A Quest of Great Importance-Developing a Broad Spectrum Escherichia coli Phage Collection

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Phage Isolation from Environmental Samples
2.3. Plaque Morphology Analysis
2.4. Host Range Determination
2.5. Phage Morphology Assessment
2.6. Phage DNA Isolation, Sequencing and Analysis of the Genomes
2.7. Proteomic Analysis of JK16
3. Results
3.1. Phage Isolation, Plaque Morphology and Host Range Determination
3.2. Phage Particle Morphology
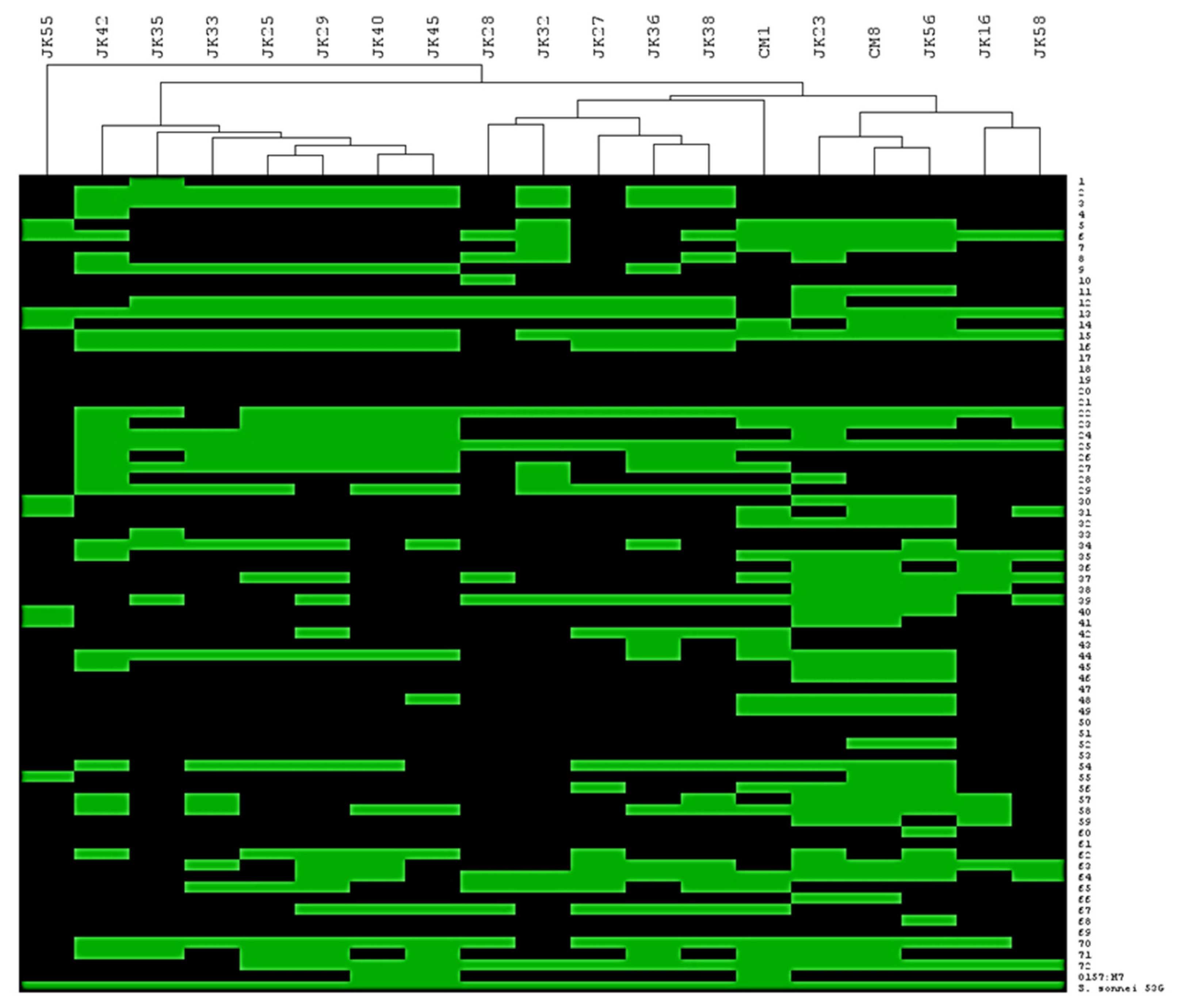
3.3. Identification of Phage Genetic Lineages
3.4. T-Even Phages: T4, RB69 and Pseudo-T-Even (RB49-like) Subgroups
3.5. rV5-Like Subgroup
3.6. Felix O1-Like Subgroup
3.7. Siphophage JK16 Genome and Structural Proteome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Johnson, H.L.; Cousens, S.; Perin, J.; Scott, S.; Lawn, J.E.; Rudan, I.; Campbell, H.; Cibulskis, R.; Li, M.; et al. Global, regional, and national causes of child mortality: An updated systematic analysis for 2010 with time trends since 2000. Lancet 2012, 379, 2151–2161. [Google Scholar] [CrossRef]
- Troeger, C.; Forouzanfar, M.; Rao, P.C.; Khalil, I.; Brown, A.; Reiner, R.C.; Fullman, N.; Thompson, R.L.; Abajobir, A.; Ahmed, M.; et al. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: A systematic analysis for the global burden of disease study 2015. Lancet Infect. Dis. 2017, 17, 909–948. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the global enteric multicenter study, gems): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for the Control of Shigellosis, Including Epidemics Due to Shigella Dysenteriae Type 1; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Sack, R.B.; Rahman, M.; Yunus, M.; Khan, E.H. Antimicrobial resistance in organisms causing diarrheal disease. Clin. Infect. Dis. 1997, 24, S102–S105. [Google Scholar] [CrossRef] [PubMed]
- Shakya, G.; Acharya, J.; Adhikari, S.; Rijal, N. Shigellosis in Nepal: 13 years review of nationwide surveillance. J. Health Popul. Nutr. 2016, 35, 36. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Nagel, T.E.; Chan, B.K.; De Vos, D.; El-Shibiny, A.; Kang’ethe, E.K.; Makumi, A.; Pirnay, J.P. The developing world urgently needs phages to combat pathogenic bacteria. Front. Microbiol. 2016, 7, 882. [Google Scholar] [CrossRef] [PubMed]
- Górski, A.; Borysowski, J.; Miedzybrodzki, R.; Weber-Dabrowska, B. Bacteriophages in Medicine; Caister Academic Press: Norfolk, UK, 2007. [Google Scholar]
- Ross, A.; Ward, S.; Hyman, P. More is better: Selecting for broad host range bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef]
- Kutateladze, M.; Adamia, R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 2010, 28, 591–595. [Google Scholar] [CrossRef]
- Sarker, S.A.; McCallin, S.; Barretto, C.; Berger, B.; Pittet, A.C.; Sultana, S.; Krause, L.; Huq, S.; Bibiloni, R.; Bruttin, A.; et al. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 2012, 434, 222–232. [Google Scholar] [CrossRef]
- Brussow, H. Bacteriophage-host interaction: From splendid isolation into a messy reality. Curr. Opin. Microbiol. 2013, 16, 500–506. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, L.D. Bacteriophages for managing Shigella in various clinical and non-clinical settings. Bacteriophage 2013, 3, e25098. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Brussow, H. Phage Therapy: The Western Perspective; Caister Academic Press: Norfolk, UK, 2007. [Google Scholar]
- Brussow, H. What is needed for phage therapy to become a reality in western medicine? Virology 2012, 434, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Lee, J.H.; Kim, H.; Choi, Y.; Heu, S.; Ryu, S. Receptor diversity and host interaction of bacteriophages infecting Salmonella enterica serovar typhimurium. PLoS ONE 2012, 7, e43392. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Sacher, J.C.; Szymanski, C.M. Development of an assay for the identification of receptor binding proteins from bacteriophages. Viruses 2016, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-J.; Lee, H.; Lee, J.-H.; Ryu, S.; Park, J.-H. Morphological features and lipopolysaccharide attachment of coliphages specific to Escherichia coli O157:H7 and to a broad range of E. coli hosts. Appl. Biol. Chem. 2016, 59, 109–116. [Google Scholar] [CrossRef]
- Breyton, C.; Flayhan, A.; Gabel, F.; Lethier, M.; Durand, G.; Boulanger, P.; Chami, M.; Ebel, C. Assessing the conformational changes of pb5, the receptor-binding protein of phage T5, upon binding to its Escherichia coli receptor FhuA. J. Biol. Chem. 2013, 288, 30763–30772. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Kanamaru, S.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure and morphogenesis of bacteriophage T4. Cell. Mol. Life Sci. CMLS 2003, 60, 2356–2370. [Google Scholar] [CrossRef]
- Jeong, H.; Barbe, V.; Lee, C.H.; Vallenet, D.; Yu, D.S.; Choi, S.H.; Couloux, A.; Lee, S.W.; Yoon, S.H.; Cattolico, L.; et al. Genome sequences of Escherichia coli b strains rel606 and BL21(de3). J. Mol. Biol. 2009, 394, 644–652. [Google Scholar] [CrossRef]
- Blattner, F.R.; Plunkett, G.; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Selander, R.K. Standard reference strains of Escherichia coli from natural populations. Am. Soc. Microbiol. 1984, 157, 690–693. [Google Scholar]
- Rangel, J.M.; Sparling, P.H.; Crowe, C.; Griffin, P.M.; Swerdlow, D.L. Epidemiology of Escherichia coli O157: H7 outbreaks, United States, 1982–2002. Emerg. Infect. Dis. 2005, 11, 603. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Plunkett III, G.; Burland, V.; Mau, B.; Glasner, J.D.; Rose, D.J.; Mayhew, G.F.; Evans, P.S.; Gregor, J.; Kirkpatrick, H.A. Genome sequence of enterohaemorrhagic Escherichia coli O157: H7. Nature 2001, 409, 529. [Google Scholar] [CrossRef] [PubMed]
- Holt, K.E.; Baker, S.; Weill, F.X.; Holmes, E.C.; Kitchen, A.; Yu, J.; Sangal, V.; Brown, D.J.; Coia, J.E.; Kim, D.W.; et al. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nat. Genet. 2012, 44, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Los, J.M.; Golec, P.; Wegrzyn, G.; Wegrzyn, A.; Los, M. Simple method for plating Escherichia coli bacteriophages forming very small plaques or no plaques under standard conditions. Appl. Environ. Microbiol. 2008, 74, 5113–5120. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Casey, E.; Mahony, J.; O’Connell-Motherway, M.; Bottacini, F.; Cornelissen, A.; Neve, H.; Heller, K.J.; Noben, J.-P.; Dal Bello, F.; van Sinderen, D. Molecular characterization of three Lactobacillus delbrueckii subsp. bulgaricus phages. Appl. Environ. Microbiol. 2014, 80, 5623–5635. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new hhpred server at its core. J. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. 5500 phages examined in the electron microscope. Arch. Virol. 2007, 152, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Krisch, H.M.; Comeau, A.M. The immense journey of bacteriophage T4—From d’herelle to delbruck and then to Darwin and beyond. Res. Microbiol. 2008, 159, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Desplats, C.; Krisch, H.M. The diversity and evolution of the T4-type bacteriophages. Res. Microbiol. 2003, 154, 259–267. [Google Scholar] [CrossRef]
- Petrov, V.M.; Ratnayaka, S.; Nolan, J.M.; Miller, E.S.; Karam, J.D. Genomes of the T4-related bacteriophages as windows on microbial genome evolution. Virol. J. 2010, 7, 292. [Google Scholar] [CrossRef] [PubMed]
- Bryson, A.L.; Hwang, Y.; Sherrill-Mix, S.; Wu, G.D.; Lewis, J.D.; Black, L.; Clark, T.A.; Bushman, F.D. Covalent modification of bacteriophage T4 DNA inhibits CRISPR-cas9. MBio 2015, 6, e00648. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Ruger, W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed]
- Nolan, J.M.; Petrov, V.; Bertrand, C.; Krisch, H.M.; Karam, J.D. Genetic diversity among five T4-like bacteriophages. Virol. J. 2006, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.B.; Kropinski, A.M.; Ceyssens, P.J.; Ackermann, H.W.; Villegas, A.; Lavigne, R.; Krylov, V.N.; Carvalho, C.M.; Ferreira, E.C.; Azeredo, J. Genomic and proteomic characterization of the broad-host-range Salmonella phage pvp-se1: Creation of a new phage genus. J. Virol. 2011, 85, 11265–11273. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Waddell, T.; Meng, J.; Franklin, K.; Ackermann, H.-W.; Ahmed, R.; Mazzocco, A.; Yates, J.; Lingohr, E.J.; Johnson, R.P. The host-range, genomics and proteomics of Escherichia coli O157: H7 bacteriophage rv5. Virol. J. 2013, 10, 76. [Google Scholar] [CrossRef]
- Garcia-Doval, C.; Caston, J.R.; Luque, D.; Granell, M.; Otero, J.M.; Llamas-Saiz, A.L.; Renouard, M.; Boulanger, P.; van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of the bacteriophage T5 l-shaped tail fibre with and without its intra-molecular chaperone. Viruses 2015, 7, 6424–6440. [Google Scholar] [CrossRef]
- Schulz, E.C.; Dickmanns, A.; Urlaub, H.; Schmitt, A.; Muhlenhoff, M.; Stummeyer, K.; Schwarzer, D.; Gerardy-Schahn, R.; Ficner, R. Crystal structure of an intramolecular chaperone mediating triple-beta-helix folding. Nat. Struct. Mol. Biol. 2010, 17, 210–215. [Google Scholar] [CrossRef]
- Whichard, J.M.; Weigt, L.A.; Borris, D.J.; Li, L.L.; Zhang, Q.; Kapur, V.; Pierson, F.W.; Lingohr, E.J.; She, Y.M.; Kropinski, A.M.; et al. Complete genomic sequence of bacteriophage felix o1. Viruses 2010, 2, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.W.; Kim, J.H.; Shin, S.P.; Han, J.E.; Chai, J.Y.; Park, S.C. Characterization and complete genome sequence of the Shigella bacteriophage psf-1. Res. Microbiol. 2013, 164, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Soffer, N.; Woolston, J.; Li, M.; Das, C.; Sulakvelidze, A. Bacteriophage preparation lytic for Shigella significantly reduces Shigella sonnei contamination in various foods. PLoS ONE 2017, 12, e0175256. [Google Scholar] [CrossRef] [PubMed]
- Mai, V.; Ukhanova, M.; Reinhard, M.K.; Li, M.; Sulakvelidze, A. Bacteriophage administration significantly reduces Shigella colonization and shedding by Shigella-challenged mice without deleterious side effects and distortions in the gut microbiota. Bacteriophage 2015, 5, e1088124. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.W.; Giri, S.S.; Kim, H.J.; Yun, S.K.; Chi, C.; Chai, J.Y.; Lee, B.C.; Park, S.C. Bacteriophage application to control the contaminated water with Shigella. Sci. Rep. 2016, 6, 22636. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.; Martinez, B.; Obeso, J.M.; Rodriguez, A. Bacteriophages and their application in food safety. Lett. Appl. Microbiol. 2008, 47, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Mendes, J.J.; Leandro, C.; Mottola, C.; Barbosa, R.; Silva, F.A.; Oliveira, M.; Vilela, C.L.; Melo-Cristino, J.; Gorski, A.; Pimentel, M.; et al. In vitro design of a novel lytic bacteriophage cocktail with therapeutic potential against organisms causing diabetic foot infections. J. Med. Microbiol. 2014, 63, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Chibani-Chennoufi, S.; Sidoti, J.; Bruttin, A.; Dillmann, M.L.; Kutter, E.; Qadri, F.; Sarker, S.A.; Brussow, H. Isolation of Escherichia coli bacteriophages from the stool of pediatric diarrhea patients in Bangladesh. J. Bacteriol. 2004, 186, 8287–8294. [Google Scholar] [CrossRef] [PubMed]
- Bourdin, G.; Navarro, A.; Sarker, S.A.; Pittet, A.C.; Qadri, F.; Sultana, S.; Cravioto, A.; Talukder, K.A.; Reuteler, G.; Brussow, H. Coverage of diarrhoea-associated Escherichia coli isolates from different origins with two types of phage cocktails. Microb. Biotechnol. 2014, 7, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef]
- Khan Mirzaei, M.; Nilsson, A.S. Isolation of phages for phage therapy: A comparison of spot tests and efficiency of plating analyses for determination of host range and efficacy. PLoS ONE 2015, 10, e0118557. [Google Scholar] [CrossRef] [PubMed]
- Michniewski, S.; Redgwell, T.; Scanlan, D.J.; Millard, A.D. Draft genome sequence of bacteriophage vb_eco_swan01. Genome Announc 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef] [PubMed]
- Rakhuba, D.; Kolomiets, E.; Dey, E.S.; Novik, G. Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Pol. J. Microbiol. 2010, 59, 145–155. [Google Scholar] [PubMed]
- Hudson, H.P.; Lindberg, A.; Stocker, B. Lipopolysaccharide core defects in Salmonella typhimurium mutants which are resistant to Felix o phage but retain smooth character. Microbiology 1978, 109, 97–112. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Source of Isolation | Isolation E. coli Host | Infectivity (% of Infected Strains) | Plaque Morphology and Diameter (mm) |
|---|---|---|---|---|
| JK16 | Cork City stream | DH5α | 22 |  3–4 |
| JK23 | Connemara National Park stream | K12 | 54 |  2–3 |
| JK25 | Glencar Waterfall | DH5α | 32 |  2–3 |
| JK27 | Glencar Waterfall | BL21 | 26 |  0.5–1 |
| JK28 | Glencar Waterfall | BL21 | 20 |  0.5–1 |
| JK29 | Glencar Waterfall | Top10 | 38 |  1–2 |
| JK32 | Glencar Waterfall | XL1 Blue | 26 |  1–2 |
| JK33 | Glencar Waterfall | K12 | 28 |  0.5–1 |
| JK35 | Sewage (Ireland) | DH5α | 27 |  1–2 |
| JK36 | Sewage (Ireland) | Top10 | 35 |  2–3 |
| JK38 | Sewage (Ireland) | BL21 | 34 |  1–2 |
| JK40 | Sewage (Ireland) | XL1 Blue | 34 |  2–3 |
| JK42 | Sewage (Ireland) | DH5α | 38 |  3–4 |
| JK45 | Sewage (Ireland) | DH5α | 34 |  2–3 |
| JK55 | Sewage (Aaalst, Belgium) | DH5α | 14 |  2–3 |
| JK56 | Sewage (Aaalst, Belgium) | DH5α | 51 |  1–2 |
| JK58 | Sewage (Aaalst, Belgium) | K12 | 19 |  2–3 |
| CM1 | Chicken meat | DH5α | 42 |  0.5–1 |
| CM8 | Chicken meat | BL21 | 53 |  0.5–1 |
| Phage | Classification | Tail (nm) | Head (nm) | Baseplate (nm) |
|---|---|---|---|---|
| JK16 | Siphoviridae | tl 151 ± 7 (8) tw 13± 1 (8) | hw 64 ± 1 (8) | - |
| JK23 | Myoviridae | tl 106 ± 3 (10) tw 22 ± 1 (10) | hl 115 ± 3 (10) hw 89 ± 5 (10) | bpw 31 ± 2 (9) bps 15 ± 1 (8) fbf 125 ± 1 (3) |
| JK32 | Myoviridae | tl 107 ± 3 (7) tw 24 ± 2 (7) | hl 114 ± 3 (7) hw 83 ± 2 (7) | bpw 32 ± 2 (6) bps 13 ± 1 (6) fbf N.A. |
| JK36 | Myoviridae | tl 111 ± 1 (7) tw 25 ± 1 (7) | hl 112 ± 4 (7) hw 82 ± 2 (7) | bpw 32 ± 3 (9) bps 13 ± 2 (7) fbf 167 ± 28 (5) |
| JK38 | Myoviridae | tl 107 ± 4 (8) tw 23 ± 1 (6) | hl 115 ± 3 (8) hw 86 ± 5 (8) | bpw 27 ± 2 (8) bps 13 ± 2 (7) fbf N.A. |
| JK42 | Myoviridae | tl 107 ± 3 (10) tw 22 ± 1 (10) | hl 112 ± 3 (10) hw 82 ± 5 (10) | bpw 33 ± 3 (11) bps 14 ± 2 (7) fbf 146 ± 13 (14) |
| JK45 | Myoviridae | tl 109 ± 2 (13) tw 23 ± 1 (13) | hl 117 ± 3 (13) hw 88 ± 2 (13) | bpw 32 ± 2 (11) bps 17 ± 1 (7) fbf 144 ± 18 (18) |
| JK55 | Myoviridae | Damaged particles; N.A. | ||
| CM1 | Myoviridae | tl 107 ± 6 (7) tw 20 ± 2 (7) | hw 85 ± 2 (7) | bpw 28 ± 5 (6) bps N.A. fbf 43 ± 5 (13) |
| CM8 | Myoviridae | tl 108 ± 1(9) tw 22 ± 1 (9) | hl 114 ± 4 (9) hw 84 ± 2 (9) | bpw 33 ± 2 (9) bps 17 ± 1 (9) fbf 126 ± 11 (8) |
| Phage | Accession Number (GenBank) | Group | Genome Size [bp] | Number of Predicted ORFs | Average GC Content [%] |
|---|---|---|---|---|---|
| JK16 | MK962751 | New siphovirus group | 51,854 | 84 | 44.55 |
| JK23 | MK962752 | T4-even | 168,349 | 272 | 35.32 |
| JK32 | MK962753 | Pseudo-T-even | 176,009 | 269 | 40.40 |
| JK36 | MK962754 | RB69-like | 168,893 | 270 | 37.73 |
| JK38 | MK962755 | T4-even | 167,852 | 268 | 35.48 |
| JK42 | MK962756 | RB69-like | 168,306 | 271 | 37.58 |
| JK45 | MK962757 | RB69-like | 170,740 | 273 | 37.64 |
| JK55 | MK962758 | FelixO1-like | 86,219 | 124 | 38.96 |
| CM1 | MK962749 | rV5-like | 139,598 | 217 | 43.54 |
| CM8 | MK962750 | T4-even | 167,247 | 269 | 35.28 |
| ORF | Putative Function | No. of Peptides | No. Identified Amino Acids | Coverage (%) | Predicted Molecular Mass (kda) |
|---|---|---|---|---|---|
| 9 | Phosphodiesterase | 3 | 31 | 7.4 | 47.8 |
| 57 | Portal | 13 | 188 | 43.7 | 48.5 |
| 58 | Head morphogenesis | 2 | 21 | 8.4 | 28.7 |
| 60 | HtjA; preneck appendage | 14 | 144 | 85.2 | 17.9 |
| 61 | Scaffolding protein | 8 | 74 | 28.5 | 28.2 |
| 62 | Major capsid | 16 | 192 | 59.4 | 35.9 |
| 64 | Hypothetical protein | 4 | 41 | 29.5 | 15.9 |
| 65 | Head-tail connector | 4 | 44 | 35.5 | 13.7 |
| 66 | Tail protein | 3 | 27 | 18.4 | 16.4 |
| 67 | Hypothetical protein | 7 | 92 | 41.8 | 15.2 |
| 70 | Tail measure protein | 25 | 305 | 34.2 | 98 |
| 71 | Tail protein | 5 | 57 | 49.1 | 12.9 |
| 72 | Tail tip assembly / minor tail protein | 3 | 41 | 16.2 | 28.4 |
| 74 | Tail assembly protein | 1 | 17 | 8.6 | 20.8 |
| 75 | Tail protein / RBP | 18 | 191 | 16.02 | 132.1 |
| 84 | Tail protein | 20 | 187 | 28.7 | 70.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczorowska, J.; Casey, E.; Neve, H.; Franz, C.M.A.P.; Noben, J.-P.; Lugli, G.A.; Ventura, M.; Sinderen, D.v.; Mahony, J. A Quest of Great Importance-Developing a Broad Spectrum Escherichia coli Phage Collection. Viruses 2019, 11, 899. https://doi.org/10.3390/v11100899
Kaczorowska J, Casey E, Neve H, Franz CMAP, Noben J-P, Lugli GA, Ventura M, Sinderen Dv, Mahony J. A Quest of Great Importance-Developing a Broad Spectrum Escherichia coli Phage Collection. Viruses. 2019; 11(10):899. https://doi.org/10.3390/v11100899
Chicago/Turabian StyleKaczorowska, Joanna, Eoghan Casey, Horst Neve, Charles M.A.P. Franz, Jean-Paul Noben, Gabriele A. Lugli, Marco Ventura, Douwe van Sinderen, and Jennifer Mahony. 2019. "A Quest of Great Importance-Developing a Broad Spectrum Escherichia coli Phage Collection" Viruses 11, no. 10: 899. https://doi.org/10.3390/v11100899
APA StyleKaczorowska, J., Casey, E., Neve, H., Franz, C. M. A. P., Noben, J.-P., Lugli, G. A., Ventura, M., Sinderen, D. v., & Mahony, J. (2019). A Quest of Great Importance-Developing a Broad Spectrum Escherichia coli Phage Collection. Viruses, 11(10), 899. https://doi.org/10.3390/v11100899