibition (LIN) collapse, particularly synchronized lysis-inhibition (LIN) collapse |
| Lysis inhibition (LIN) | Multiple virion-adsorption- (secondary adsorption-) associated, inducible lytic cycle lysis delay | Lysis inhibition results in an extended primary infection lytic cycle and resulting increase in infection burst size; LIN is an example of phage-associated intercellularly mediated communication |
| Lysis-inhibition (LIN) collapse | Lysis of lysis-inhibited phage infections (see possible mechanistic underpinnings, 1 through 4, immediately below) | LIN collapse does not imply substantial synchronization of lysis across a LINed culture nor necessarily a lack of lysis synchronization (unsynchronized LIN collapse); synchronized LIN collapse is a possible example of phage-associated intercellularly mediated communication |
| Lysis-inhibition collapse, proposed mechanism 1 | As associated especially with lysis from within (LI) | Reversal of R-protein associated inhibition of T-hole formation |
| Lysis-inhibition collapse, proposed mechanism 2 | As associated especially with membrane deterioration (MD) | Spontaneous loss of plasma membrane stability as potentially leading to lysis from within (LI) |
| Lysis-inhibition collapse, proposed mechanism 3 | As associated especially with lysis from without (LO) | Secondary adsorption-associated loss of cell-wall stability as potentially leading to LI |
| Lysis-inhibition collapse, proposed mechanism 4 | As associated especially with secondary traumatization (ST) | Secondary adsorption-associated loss of plasma membrane stability as potentially leading to LI |
| Lysogenic cycle | Non virion-productive, but otherwise phage-genome replicative temperate phage latent infection | During lysogenic cycles phages exist as prophages and do not produce virion progeny; both the occurrence and extensions of lysogenic cycles constitute lysis delays |
| Lytic-lysogeny decision | Choice that must be made at the start of temperate phage infections | Depending on conditions, this choice may be biased either towards or away from display of lysogenic cycles (as representing delayed lysis), though lytic cycles (representing accelerated lysis) tend to be the default decisions |
| Lytic cycle | Productive phage infection which ends in infection lysis | During lytic cycles, phages are committed to producing phage virions and, if successful, then infected host bacteria do not survive; extensions of the duration of lytic cycles represent lysis delays, whereas earlier lysis represents lysis acceleration |
| Membrane deterioration (MD) | Nonspecific spontaneous deterioration of plasma membranes as potentially inducing lysis from within (LI) | A mechanism (mechanism 2) potentially underlying certain aspects of lysis-inhibition (LIN) collapse, particularly unsynchronized LIN collapse |
| Premature lysis-inhibition (LIN) collapse | Earlier than expected lysis of LINed culture (accelerated lysis) | Such as might be caused by excessive lysis from without- (LO-) like secondary adsorption-associated damage to otherwise lysis-inhibited (LINed) bacteria infected with sp or imm mutant phages |
| Primary infection or phage | Infection of a cell by only a single phage or referring to the first phage to reach and infect a cell | Primary infections may display superinfection exclusion or superinfection immunity against secondarily adsorbing phages; it is primary infections that both encode and display lysis inhibition (LIN) |
| Productive infection | Phage infection in which virion progeny are both produced and released | Both rapid lysis lytic cycles and lysis-inhibited (LINed) infections are productive infections, while lysogenic cycles by definition are not virion productive |
| Prophage | Temperate phage genome as observed during lysogenic cycles | Prophages are generated following lytic-lysogeny decisions (given a lysogeny decision) and are lost given prophage induction |
| Prophage induction | Productive termination of a lysogenic cycle | This can be viewed as lysis acceleration as observed in a context of a temperate phage lysogenic cycle; see also induction (prophage) |
| Rapid lysis | Constitutively non-lysis inhibited latent period | The phenotype associated with a genetic inability to display lysis inhibition (LIN) but an ability to still display lytic cycles is described as rapid lysis |
| R (r) | Rapid lysis, as mutated in rapid-lysis (r) phage mutants | Products of rapid-lysis (r) genes include the RI, RIIA, RIIB, and RIII proteins, as required for lysis-inhibition (LIN) expression |
| Resistance to lysis from without | Phage-encoded minimization of cell wall damage caused by phage secondary adsorption | In T4 phages this resistance is associated with gene imm and especially with gene sp |
| Restrict | The killing of a phage upon its adsorption or infection of a bacterium | Phage restriction is mediated by superinfection exclusion as well as by superinfection immunity |
| Secondary adsorption (SA) | Attachment of a virion to an already phage-infected cell | Typically described as superinfection, but secondary adsorption as a term is used here instead to avoid implying that secondary infection necessarily always occurs following secondary adsorption |
| Secondary infection | Infection by a virion of an already phage-infected cell | Infection here is defined as successful phage genome entry into an adsorbed cell’s cytoplasm; superinfection exclusion specifically blocks the initiation of secondary infections by secondarily adsorbing virions |
| Secondary traumatization (ST) | Death of T4-infected bacteria due to excessive secondary adsorption but not as due to lysis from without | Not strictly associated with phage-infection lysis and thereby not strictly equivalent to lysis from without (LO); ST is a possible mechanism (mechanism 4) underlying at least certain aspects of lysis-inhibition (LIN) collapse, particularly synchronized lysis-inhibition (LIN) collapse |
| Sp (sp) | Spackle | Phage T4 protein, as encoded by gene sp, associated with expressing both superinfection exclusion and resistance to lysis from without (LO); likely also associated with expression of a resistance to premature lysis-inhibition (LIN) collapse |
| Strictly lytic | Description of a lytic phage which is unable to display lysogenic cycles | Also known as obligately lytic, professionally lytic, or virulent; contrast with temperate phage |
| Superinfection | Virion infection of an already phage-infected cell | Often in the literature superinfection is not rigorously distinguished from simply secondary adsorption |
| Superinfection exclusion | Block on phage infection, but not on phage adsorption | Superinfection exclusion is imposed post virion attachment but prior to successful phage DNA translocation into the bacterial cytoplasm; it is a form of phage restriction; contrast with superinfection immunity |
| Superinfection immunity | Block on phage infection which occurs post successful phage DNA translocation into the bacterial cytoplasm | Superinfection immunity particularly is as associated with superinfection, by temperate phages, of homoimmune phage lysogens, and is a form of phage restriction; contrast with superinfection exclusion |
| Synchronized lysis-inhibition (LIN) collapse | Multiple virion secondary adsorption-associated, coerced, accelerated LIN collapse | As resulting in faster-than-may-otherwise-be-expected lysis of a lysis-inhibited (LINed) culture once LIN collapse has begun; see lysis-inhibition (LIN) collapse proposed mechanisms 3 and 4; contrast with unsynchronized lysis-inhibition (LIN) collapse |
| T (t) | Tithonus (of Greek mythology, who was to become immortally old, as so too, arguably, do the infections of never-lysing gene t knock-out mutants) | The phage T4 holin protein as encoded by gene t is responsible for controlling the timing of infection lysis as well as allowing otherwise cytoplasmic E protein to access the bacterial cell wall from within, resulting in lysis from within (LI) |
| T-hole | T protein-associated plasma membrane hole | Product of holin activation, resulting in a hole in an infected bacterium’s plasma membrane through which otherwise cytoplasmic E-lysozyme protein can diffuse |
| T-holin | T protein, which is a holin | This construct is used here simply to clarify the function of T protein |
| Temperate phage | Lysogenic cycle-capable bacteriophage | Lytic temperate phages can display both lytic cycles and lysogenic cycles, but not both simultaneously; contrast with phages that are strictly lytic |
| Unsynchronized lysis-inhibition (LIN) collapse | LIN collapse that is not directly associated with a multiple virion secondary adsorption-coerced lysis acceleration | See lysis-inhibition (LIN) collapse proposed mechanisms 1 and 2; contrast with synchronized lysis-inhibition (LIN) collapse |
| Zone of infection | Area associated with a phage plaque that contains either phage virions or phage-infected bacteria but is not necessarily visible to the eye | The zones of infection of wild-type phage T4 plaques may be somewhat larger than the visible clearings associated with these plaques |
| Specific Experimental Results | Phage | Over-Expression from a Plasmid of | Effect | |||
|---|---|---|---|---|---|---|
| gene rI | gene rIII | gene rI.1 | gene rI.-1 | |||
| A | WT | ✓ | Slightly delayed LIN collapse | |||
| A | WT | ✓ | Slightly delayed LIN collapse | |||
| B | WT | ✓ | Greatly delayed LIN collapse | |||
| C | WT | ✓ | Little impact | |||
| D | WT | ✓ | ✓ | Slightly delayed LIN collapse | ||
| E | WT | ✓ | ✓ | Somewhat delayed LIN collapse | ||
| F | rIII | ✓ | Greatly delayed LIN collapse | |||
| G | rIII | ✓ | ✓ | Slightly delayed LIN collapse | ||
| H | rIII | ✓ | ✓ | Less than greatly delayed LIN collapse | ||
| I | none | ✓ | Toxicity to bacteria | |||
| J | none | ✓ | ✓ | Absence of toxicity to bacteria | ||
| K | none | ✓ | ✓ | ✓ | Toxicity to bacteria | |
| L | none | ✓ | Slowed bacterial growth | |||
| M | none | ✓ | ✓ | Absence of toxicity to bacteria | ||
| M | none | ✓ | ✓ | Absence of toxicity to bacteria | ||
| 1 | none | ✓ | Absence of toxicity to bacteria | |||
| Associated with… | Hypothesized LIN Collapse Mechanisms | |||
|---|---|---|---|---|
| 1 (LI) | 2 (MD) | 3 (LO) | 4 (ST) | |
| LIN collapse | Yes | Yes | Yes | Yes |
| Synchronized LIN collapse | No | No | Yes | Yes |
| Unsynchronized LIN collapse | Yes | Yes | No | No |
| E lysozyme | Yes | Yes | No 1 | Yes |
| T holin | Yes | Yes | No 1 | Yes |
| RI antiholin inactivation | Yes | No | No | No |
| Membrane deterioration (MD) 2 | No | Yes | No | Yes |
| Secondary adsorption (SA) 3 | No | No | Yes | Yes |
| Lysis from within (LI) | Yes | Yes 4 | No 1 | Yes 5 |
| Lysis from without (LO) | No | No | Yes | No |
| Secondary traumatization (ST) 6 | No | No | No | Yes 7 |
| Well-established mechanism 8 | Yes | No | Yes | No |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abedon, S.T. Look Who’s Talking: T-Even Phage Lysis Inhibition, the Granddaddy of Virus-Virus Intercellular Communication Research. Viruses 2019, 11, 951. https://doi.org/10.3390/v11100951
Abedon ST. Look Who’s Talking: T-Even Phage Lysis Inhibition, the Granddaddy of Virus-Virus Intercellular Communication Research. Viruses. 2019; 11(10):951. https://doi.org/10.3390/v11100951
Chicago/Turabian StyleAbedon, Stephen T. 2019. "Look Who’s Talking: T-Even Phage Lysis Inhibition, the Granddaddy of Virus-Virus Intercellular Communication Research" Viruses 11, no. 10: 951. https://doi.org/10.3390/v11100951
APA StyleAbedon, S. T. (2019). Look Who’s Talking: T-Even Phage Lysis Inhibition, the Granddaddy of Virus-Virus Intercellular Communication Research. Viruses, 11(10), 951. https://doi.org/10.3390/v11100951