Differential Pathogenicity of SHIV KB9 and 89.6 Env Correlates with Bystander Apoptosis Induction in CD4+ T cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Plasmid Constructs
2.3. Expression and Shedding of SHIV Envs
2.4. Measurement of Apoptosis Induction
2.5. Pseudotyped Virus Studies
2.6. Immunoblotting
2.7. Statistical Analysis
3. Results
3.1. HIV KB9 is More Potent at InducingBbystander Apoptosis in SupT1 Cell Lines in Coculture Model
3.2. Bystander Apoptosis Induction Mediated by SHIV-KB9 Env is gp41 Dependent
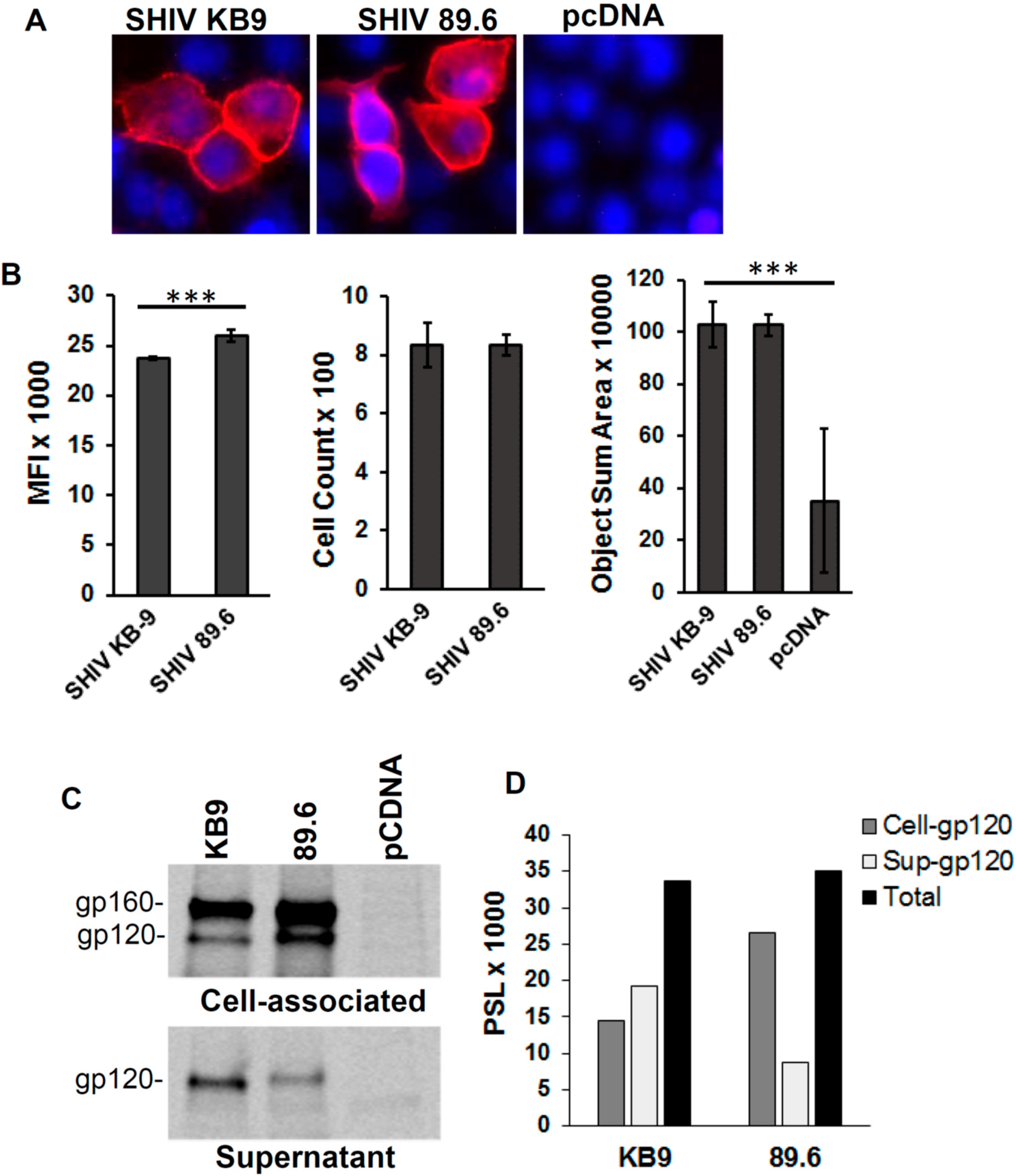
3.3. Differences in Bystander Apoptosis Between the KB9 and 89.6 Envs are Not Due to Differences in Env Expression or Env Shedding
3.4. SHIV 89.6 Prefers CXCR4 Co-Receptor Usage While KB9 Utilizes CXCR4 or CCR5 with Equal Efficacy for Viral Entry
3.5. SHIV-KB9 Env Utilizes both CCR5 and CXCR4 Equally for Apoptosis Induction
3.6. SHIV KB9 Mediated Bystander Apoptosis is Partially Caspase Dependent
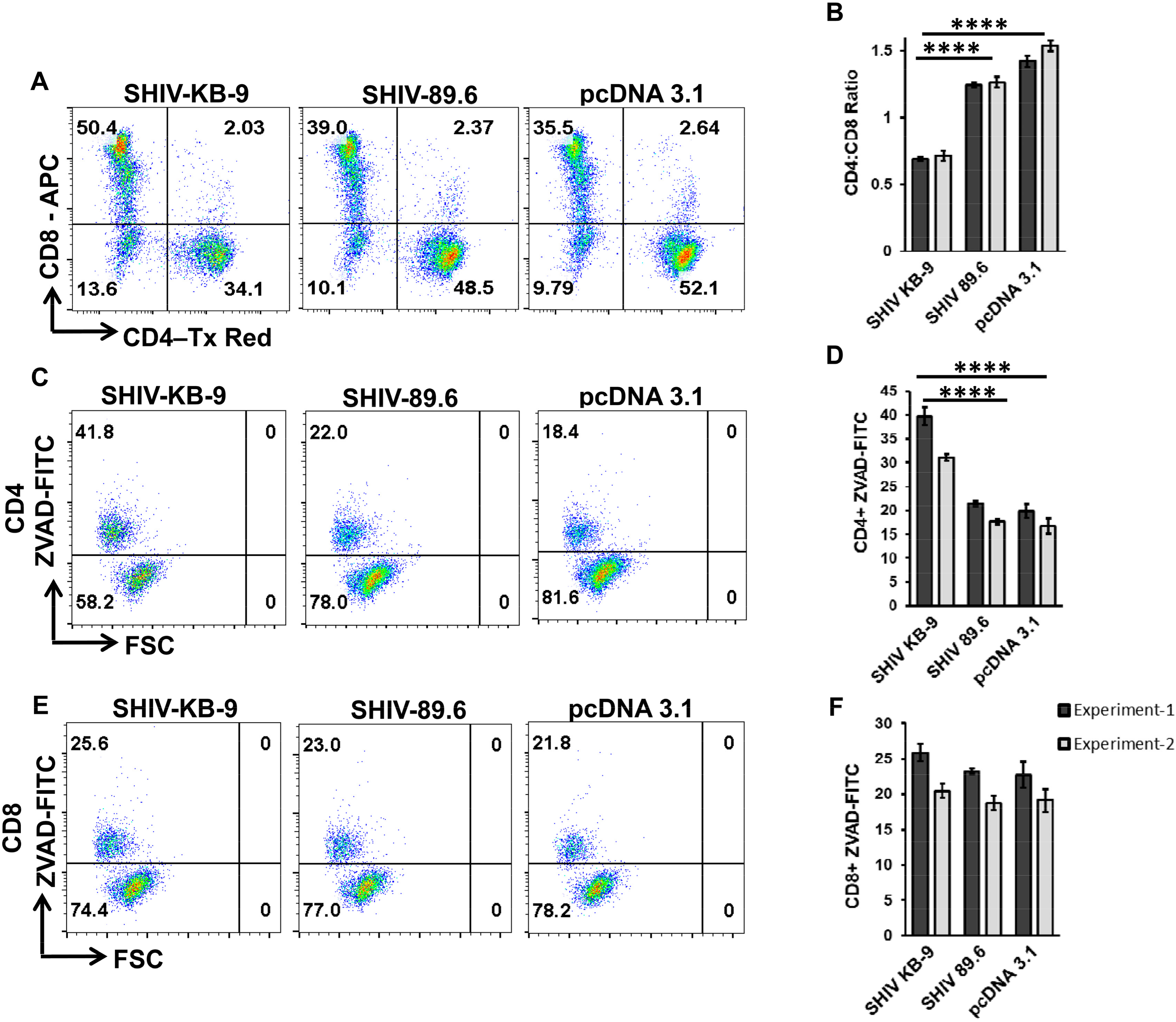
3.7. Coculture of SHIV KB9 Env Expressing Cells with Unstimulated PBMCs Results in a Specific Loss of CD4+ T cells Via Apoptosis
3.8. Coculture of SHIV KB9 Env Expressing Cells with Rhesus PBMCs Also Results in a Specific Loss of CD4 + T cells Via Apoptosis
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Silvestri, G. AIDS pathogenesis: A tale of two monkeys. J. Med. Primatol. 2008, 37, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.; Sodora, D.L.; Koup, R.A.; Paiardini, M.; O’Neil, S.P.; McClure, H.M.; Staprans, S.I.; Feinberg, M.B. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 2003, 18, 441–452. [Google Scholar] [CrossRef]
- Monceaux, V.; Estaquier, J.; Fevrier, M.; Cumont, M.C.; Riviere, Y.; Aubertin, A.M.; Ameisen, J.C.; Hurtrel, B. Extensive apoptosis in lymphoid organs during primary SIV infection predicts rapid progression towards AIDS. AIDS 2003, 17, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Viollet, L.; Monceaux, V.; Petit, F.; Ho Tsong Fang, R.; Cumont, M.C.; Hurtrel, B.; Estaquier, J. Death of CD4+ T cells from lymph nodes during primary SIVmac251 infection predicts the rate of AIDS progression. J. Immunol. 2006, 177, 6685–6694. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.M.; Earl, P.L.; Moss, B.; Reimann, K.A.; Wyand, M.S.; Manson, K.H.; Bilska, M.; Zhou, J.T.; Pauza, C.D.; Parren, P.W.; et al. Characterization of primary isolate-like variants of simian-human immunodeficiency virus. J. Virol. 1999, 73, 10199–10207. [Google Scholar] [PubMed]
- Reimann, K.A.; Li, J.T.; Veazey, R.; Halloran, M.; Park, I.W.; Karlsson, G.B.; Sodroski, J.; Letvin, N.L. A chimeric simian/human immunodeficiency virus expressing a primary patient human immunodeficiency virus type 1 isolate env causes an AIDS-like disease after in vivo passage in rhesus monkeys. J. Virol. 1996, 70, 6922–6928. [Google Scholar] [PubMed]
- Reimann, K.A.; Watson, A.; Dailey, P.J.; Lin, W.; Lord, C.I.; Steenbeke, T.D.; Parker, R.A.; Axthelm, M.K.; Karlsson, G.B. Viral burden and disease progression in rhesus monkeys infected with chimeric simian-human immunodeficiency viruses. Virology 1999, 256, 15–21. [Google Scholar] [CrossRef][Green Version]
- Karlsson, G.B.; Halloran, M.; Li, J.; Park, I.W.; Gomila, R.; Reimann, K.A.; Axthelm, M.K.; Iliff, S.A.; Letvin, N.L.; Sodroski, J. Characterization of molecularly cloned simian-human immunodeficiency viruses causing rapid CD4+ lymphocyte depletion in rhesus monkeys. J. Virol. 1997, 71, 4218–4225. [Google Scholar]
- Etemad-Moghadam, B.; Sun, Y.; Nicholson, E.K.; Fernandes, M.; Liou, K.; Gomila, R.; Lee, J.; Sodroski, J. Envelope glycoprotein determinants of increased fusogenicity in a pathogenic simian-human immunodeficiency virus (SHIV-KB9) passaged in vivo. J. Virol. 2000, 74, 4433–4440. [Google Scholar] [CrossRef]
- Etemad-Moghadam, B.; Rhone, D.; Steenbeke, T.; Sun, Y.; Manola, J.; Gelman, R.; Fanton, J.W.; Racz, P.; Tenner-Racz, K.; Axthelm, M.K.; et al. Membrane-fusing capacity of the human immunodeficiency virus envelope proteins determines the efficiency of CD+ T-cell depletion in macaques infected by a simian-human immunodeficiency virus. J. Virol. 2001, 75, 5646–5655. [Google Scholar] [CrossRef]
- Etemad-Moghadam, B.; Rhone, D.; Steenbeke, T.; Sun, Y.; Manola, J.; Gelman, R.; Fanton, J.W.; Racz, P.; Tenner-Racz, K.; Axthelm, M.K.; et al. Understanding the basis of CD4(+) T-cell depletion in macaques infected by a simian-human immunodeficiency virus. Vaccine 2002, 20, 1934–1937. [Google Scholar] [CrossRef]
- Garg, H.; Mohl, J.; Joshi, A. HIV-1 induced bystander apoptosis. Viruses 2012, 4, 3020. [Google Scholar] [CrossRef]
- Ahr, B.; Robert-Hebmann, V.; Devaux, C.; Biard-Piechaczyk, M. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology 2004, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1995, 1, 129–134. [Google Scholar] [CrossRef]
- Garg, H.; Joshi, A.; Ye, C.; Shankar, P.; Manjunath, N. Single amino acid change in gp41 region of HIV-1 alters bystander apoptosis and CD4 decline in humanized mice. Virol. J. 2011, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Sedano, M.; Beauchamp, B.; Punke, E.B.; Mulla, Z.D.; Meza, A.; Alozie, O.K.; Mukherjee, D.; Garg, H. HIV-1 Env Glycoprotein Phenotype along with Immune Activation Determines CD4 T Cell Loss in HIV Patients. J. Immunol. 2016, 196, 1768–1779. [Google Scholar] [CrossRef]
- Garg, H.; Joshi, A.; Freed, E.O.; Blumenthal, R. Site-specific mutations in HIV-1 gp41 reveal a correlation between HIV-1-mediated bystander apoptosis and fusion/hemifusion. J. Biol. Chem. 2007, 282, 16899–16906. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.; Barretina, J.; Ferri, K.F.; Jacotot, E.; Gutierrez, A.; Armand-Ugon, M.; Cabrera, C.; Kroemer, G.; Clotet, B.; Este, J.A. Cell-surface-expressed HIV-1 envelope induces the death of CD4 T cells during GP41-mediated hemifusion-like events. Virology 2003, 305, 318–329. [Google Scholar] [CrossRef]
- Joshi, A.; Nyakeriga, A.M.; Ravi, R.; Garg, H. HIV ENV glycoprotein-mediated bystander apoptosis depends on expression of the CCR5 co-receptor at the cell surface and ENV fusogenic activity. J. Biol. Chem. 2011, 286, 36404–36413. [Google Scholar] [CrossRef]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar]
- Denizot, M.; Varbanov, M.; Espert, L.; Robert-Hebmann, V.; Sagnier, S.; Garcia, E.; Curriu, M.; Mamoun, R.; Blanco, J.; Biard-Piechaczyk, M. HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 2008, 4, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Blumenthal, R. HIV gp41-induced apoptosis is mediated by caspase-3-dependent mitochondrial depolarization, which is inhibited by HIV protease inhibitor nelfinavir. J.Leukoc. Biol. 2006, 79, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Biard-Piechaczyk, M.; Robert-Hebmann, V.; Richard, V.; Roland, J.; Hipskind, R.A.; Devaux, C. Caspase-dependent apoptosis of cells expressing the chemokine receptor CXCR4 is induced by cell membrane-associated human immunodeficiency virus type 1 envelope glycoprotein (gp120). Virology 2000, 268, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Ohnimus, H.; Heinkelein, M.; Jassoy, C. Apoptotic cell death upon contact of CD4+ T lymphocytes with HIV glycoprotein-expressing cells is mediated by caspases but bypasses CD95 (Fas/Apo-1) and TNF receptor 1. J. Immunol. 1997, 159, 5246–5252. [Google Scholar]
- Joshi, A.; Lee, R.T.; Mohl, J.; Sedano, M.; Khong, W.X.; Ng, O.T.; Maurer-Stroh, S.; Garg, H. Genetic signatures of HIV-1 envelope-mediated bystander apoptosis. J. Biol. Chem. 2014, 289, 2497–2514. [Google Scholar] [CrossRef]
- Jacobs, A.; Garg, H.; Viard, M.; Raviv, Y.; Puri, A.; Blumenthal, R. HIV-1 envelope glycoprotein-mediated fusion and pathogenesis: Implications for therapy and vaccine development. Vaccine 2008, 26, 3026–3035. [Google Scholar] [CrossRef] [PubMed][Green Version]
- LaBonte, J.A.; Patel, T.; Hofmann, W.; Sodroski, J. Importance of membrane fusion mediated by human immunodeficiency virus envelope glycoproteins for lysis of primary CD4-positive T cells. J. Virol. 2000, 74, 10690–10698. [Google Scholar] [CrossRef]
- Karlsson, G.B.; Halloran, M.; Schenten, D.; Lee, J.; Racz, P.; Tenner-Racz, K.; Manola, J.; Gelman, R.; Etemad-Moghadam, B.; Desjardins, E.; et al. The envelope glycoprotein ectodomains determine the efficiency of CD4+ T lymphocyte depletion in simian-human immunodeficiency virus-infected macaques. J. Exp. Med. 1998, 188, 1159–1171. [Google Scholar] [CrossRef]
- Garg, H.; Joshi, A. Host and Viral Factors in HIV-Mediated Bystander Apoptosis. Viruses 2017, 9, 237. [Google Scholar] [CrossRef]
- Koot, M.; Keet, I.P.; Vos, A.H.; de Goede, R.E.; Roos, M.T.; Coutinho, R.A.; Miedema, F.; Schellekens, P.T.; Tersmette, M. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann. Intern. Med. 1993, 118, 681–688. [Google Scholar] [CrossRef]
- Garg, H.; Joshi, A.; Blumenthal, R. Altered bystander apoptosis induction and pathogenesis of enfuvirtide-resistant HIV type 1 Env mutants. AIDS Res. Hum. Retrovir. 2009, 25, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Cunyat, F.; Curriu, M.; Marfil, S.; Garcia, E.; Clotet, B.; Blanco, J.; Cabrera, C. Evaluation of the cytopathicity (fusion/hemifusion) of patient-derived HIV-1 envelope glycoproteins comparing two effector cell lines. J. Biomol. Screen. 2012, 17, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.; Sodroski, J. The HIV-1 envelope glycoproteins: Fusogens, antigens, and immunogens. Science 1998, 280, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.A.; Finnegan, C.M.; Viard, M.; Raviv, Y.; Dimitrov, A.; Rawat, S.S.; Puri, A.; Durell, S.; Blumenthal, R. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 2003, 1614, 36–50. [Google Scholar] [CrossRef]
- Holm, G.H.; Zhang, C.; Gorry, P.R.; Peden, K.; Schols, D.; De Clercq, E.; Gabuzda, D. Apoptosis of bystander T cells induced by human immunodeficiency virus type 1 with increased envelope/receptor affinity and coreceptor binding site exposure. J. Virol. 2004, 78, 4541–4551. [Google Scholar] [CrossRef] [PubMed]
- Sterjovski, J.; Churchill, M.J.; Roche, M.; Ellett, A.; Farrugia, W.; Wesselingh, S.L.; Cunningham, A.L.; Ramsland, P.A.; Gorry, P.R. CD4-binding site alterations in CCR5-using HIV-1 envelopes influencing gp120-CD4 interactions and fusogenicity. Virology 2011, 410, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Sterjovski, J.; Churchill, M.J.; Ellett, A.; Gray, L.R.; Roche, M.J.; Dunfee, R.L.; Purcell, D.F.; Saksena, N.; Wang, B.; Sonza, S.; et al. Asn 362 in gp120 contributes to enhanced fusogenicity by CCR5-restricted HIV-1 envelope glycoprotein variants from patients with AIDS. Retrovirology 2007, 4, 89. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, V.; Zhang, L.; Meissner, E.G.; Jeffrey, J.L.; Su, L. The heptad repeat 2 domain is a major determinant for enhanced human immunodeficiency virus type 1 (HIV-1) fusion and pathogenicity of a highly pathogenic HIV-1 Env. J. Virol. 2009, 83, 11715–11725. [Google Scholar] [CrossRef]
- Wade, J.; Sterjovski, J.; Gray, L.; Roche, M.; Chiavaroli, L.; Ellett, A.; Jakobsen, M.R.; Cowley, D.; Pereira Cda, F.; Saksena, N.; et al. Enhanced CD4+ cellular apoptosis by CCR5-restricted HIV-1 envelope glycoprotein variants from patients with progressive HIV-1 infection. Virology 2010, 396, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Repits, J.; Sterjovski, J.; Badia-Martinez, D.; Mild, M.; Gray, L.; Churchill, M.J.; Purcell, D.F.; Karlsson, A.; Albert, J.; Fenyo, E.M.; et al. Primary HIV-1 R5 isolates from end-stage disease display enhanced viral fitness in parallel with increased gp120 net charge. Virology 2008, 379, 125–134. [Google Scholar] [CrossRef]
- Tsao, L.C.; Guo, H.; Jeffrey, J.; Hoxie, J.A.; Su, L. CCR5 interaction with HIV-1 Env contributes to Env-induced depletion of CD4 T cells in vitro and in vivo. Retrovirology 2016, 13, 22. [Google Scholar] [CrossRef] [PubMed]
- Meissner, E.G.; Zhang, L.; Jiang, S.; Su, L. Fusion-induced apoptosis contributes to thymocyte depletion by a pathogenic human immunodeficiency virus type 1 envelope in the human thymus. J. Virol. 2006, 80, 11019–11030. [Google Scholar] [CrossRef] [PubMed]
- Aquaro, S.; D’Arrigo, R.; Svicher, V.; Perri, G.D.; Caputo, S.L.; Visco-Comandini, U.; Santoro, M.; Bertoli, A.; Mazzotta, F.; Bonora, S.; et al. Specific mutations in HIV-1 gp41 are associated with immunological success in HIV-1-infected patients receiving enfuvirtide treatment. J. Antimicrob. Chemother. 2006, 58, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Hurtrel, B.; Petit, F.; Arnoult, D.; Muller-Trutwin, M.; Silvestri, G.; Estaquier, J. Apoptosis in SIV infection. Cell Death Differ. 2005, 12, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Meythaler, M.; Martinot, A.; Wang, Z.; Pryputniewicz, S.; Kasheta, M.; Ling, B.; Marx, P.A.; O’Neil, S.; Kaur, A. Differential CD4+ T-lymphocyte apoptosis and bystander T-cell activation in rhesus macaques and sooty mangabeys during acute simian immunodeficiency virus infection. J. Virol. 2009, 83, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Cumont, M.C.; Diop, O.; Vaslin, B.; Elbim, C.; Viollet, L.; Monceaux, V.; Lay, S.; Silvestri, G.; Le Grand, R.; Muller-Trutwin, M.; et al. Early divergence in lymphoid tissue apoptosis between pathogenic and nonpathogenic simian immunodeficiency virus infections of nonhuman primates. J. Virol. 2008, 82, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Idziorek, T.; de Bels, F.; Barre-Sinoussi, F.; Hurtrel, B.; Aubertin, A.M.; Venet, A.; Mehtali, M.; Muchmore, E.; Michel, P.; et al. Programmed cell death and AIDS: Significance of T-cell apoptosis in pathogenic and nonpathogenic primate lentiviral infections. Proc. Natl. Acad. Sci. USA 1994, 91, 9431–9435. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Petit, F.; Lelievre, J.D.; Lecossier, D.; Hance, A.; Monceaux, V.; Hurtrel, B.; Ho Tsong Fang, R.; Ameisen, J.C.; Estaquier, J. Caspase-dependent and -independent T-cell death pathways in pathogenic simian immunodeficiency virus infection: Relationship to disease progression. Cell Death Differ. 2003, 10, 1240–1252. [Google Scholar] [CrossRef]
- Laforge, M.; Silvestre, R.; Rodrigues, V.; Garibal, J.; Campillo-Gimenez, L.; Mouhamad, S.; Monceaux, V.; Cumont, M.C.; Rabezanahary, H.; Pruvost, A.; et al. The anti-caspase inhibitor Q-VD-OPH prevents AIDS disease progression in SIV-infected rhesus macaques. J. Clin. Investig. 2018, 128, 1627–1640. [Google Scholar] [CrossRef]
- Matrajt, L.; Younan, P.M.; Kiem, H.P.; Schiffer, J.T. The majority of CD4+ T-cell depletion during acute simian-human immunodeficiency virus SHIV89.6P infection occurs in uninfected cells. J. Virol. 2014, 88, 3202–3212. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmetoglu-Gurbuz, T.; Joshi, A.; Garg, H. Differential Pathogenicity of SHIV KB9 and 89.6 Env Correlates with Bystander Apoptosis Induction in CD4+ T cells. Viruses 2019, 11, 911. https://doi.org/10.3390/v11100911
Mehmetoglu-Gurbuz T, Joshi A, Garg H. Differential Pathogenicity of SHIV KB9 and 89.6 Env Correlates with Bystander Apoptosis Induction in CD4+ T cells. Viruses. 2019; 11(10):911. https://doi.org/10.3390/v11100911
Chicago/Turabian StyleMehmetoglu-Gurbuz, Tugba, Anjali Joshi, and Himanshu Garg. 2019. "Differential Pathogenicity of SHIV KB9 and 89.6 Env Correlates with Bystander Apoptosis Induction in CD4+ T cells" Viruses 11, no. 10: 911. https://doi.org/10.3390/v11100911
APA StyleMehmetoglu-Gurbuz, T., Joshi, A., & Garg, H. (2019). Differential Pathogenicity of SHIV KB9 and 89.6 Env Correlates with Bystander Apoptosis Induction in CD4+ T cells. Viruses, 11(10), 911. https://doi.org/10.3390/v11100911