Host Range of Bacteriophages Against a World-Wide Collection of Erwinia amylovora Determined Using a Quantitative PCR Assay


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates
2.2. Bacteriophage Propagation
2.3. Probe and Primer Creation
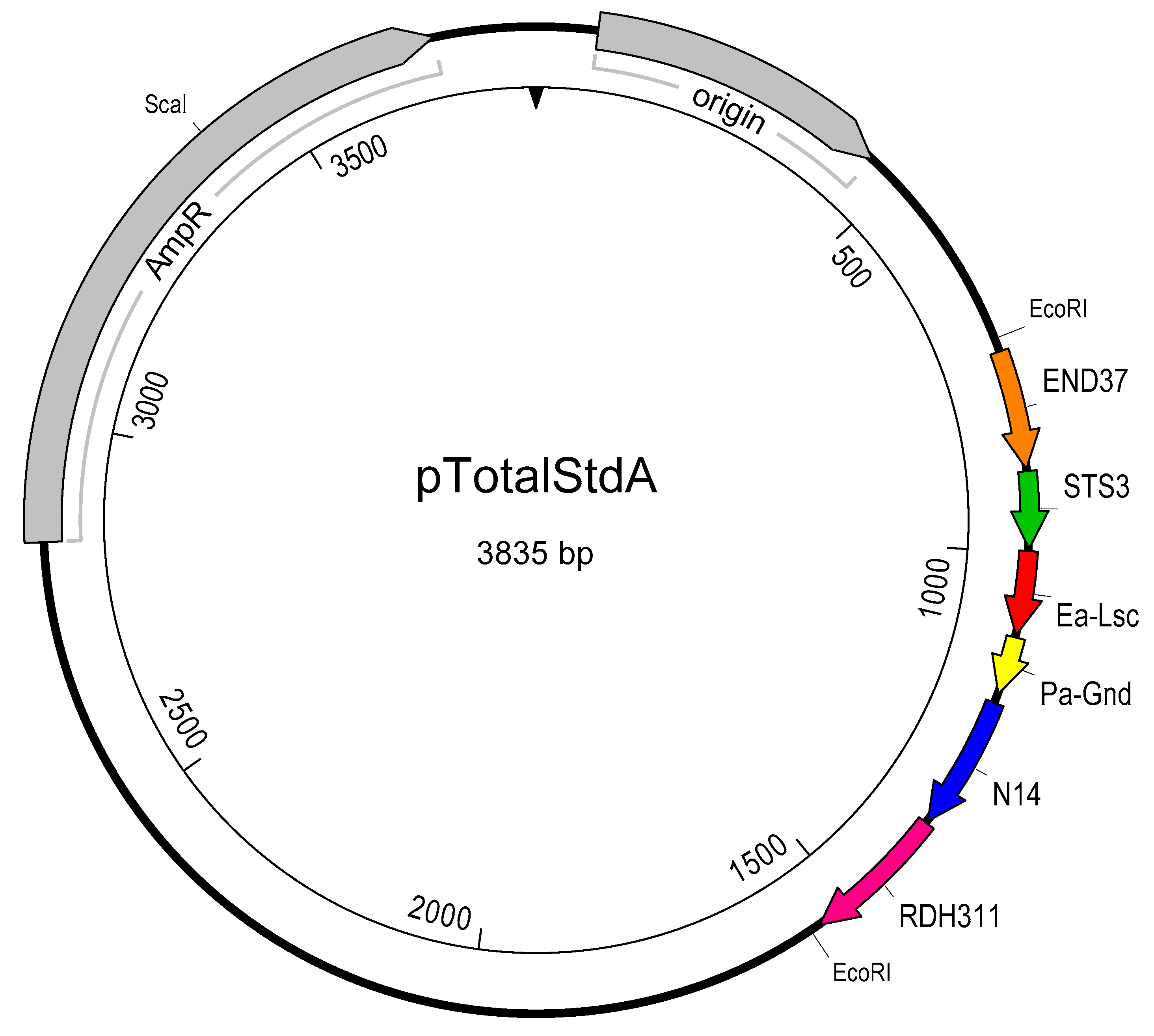
2.4. Creation of pTotalStdA
2.5. Quantitative Real-Time PCR (qPCR)
2.6. Genomic DNA Isolation and Quantification Accuracy of pTotalStdA
2.7. Host Range Assay
2.8. Amylovoran Quantification
2.9. Data Analysis
3. Results
3.1. Standardization of qPCR
3.2. Phage Host Range Assay
3.3. Identifying Determinants of Phage Preference
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vanneste, J.L. Fire Blight: The Disease and Its Causative Agent, Erwinia Amylovora; CABI Publishing: Wallingford, UK, 2000; p. 370. [Google Scholar]
- Van der Zwet, T.; Orolaza-Halbrendt, N.; Zeller, W. Losses due to fire blight and economic importance of the disease. In Fire Blight: History, Biology, and Management; APS Press: St. Paul, MN, USA, 2012; pp. 37–41. [Google Scholar]
- Van der Zwet, T.; Orolaza-Halbrendt, N.; Zeller, W. Early theories and discoveries regarding fire blight. In Fire Blight: History, Biology, and Management; APS Press: St. Paul, MN, USA, 2012; pp. 3–14. [Google Scholar]
- Denning, W. On the decay of apple trees. N. Y. Soc. Prom. Agric. Arts Manuf. Trans. 1794, 2, 219–222. [Google Scholar]
- Eastham, J. The relation between climate and the incidence of some orchard diseases in British Columbia. Pac. Sci. Congr. Proc. 1935, 5, 3229–3232. [Google Scholar]
- Harrison, F.C.; Barlow, B. Some bacterial diseases of plants prevalent in Ontario: Fire blight or twig blight. Ont. Agric. Coll. Bull. 1904, 136, 1–9. [Google Scholar]
- Pierce, N.B. Pear blight in California. Science 1902, 16, 193–194. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Campbell, J. The orchard: The outbreak of fire blight. N. Z. J. Agric. 1920, 20, 181–182. [Google Scholar]
- Cockayne, A. Fire blight and its control: The hawthorn question. N. Z. J. Agric. 1921, 23, 30–36. [Google Scholar]
- Uyeda, E. The causal bacterium of apple blight. Dai-Nippon No-Kaiho 1903, 260, 1–3. [Google Scholar]
- Crosse, J.; Bennett, M.; Garrett, C.M. Fire-blight of pear in England. Nature 1958, 182, 1530. [Google Scholar] [CrossRef]
- El-Helaly, A.F.; Abo-el-Dahab, M.K.; El-Goorani, M.A. The occurrence of the fire blight disease of pear in Egypt. Phytopathol. Mediterr. 1964, 3, 156–163. [Google Scholar]
- Vanneste, J. Distribution maps of plant diseases. In Erwinia Amylovora, 10th ed.; CABI: Wallingford, CT, USA, 2017; Volume 2. [Google Scholar]
- Borecki, Z.; Basak, W.; Zawadzka, B.; Millikan, D. Fire blight in continental Europe. Plant Dis. Rep. 1967, 51, 3. [Google Scholar]
- Grimm, R.; Vogelsanger, J. Erster Nachweis von Feuerbrand in der Schweiz. Schweizerische Z. Obst Weinbau 1989, 125, 514–516. [Google Scholar]
- Meijneke, C. Perevuur en zijn verspreiding. Gewasbescherming 1972, 3, 128–136. [Google Scholar]
- Saad, A.; Hanna, L.; Asly, O.; Choueiri, E. The distribution and host range of the first serious outbreak of fire blight in Lebanon. Acta Hortic. 1999, 489, 65–70. [Google Scholar] [CrossRef]
- Yaich, M.; Fatmi, M.B.; Bougsiba, M.; Valentini, F.; Scuderi, G.; D’Onghia, A.M.; Cirvilleri, G. Fire blight (Erwinia amylovora [Burrill] Winslow) in Morocco: Importance, geographical distribution and characterization. Phytopathol. Mediterr. 2011, 50, 212–227. [Google Scholar]
- Zutra, D.; Shabi, E. First evidence of fire blight in Israel. Phytoparasitica 1985, 13, 151. [Google Scholar]
- Svircev, A.; Roach, D.; Castle, A. Framing the future with bacteriophages in agriculture. Viruses 2018, 10, 218. [Google Scholar] [CrossRef]
- Ngugi, H.K.; Lehman, B.L.; Madden, L.V. Multiple treatment meta-analysis of products evaluated for control of fire blight in the eastern United States. Phytopathology 2011, 101, 512–522. [Google Scholar] [CrossRef]
- Smits, T.H.M.; Duffy, B.; Sundin, G.W.; Zhao, Y.F.; Rezzonico, F. Erwinia amylovora in the genomics era: From genomes to pathogen virulence, regulation, and disease control strategies. J. Plant Pathol. 2017, 99, 7–23. [Google Scholar]
- Stockwell, V.O.; Duffy, B. Use of antibiotics in plant agriculture. OIE Rev. Sci. Tech. 2012, 31, 199–210. [Google Scholar] [CrossRef]
- Förster, H.; McGhee, G.C.; Sundin, G.W.; Adaskaveg, J.E. Characterization of streptomycin resistance in isolates of Erwinia amylovora in California. Phytopathology 2015, 105, 1302–1310. [Google Scholar] [CrossRef]
- McManus, P.S.; Stockwell, V.O.; Sundin, G.W.; Jones, A.L. Antibiotic use in plant agriculture. Ann. Rev. Phytopathol. 2002, 40, 443–465. [Google Scholar] [CrossRef] [PubMed]
- Sholberg, P.; Bedford, K.; Haag, P.; Randall, P. Survey of Erwinia amylovora isolates from British Columbia for resistance to bactericides and virulence on apple. Can. J. Plant Pathol. 2001, 23, 60–67. [Google Scholar] [CrossRef]
- Tancos, K.; Villani, S.; Kuehne, S.; Borejsza-Wysocka, E.; Breth, D.; Carol, J.; Aldwinckle, H.; Cox, K. Prevalence of streptomycin-resistant Erwinia amylovora in New York apple orchards. Plant Dis. 2016, 100, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Doss, J.; Culbertson, K.; Hahn, D.; Camacho, J.; Barekzi, N. A review of phage therapy against bacterial pathogens of aquatic and terrestrial organisms. Viruses 2017, 9, 50. [Google Scholar] [CrossRef]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Gill, J.J.; Svircev, A.M.; Smith, R.; Castle, A.J. Bacteriophages of Erwinia amylovora. Appl. Environ. Microbiol. 2003, 69, 2133–2138. [Google Scholar] [CrossRef]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef]
- Carstens, E. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2009). Arch. Virol. 2010, 155, 133–146. [Google Scholar] [CrossRef]
- Mirzaei, M.K.; Nilsson, A.S. Isolation of phages for phage therapy: A comparison of spot tests and efficiency of plating analyses for determination of host range and efficacy. PLoS ONE 2015, 10, e0118557. [Google Scholar] [CrossRef]
- Roach, D.R.; Sjaarda, D.R.; Castle, A.J.; Svircev, A.M. Host exopolysaccharide quantity and composition impact Erwinia amylovora bacteriophage pathogenesis. Appl. Environ. Microbiol. 2013, 79, 3249–3256. [Google Scholar] [CrossRef]
- Rohde, C.; Resch, G.; Pirnay, J.P.; Blasdel, B.G.; Debarbieux, L.; Gelman, D.; Górski, A.; Hazan, R.; Huys, I.; Kakabadze, E.; et al. Expert opinion on three phage therapy related topics: Bacterial phage resistance, phage training and prophages in bacterial production strains. Viruses 2018, 10, 178. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, P.A.; Nobrega, F.L.; Brouns, S.J.J.; Dutilh, B.E. Molecular and evolutionary determinants of bacteriophage host range. Trends Microbiol. 2019, 27, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.; Ward, S.; Hyman, P. More is better: Selecting for broad host range bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar] [PubMed]
- Abedon, S.T.; Yin, J. Bacteriophage Plaques: Theory and Analysis. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 161–174. [Google Scholar]
- Xie, Y.; Wahab, L.; Gill, J.J. Development and validation of a microtiter plate-based assay for determination of bacteriophage host range and virulence. Viruses 2018, 10, 189. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Duhaime, M.B.; Koutra, D.; Schloss, P.D. Biogeography and environmental conditions shape bacteriophage-bacteria networks across the human microbiome. PLoS Comput. Biol. 2018, 14, e1006099. [Google Scholar] [CrossRef]
- Leite, D.M.C.; Brochet, X.; Resch, G.; Que, Y.-A.; Neves, A.; Peña-Reyes, C. Computational prediction of host-pathogen interactions through omics data analysis and machine learning. In Proceedings of the International Conference on Bioinformatics and Biomedical Engineering, Granada, Spain, 26–28 April 2017; pp. 360–371. [Google Scholar]
- Holmfeldt, K.; Solonenko, N.; Howard-Varona, C.; Moreno, M.; Malmstrom, R.R.; Blow, M.J.; Sullivan, M.B. Large-scale maps of variable infection efficiencies in aquatic Bacteroidetes phage-host model systems. Environ. Microbiol. 2016, 18, 3949–3961. [Google Scholar] [CrossRef]
- Silva, Y.J.; Costa, L.; Pereira, C.; Mateus, C.; Cunha, Â.; Calado, R.; Gomes, N.C.; Pardo, M.A.; Hernandez, I.; Almeida, A. Phage therapy as an approach to prevent Vibrio anguillarum infections in fish larvae production. PLoS ONE 2014, 9, e114197. [Google Scholar] [CrossRef]
- Anderson, B.; Rashid, M.H.; Carter, C.; Pasternack, G.; Rajanna, C.; Revazishvili, T.; Dean, T.; Senecal, A.; Sulakvelidze, A. Enumeration of bacteriophage particles: Comparative analysis of the traditional plaque assay and real-time QPCR- and nanosight-based assays. Bacteriophage 2011, 1, 86–93. [Google Scholar] [CrossRef]
- Del Rio, B.; Martín, M.C.; Martínez, N.; Magadán, A.H.; Alvarez, M.A. Multiplex fast real-time PCR for quantitative detection and identification of cos- and pac-type Streptococcus thermophilus bacteriophages. Appl. Environ. Microbiol. 2008, 74, 4779–4781. [Google Scholar] [CrossRef]
- Dreo, T.; Pirc, M.; Ravnikar, M. Real-time PCR, a method fit for detection and quantification of Erwinia amylovora. Trees Struct. Funct. 2012, 26, 165–178. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ko, G. Using propidium monoazide to distinguish between viable and nonviable bacteria, MS2 and murine norovirus. Lett. Appl. Microbiol. 2012, 55, 182–188. [Google Scholar] [CrossRef] [PubMed]
- McLellan, N.L.; Lee, H.; Habash, M.B. Evaluation of propidium monoazide and long-amplicon qPCR as an infectivity assay for coliphage. J. Virol. Methods 2016, 238, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.K.; Krych, L.; Nielsen, D.S.; Vogensen, F.K. A high-throughput qPCR system for simultaneous quantitative detection of dairy Lactococcus lactis and Leuconostoc bacteriophages. PLoS ONE 2017, 12, e0174223. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Nguyen, A.; Ghosh, D. Quantification of M13 and T7 bacteriophages by TaqMan and SYBR green qPCR. J. Virol. Methods 2018, 252, 100–107. [Google Scholar] [CrossRef]
- Refardt, D. Real-time quantitative PCR to discriminate and quantify lambdoid bacteriophages of Escherichia coli K-12. Bacteriophage 2012, 2, 98–104. [Google Scholar] [CrossRef][Green Version]
- Ritchie, D.F.; Klos, E.J. Isolation of Erwinia amylovora bacteriophage from aerial parts of apple trees. Phytopathology 1977, 67, 101–104. [Google Scholar] [CrossRef]
- Chiou, C.S.; Jones, A.L. The analysis of plasmid-mediated streptomycin resistance in Erwinia amylovora. Phytopathology 1991, 81, 710–714. [Google Scholar] [CrossRef]
- Bauer, D.W.; Beer, S.V. Further characterization of an hrp gene cluster of Erwinia amylovora. Mol. Plant. Microbe Interact. 1991, 4, 493–499. [Google Scholar] [CrossRef]
- Valinsky, L.; Manulis, S.; Nizan, R.; Ezra, D.; Barash, I. A pathogenicity gene isolated from the pPATH plasmid of Erwinia herbicola pv. gypsophilae determines host specificity. Mol. Plant Microbe Interact. 1998, 11, 753–762. [Google Scholar] [CrossRef]
- Manulis, S.; Zutra, D.; Kleitman, F.; Dror, O.; David, I.; Zilberstaine, M.; Shabi, E. Distribution of streptomycin-resistant strains of Erwinia amylovora in Israel and occurrence of blossom blight in the autumn. Phytoparasitica 1998, 26, 223–230. [Google Scholar] [CrossRef]
- Braun-Kiewnick, A.; Altenbach, D.; Oberhansli, T.; Bitterlin, W.; Duffy, B. A rapid lateral-flow immunoassay for phytosanitary detection of Erwinia amylovora and on-site fire blight diagnosis. J. Microbiol. Methods 2011, 87, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jeng, R.S.; Svircev, A.M.; Myers, A.L.; Beliaeva, L.; Hunter, D.M.; Hubbes, M. The use of 16S and 16S–23S rDNA to easily detect and differentiate common Gram-negative orchard epiphytes. J. Microbiol. Methods 2001, 44, 69–77. [Google Scholar] [CrossRef]
- Kim, W.S.; Gardan, L.; Rhim, S.L.; Geider, K. Erwinia pyrifoliae sp. nov., a novel pathogen that affects Asian pear trees (Pyrus pyrifolia Nakai). Int. J. Syst. Bacteriol. 1999, 49, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Hildebrand, M.; Jock, S.; Geider, K. Molecular comparison of pathogenic bacteria from pear trees in Japan and the fire blight pathogen Erwinia amylovora. Microbiology 2001, 147, 2951–2959. [Google Scholar] [CrossRef]
- Lehman, S.M.; Kropinski, A.M.; Castle, A.J.; Svircev, A.M. Complete genome of the broad-host-range Erwinia amylovora phage φEa21-4 and its relationship to Salmonella phage Felix O1. Appl. Environ. Microbiol. 2009, 75, 2139–2147. [Google Scholar] [CrossRef]
- Yagubi, A.I.; Castle, A.J.; Kropinski, A.M.; Banks, T.W.; Svircev, A.M. Complete genome sequence of Erwinia amylovora bacteriophage vB_EamM_Ea35-70. Genome Announc. 2014, 2, e00413–e00414. [Google Scholar] [CrossRef]
- Roach, D.R. Erwinia Amylovora Bacteriophage Resistance; Brock University: St. Catharines, ON, Canada, 2011. [Google Scholar]
- Lehman, S.M. Development of a Bacteriophage-Based Biopesticide for Fire Blight; Brock University: St. Catharines, ON, Canada, 2007. [Google Scholar]
- Santos, M.A. An improved method for the small scale preparation of bacteriophage DNA based on phage precipitation by zinc chloride. Nucleic Acids Res. 1991, 19, 5442. [Google Scholar] [CrossRef]
- Adams, M.H. Bacteriophages; Interscience Publishers, Inc.: New York, NY, USA, 1959. [Google Scholar]
- Bellemann, P.; Bereswill, S.; Berger, S.; Geider, K. Visualization of capsule formation by Erwinia amylovora and assays to determine amylovoran synthesis. Int. J. Biol. Macromol. 1994, 16, 290–296. [Google Scholar] [CrossRef]
- Imamovic, L.; Balleste, E.; Jofre, J.; Muniesa, M. Quantification of Shiga toxin-converting bacteriophages in wastewater and in fecal samples by real-time quantitative PCR. Appl. Environ. Microbiol. 2010, 76, 5693–5701. [Google Scholar] [CrossRef]
- Morella, N.M.; Yang, S.C.; Hernandez, C.A.; Koskella, B. Rapid quantification of bacteriophages and their bacterial hosts in vitro and in vivo using droplet digital PCR. J. Virol. Methods 2018, 259, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Baran, N.; Goldin, S.; Maidanik, I.; Lindell, D. Quantification of diverse virus populations in the environment using the polony method. Nat. Microbiol. 2018, 3, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Payyappat, S.; Cassidy, M.; Besley, C. A duplex PCR assay for the simultaneous quantification of Bacteroides HF183 and crAssphage CPQ_056 marker genes in untreated sewage and stormwater. Environ. Int. 2019, 126, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Lurz, R.; Kube, M.; Quedenau, C.; Jelkmann, W.; Geider, K. Molecular and physiological properties of bacteriophages from North America and Germany affecting the fire blight pathogen Erwinia amylovora. Microbial. Biotechnol. 2011, 4, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Koczan, J.M.; McGrath, M.J.; Zhao, Y.; Sundin, G.W. Contribution of Erwinia amylovora exopolysaccharides amylovoran and levan to biofilm formation: Implications in pathogenicity. Phytopathology 2009, 99, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Jonczyk, E.; Klak, M.; Miedzybrodzki, R.; Gorski, A. The influence of external factors on bacteriophages-review. Folia Microbiol. 2011, 56, 191–200. [Google Scholar] [CrossRef] [PubMed]
- McGhee, G.C.; Sundin, G.W. Erwinia amylovora CRISPR elements provide new tools for evaluating strain diversity and for microbial source tracking. PLoS ONE 2012, 7, e41706. [Google Scholar] [CrossRef]
- Rezzonico, F.; Smits, T.H.M.; Duffy, B. Diversity, evolution, and functionality of clustered regularly interspaced short palindromic repeat (CRISPR) regions in the fire blight pathogen Erwinia amylovora. Appl. Environ. Microbiol. 2011, 77, 3819–3829. [Google Scholar] [CrossRef]
- Zeng, Q.; Cui, Z.; Wang, J.; Childs, K.L.; Sundin, G.W.; Cooley, D.R.; Yang, C.H.; Garofalo, E.; Eaton, A.; Huntley, R.B. Comparative genomics of Spiraeoideae-infecting Erwinia amylovora strains provides novel insight to genetic diversity and identifies the genetic basis of a low-virulence strain. Mol. Plant Pathol. 2018, 19, 1652–1666. [Google Scholar] [CrossRef]
- Power, R.A.; Parkhill, J.; de Oliveira, T. Microbial genome-wide association studies: Lessons from human GWAS. Nat. Rev. Genet. 2017, 18, 41–50. [Google Scholar] [CrossRef]
- Azam, A.H.; Tanji, Y. Bacteriophage-host arm race: An update on the mechanism of phage resistance in bacteria and revenge of the phage with the perspective for phage therapy. Appl. Microbiol. Biotechnol. 2019, 103, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Sjaarda, D.R.; Sjaarda, C.P.; Ayala, C.J.; Howcroft, B.; Castle, A.J.; Svircev, A.M. Absence of lysogeny in wild populations of Erwinia amylovora and Pantoea agglomerans. Microb. Biotechnol. 2015, 8, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Yagubi, A. Phage-Mediated Biological Control of Erwinia Amylovora: The Role of CRISPRs and Exopolysaccharide. Ph.D. Thesis, Brock University, St. Catharines, ON, Canada, 2016. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type Isolate (Synonym) | EPS (OD600) | Host | Isolation Location | Reference | Researcher |
|---|---|---|---|---|---|
| Erwinia amylovora | |||||
| Ea 432 (547) | 0.159 ± 0.006 | Apple | QC, Canada | L. Vézina a | |
| Ea 433 (998) | 0.108 ± 0.015 | Apple | |||
| Ea 434 (1582) | 0.068 ± 0.002 | Apple | |||
| Ea 435 (1585) | 0.183 ± 0.014 | Apple | |||
| Ea 436 (1588) | 0.164 ± 0.028 | Apple | |||
| Ea 438 (1024) | 0.153 ± 0.023 | Raspberry | |||
| C-GG-11 | 0.274 ± 0.005 | Apple | NY, USA | H. Aldwinckle b | |
| Ea 273 (ATCC 49946) | 0.165 ± 0.013 | Apple | |||
| O-TO-1b | 0.090 ± 0.036 | Apple | NY, USA | S.V. Beer c | |
| O-RG-11 | 0.178 ± 0.007 | Apple | |||
| O-RG-21 | 0.234 ± 0.013 | Apple | |||
| P-C-3b | 0.112 ± 0.018 | Apple | |||
| Ea G5 | 0.056 ± 0.002 | Pear | ON, Canada | A.M. Svircev d | |
| Ea 6-4 (E2005A) | 0.055 ± 0.001 | Apple | |||
| Ea 17-1-1 (E2006A) | 0.071 ± 0.023 | Apple | |||
| Ea 29-7 | 0.257 ± 0.032 | Apple | |||
| Ea D7 (E2030A) | 0.249 ± 0.016 | Apple | |||
| Ea 110 (ATCC29780) | 0.203 ± 0.031 | Apple | MI, USA | [53] | A. Jones e |
| BJN | 0.195 ± 0.004 | Apple | G. Sundin e | ||
| CA11 | 0.039 ± 0.002 | Apple | [54] | ||
| GM-1 | 0.196 ± 0.013 | Apple | |||
| 20060013 | 0.044 ± 0.002 | Pear | UT, USA | C.K. Evans f | |
| 20070025 | 0.152 ± 0.011 | Apple | |||
| 20070126 | 0.161 ± 0.020 | Apple | |||
| 20070245 | 0.130 ± 0.007 | Apple | |||
| 20070270 | 0.082 ± 0.010 | Pear | |||
| 20070276 | 0.145 ± 0.005 | Pear | |||
| Ea Apl 1 | 0.241 ± 0.038 | Apple | CA, USA | S. Lindow g | |
| Ea IF | 0.038 ± 0.003 | Apple | |||
| Ea 12 | 0.103 ± 0.004 | Pear | |||
| Ea 75 | 0.111 ± 0.007 | Pear | |||
| Ea 120 | 0.091 ± 0.003 | Pear | |||
| LA469 (Ea 138) | 0.173 ± 0.014 | Apple | OR, USA | V. Stockwell h | |
| LA470 (Ea 144) | 0.198 ± 0.004 | Apple | |||
| LA472 (HR 11) | 0.040 ± 0.001 | Pear | |||
| LA474 | 0.102 ± 0.006 | Asian pear | |||
| LA476 | 0.104 ± 0.019 | Pear | |||
| LA478 | 0.110 ± 0.001 | Pear | |||
| BC Ea 5 | 0.201 ± 0.009 | Apple | BC, Canada | P. Sholberg i | |
| BC Ea 20A | 0.185 ± 0.010 | Crabapple | |||
| BC Ea 23A (1598) | 0.090 ± 0.010 | Pear | [26] | ||
| BC Ea 29 (1611) | 0.079 ± 0.003 | Pear | [26] | ||
| BC Ea 31b (1615) | 0.091 ± 0.016 | Pear | [26] | ||
| BC Ea 1337 (1337) | 0.170 ± 0.008 | Apple | [26] | ||
| Ea 315-1 | 0.191 ± 0.010 | Apple | New Zealand | J. Vanneste j | |
| Ea 412-1 | 0.138 ± 0.011 | Pear | |||
| Ea 501 | 0.233 ± 0.014 | Apple | |||
| FB 29 | 0.114 ± 0.018 | Pear | |||
| FB 78a | 0.162 ± 0.009 | Apple | |||
| FB 97b | 0.123 ± 0.007 | Apple | |||
| 367 | 0.040 ± 0.002 | Firethorn | Poland | P. Sobiczewski k | |
| 464 | 0.225 ± 0.019 | Pear | |||
| 616a | 0.243 ± 0.003 | Apple | |||
| 633a | 0.185 ± 0.014 | Apple | |||
| 650 | 0.123 ± 0.004 | Hawthorn | |||
| 661 | 0.081 ± 0.008 | Rowan | |||
| 214/07 | 0.286 ± 0.011 | Apple | Germany | E. Moltmann l | |
| 224/07 | 0.185 ± 0.026 | Quince | |||
| 234/07 | 0.358 ± 0.037 | Apple | |||
| 245/07 | 0.220 ± 0.030 | Apple | |||
| 294/07 | 0.150 ± 0.024 | Hawthorn | |||
| Ea 321 (ATCC49947, CFBP 1367) | 0.033 ± 0.002 | Hawthorn | France | [55] | J.P. Paulin m |
| CFBP 1987 | 0.068 ± 0.004 | Apple | |||
| CFBP 1990 | 0.121 ± 0.006 | Apple | |||
| CFBP 1995 | 0.043 ± 0.001 | Apple | |||
| CFBP 2305 | 0.064 ± 0.029 | Apple | |||
| CFBP 2313 | 0.126 ± 0.015 | Apple | |||
| Ea 169 | 0.034 ± 0.000 | Pear | Israel | S. Manulis n | |
| Ea 209 | 0.065 ± 0.003 | Pear | [56] | ||
| Ea 238 | 0.040 ± 0.009 | Pear | |||
| Ea 241 | 0.111 ± 0.008 | Pear | [57] | ||
| Ea 249 | 0.057 ± 0.002 | Pear | |||
| Ea 328 | 0.113 ± 0.004 | Pear | |||
| A-11 | 0.037 ± 0.002 | Apple | Lebanon | G. Sundin | |
| A-24 | 0.043 ± 0.002 | Apple | |||
| B-58 | 0.053 ± 0.003 | Apple | |||
| B-65 | 0.045 ± 0.002 | Apple | |||
| B-95 | 0.105 ± 0.008 | Apple | |||
| B-110 | 0.048 ± 0.001 | Apple | |||
| ACW 35260 | 0.051 ± 0.002 | Hawthorn | Switzerland | [58] | B. Duffy o |
| ACW 42107 | 0.264 ± 0.019 | Unknown | |||
| ACW 42287 | 0.086 ± 0.011 | Unknown | |||
| ACW 42288 | 0.149 ± 0.007 | Pear | [58] | ||
| IVIA 1525-1 | 0.091 ± 0.011 | Cotoneaster | Spain | M. Lopez p | |
| IVIA 1626 | 0.099 ± 0.006 | Apple | |||
| IVIA 1777-1 | 0.160 ± 0.014 | Firethorn | |||
| IVIA 1892-1 | 0.065 ± 0.002 | Pear | |||
| IVIA 2303-3 | 0.044 ± 0.004 | Pear | |||
| IVIA 2397-5.4 | 0.098 ± 0.015 | Pear | |||
| CFBP 7130 | 0.097 ± 0.011 | Pear | Morocco | J.P. Paulin | |
| CFBP 7131 | 0.174 ± 0.020 | Pear | |||
| CFBP 7133 | 0.085 ± 0.009 | Pear | |||
| CFBP 7134 | 0.093 ± 0.017 | Pear | |||
| CFBP 7135 | 0.179 ± 0.015 | Pear | |||
| CFBP 7140 | 0.163 ± 0.023 | Pear | |||
| Ea 1-97 | 0.134 ± 0.013 | Raspberry | NS, Canada | [59] | G. Braun q |
| Ea 2-95 | 0.061 ± 0.006 | Raspberry | [59] | ||
| Ea 2-97 | 0.113 ± 0.006 | Raspberry | [59] | ||
| Ea 3-97 | 0.103 ± 0.010 | Raspberry | [59] | ||
| Ea 6-96b | 0.216 ± 0.022 | Raspberry | [59] | ||
| Ea 7-96b | 0.111 ± 0.007 | Raspberry | [59] | ||
| Erwinia pyrifoliae | |||||
| Ep 16/96 | - | Asian Pear | South Korea | [60] | K. Geider r |
| Ep 28/96 | - | Asian Pear | [60] | ||
| Ep 4/97 | - | Asian Pear | [60] | ||
| JapaneseErwinia spp. | |||||
| EJp 556 | - | Asian Pear | Japan | [61] | S. V. Beer |
| EJp 617 | - | Asian Pear | [61] |
| Phage | Detection Primer | Species | Family | Isolation Host | Phage Source | Accession Number a | Reference |
|---|---|---|---|---|---|---|---|
| ɸEa10-1 | END37 | Erwinia virus Ea214 | Myoviridae | Ea 17-1-1 | Apple | [30] | |
| ɸEa10-2 | Ea 6-4 | Apple | [30] | ||||
| ɸEa21-4 | Ea 6-4 | Pear | NC_011811.1 | [30,31,62] | |||
| ɸEa45-1B | Ea 29-7 | Pear | [30] | ||||
| ɸEa35-70 | RDH311 | Erwinia virus Ea35-70 | Ea 29-7 | Pear | NC_023557.1 | [30,31,63] | |
| ɸEa10-7 | STS3 | Erwinia virus Era103 | Podoviridae | Ea 29-7 | Apple | [30] | |
| ɸEa31-3 | Ea 29-7 | Apple | [30] | ||||
| ɸEa46-1-A1 | Ea D7 | Apple | [30] | ||||
| ɸEa46-1-A2 | Ea D7 | Apple | [30] | ||||
| ɸEa9-2 | N14 | Erwinia virus Ea9-2 | Ea 17-1-1 | Pear | NC_023579.1 | [30,31] |
| Name | Species | Amplicon Size (bp) | Sequence (5′-3′) | Reference |
|---|---|---|---|---|
| END37-F | Erwinia virus Ea214 | 149 | TTCAGCTTTAGCGGCTTCGAGA | This study |
| END37-R | AGCAAGCCCTTGAGGTAATGGA | |||
| END37-P | /56-ROXN/AGTCGGTACACCTGCAACGTCAAGAT/3IAbRQSp/ | |||
| STS3-F | Erwinia virus Era103 | 96 | GACAAACAAGAACGCGGCAACTGA | [64] |
| STS3-R | ATACCCAGCAAGGCGTCAACCTTA | |||
| STS3-P | /56-FAM/AGATGAAGTAGGTTATCTTCACAGTGCCCT/3BHQ_1/ | |||
| N14-F | Erwinia virus Ea9-2 | 168 | CATTGGGTAATCCCTTTGAG | This study |
| N14-R | GATAGACTGGTTCCCCTGTG | |||
| N14-P | /56-FAM/TCTGGTGGA/ZEN/CAGAGACGATGTAAT/3IABkFQ/ | |||
| RDH311-F | Erwinia virus Ea35-70 | 183 | TGGAAGGTCTTCTTCGAGAC | This study |
| RDH311-R | GACTACCTGGGGATGTTCAG | |||
| RDH311-P | /56-ROXN/GACGGAAAAGATCACGGTACTCTT/3IAbRQSp/ | |||
| Ea-Lsc-F | E. amylovora | 105 | CGCTAACAGCAGATCGCA | [65] |
| Ea-Lsc-R | AAATACGCGCACGACCAT | |||
| Ea-Lsc-P | /5Cy5/CTGATAATCCGCAATTCCAGGATG/3IAbRQsp/ | |||
| Pa-Gnd-F | P. agglomerans | 73 | TGGATGAAGCAGCGAACA | [65] |
| Pa-Gnd-R | GACAGAGGTTCGCCGAGA | |||
| Pa-Gnd-P | /5HEX/AAATGGACCAGCCAGAGCTCACTG/3BHQ_1/ |
| Primer | R2 | Slope | Efficiency (%) | Ratio a |
|---|---|---|---|---|
| END37 | 1.00 ± 0.00 | −3.22 ± 0.16 | 104.9 ± 6.9 | 3.47 ± 2.01 |
| STS3 | 0.99 ± 0.01 | −3.22 ± 0.12 | 104.6 ± 5.3 | 1.79 ± 0.60 |
| N14 | 0.99 ± 0.01 | −3.24 ± 0.06 | 103.7 ± 2.5 | 5.19 ± 2.42 |
| RDH311 | 0.99 ± 0.01 | −3.38 ± 0.11 | 97.7 ± 4.4 | 0.61 ± 0.25 |
| Ea-Lsc | 1.00 ± 0.00 | −3.49 ± 0.30 | 94.5 ± 11.2 | 1.43 ± 0.60 |
| Pa-Gnd | 0.99 ± 0.00 | −3.31 ± 0.09 | 100.5 ± 4.2 | 1.63 ± 0.23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gayder, S.; Parcey, M.; Castle, A.J.; Svircev, A.M. Host Range of Bacteriophages Against a World-Wide Collection of Erwinia amylovora Determined Using a Quantitative PCR Assay. Viruses 2019, 11, 910. https://doi.org/10.3390/v11100910
Gayder S, Parcey M, Castle AJ, Svircev AM. Host Range of Bacteriophages Against a World-Wide Collection of Erwinia amylovora Determined Using a Quantitative PCR Assay. Viruses. 2019; 11(10):910. https://doi.org/10.3390/v11100910
Chicago/Turabian StyleGayder, Steven, Michael Parcey, Alan J. Castle, and Antonet M. Svircev. 2019. "Host Range of Bacteriophages Against a World-Wide Collection of Erwinia amylovora Determined Using a Quantitative PCR Assay" Viruses 11, no. 10: 910. https://doi.org/10.3390/v11100910
APA StyleGayder, S., Parcey, M., Castle, A. J., & Svircev, A. M. (2019). Host Range of Bacteriophages Against a World-Wide Collection of Erwinia amylovora Determined Using a Quantitative PCR Assay. Viruses, 11(10), 910. https://doi.org/10.3390/v11100910