A New High-Throughput Tool to Screen Mosquito-Borne Viruses in Zika Virus Endemic/Epidemic Areas

 ,
,  , , , , , , add
Show full author list
, , , , , , add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Mosquitoes
2.2. RNA Extraction
2.3. Reverse Transcription and cDNA Pre-Amplification
2.4. Assay Design
2.5. High-Throughput Real-Time PCR
2.6. Validation of the Results by Real-Time PCR, Virus Isolation, and Genome Sequencing
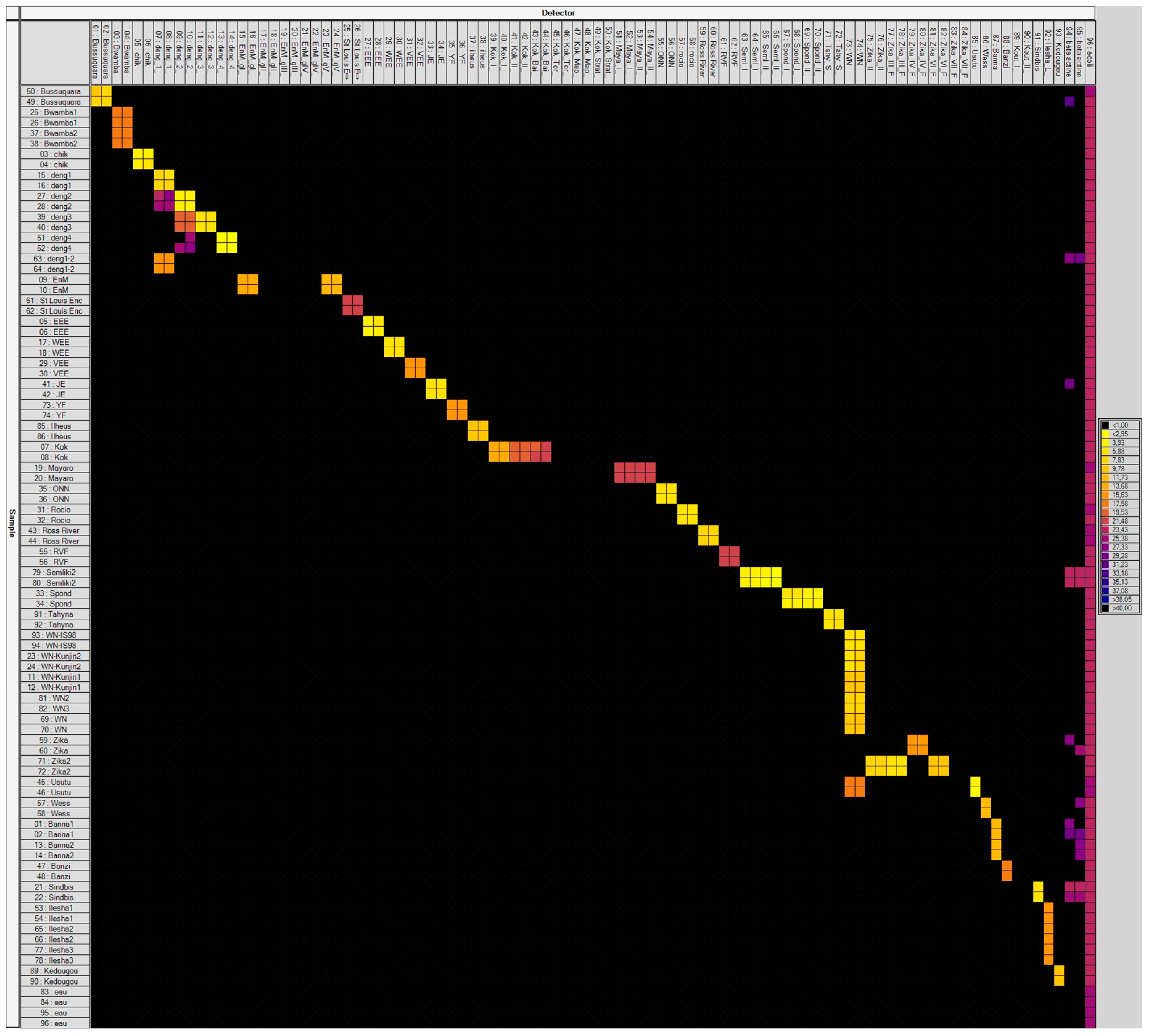
3. Results
3.1. Laboratory-Infected Mosquitoes
3.2. Field-Collected Mosquitoes from Endemic and Epidemic Areas
3.3. Endemic Areas
3.3.1. Senegal
3.3.2. Cambodia
3.4. Endemic/Epidemic area, Brazil
3.5. Epidemic Areas
3.5.1. Guadeloupe
3.5.2. French Guiana
3.5.3. Suriname
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bonaldo, M.C.; Gomez, M.M.; Dos Santos, A.A.; Abreu, F.V.S.; Ferreira-de-Brito, A.; Miranda, R.M.; Castro, M.G.; Lourenco-de-Oliveira, R. Genome analysis of yellow fever virus of the ongoing outbreak in brazil reveals polymorphisms. Memórias Inst. Oswaldo Cruz 2017, 112, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Kramer, L.D.; Ebel, G.D. Dynamics of flavivirus infection in mosquitoes. Adv. Virus Res. 2003, 60, 187–232. [Google Scholar]
- Jupille, H.; Seixas, G.; Mousson, L.; Sousa, C.A.; Failloux, A.B. Zika virus, a new threat for europe? PLoS Negl. Trop. Dis. 2016, 10, e0004901. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Barrett, A.D. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2004, 2, 789–801. [Google Scholar] [CrossRef]
- Weaver, S.C.; Charlier, C.; Vasilakis, N.; Lecuit, M. Zika, chikungunya, and other emerging vector-borne viral diseases. Annu. Rev. Med. 2018, 69, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Dupont-Rouzeyrol, M.; O’Connor, O.; Calvez, E.; Daures, M.; John, M.; Grangeon, J.P.; Gourinat, A.C. Co-infection with zika and dengue viruses in 2 patients, new caledonia, 2014. Emerg. Infect. Dis. 2015, 21, 381–382. [Google Scholar] [CrossRef]
- Villamil-Gomez, W.E.; Gonzalez-Camargo, O.; Rodriguez-Ayubi, J.; Zapata-Serpa, D.; Rodriguez-Morales, A.J. Dengue, chikungunya and zika co-infection in a patient from colombia. J. Infect. Public Health 2016, 9, 684–686. [Google Scholar] [CrossRef]
- Caron, M.; Paupy, C.; Grard, G.; Becquart, P.; Mombo, I.; Nso, B.B.; Kassa Kassa, F.; Nkoghe, D.; Leroy, E.M. Recent introduction and rapid dissemination of chikungunya virus and dengue virus serotype 2 associated with human and mosquito coinfections in gabon, central africa. Clin. Infect. Dis. 2012, 55, e45–e53. [Google Scholar] [CrossRef]
- Vogels, C.B.F.; Ruckert, C.; Cavany, S.M.; Perkins, T.A.; Ebel, G.D.; Grubaugh, N.D. Arbovirus coinfection and co-transmission: A neglected public health concern? PLoS Biol. 2019, 17, e3000130. [Google Scholar] [CrossRef]
- Michelet, L.; Delannoy, S.; Devillers, E.; Umhang, G.; Aspan, A.; Juremalm, M.; Chirico, J.; van der Wal, F.J.; Sprong, H.; Boye Pihl, T.P.; et al. High-throughput screening of tick-borne pathogens in europe. Front. Cell. Infect. Microbiol. 2014, 4, 103. [Google Scholar] [CrossRef]
- Gondard, M.; Michelet, L.; Nisavanh, A.; Devillers, E.; Delannoy, S.; Fach, P.; Aspan, A.; Ullman, K.; Chirico, J.; Hoffmann, B.; et al. Prevalence of tick-borne viruses in ixodes ricinus assessed by high-throughput real-time pcr. Pathog. Dis. 2018, 76, fty083. [Google Scholar] [CrossRef]
- Vazeille-Falcoz, M.; Mousson, L.; Rodhain, F.; Chungue, E.; Failloux, A.-B. Variation in oral susceptibility to dengue type 2 virus of populations of aedes aegypti from the islands of tahiti and moorea, french polynesia. Am. J. Trop. Med. Hyg. 1999, 60, 292–299. [Google Scholar] [CrossRef]
- Nielsen, E.M.; Andersen, M.T. Detection and characterization of verocytotoxin-producing escherichia coli by automated 5’ nuclease pcr assay. J. Clin. Microbiol. 2003, 41, 2884–2893. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef]
- Vaddadi, K.; Gandikota, C.; Jain, P.K.; Prasad, V.S.V.; Venkataramana, M. Co-circulation and co-infections of all dengue virus serotypes in hyderabad, india 2014. Epidemiol. Infect. 2017, 145, 2563–2574. [Google Scholar] [CrossRef]
- Vazeille, M.; Gaborit, P.; Mousson, L.; Girod, R.; Failloux, A.B. Competitive advantage of a dengue 4 virus when co-infecting the mosquito Aedes aegypti with a dengue 1 virus. BMC Infect. Dis. 2016, 16, 318. [Google Scholar] [CrossRef]
- Guzman, M.G.; Harris, E. Dengue. Lancet 2015, 385, 453–465. [Google Scholar] [CrossRef]
- Ciota, A.T.; Kramer, L.D. Vector-virus interactions and transmission dynamics of west nile virus. Viruses 2013, 5, 3021–3047. [Google Scholar] [CrossRef]
- Weissenbock, H.; Kolodziejek, J.; Fragner, K.; Kuhn, R.; Pfeffer, M.; Nowotny, N. Usutu virus activity in austria, 2001–2002. Microbes Infect. 2003, 5, 1132–1136. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on yap island, federated states of micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef]
- Musso, D.; Nilles, E.J.; Cao-Lormeau, V.M. Rapid spread of emerging zika virus in the pacific area. Clin. Microbiol. Infect. 2014, 20, O595–O596. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-barre syndrome outbreak associated with zika virus infection in french polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Cauchemez, S.; Besnard, M.; Bompard, P.; Dub, T.; Guillemette-Artur, P.; Eyrolle-Guignot, D.; Salje, H.; Van Kerkhove, M.D.; Abadie, V.; Garel, C.; et al. Association between zika virus and microcephaly in french polynesia, 2013–15: A retrospective study. Lancet 2016, 387, 2125–2132. [Google Scholar] [CrossRef]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika virus outbreak, bahia, brazil. Emerg. Infect. Dis. 2015, 21, 1885–1886. [Google Scholar] [CrossRef]
- Enfissi, A.; Codrington, J.; Roosblad, J.; Kazanji, M.; Rousset, D. Zika virus genome from the americas. Lancet 2016, 387, 227–228. [Google Scholar] [CrossRef]
- Zanluca, C.; Melo, V.C.; Mosimann, A.L.; Santos, G.I.; Santos, C.N.; Luz, K. First report of autochthonous transmission of zika virus in brazil. Memórias Inst. Oswaldo Cruz 2015, 110, 569–572. [Google Scholar] [CrossRef]
- Roundy, C.M.; Azar, S.R.; Brault, A.C.; Ebel, G.D.; Failloux, A.B.; Fernandez-Salas, I.; Kitron, U.; Kramer, L.D.; Lourenco-de-Oliveira, R.; Osorio, J.E.; et al. Lack of evidence for zika virus transmission by culex mosquitoes. Emerg. Microbes Infect. 2017, 6, e90. [Google Scholar] [CrossRef]
- Shannon, R.C.; Whitman, L.; Franca, M. Yellow fever virus in jungle mosquitoes. Science 1938, 88, 110–111. [Google Scholar] [CrossRef]
- Mascheretti, M.; Tengan, C.H.; Sato, H.K.; Suzuki, A.; de Souza, R.P.; Maeda, M.; Brasil, R.; Pereira, M.; Tubaki, R.M.; Wanderley, D.M.; et al. Yellow fever: Reemerging in the state of sao paulo, brazil, 2009. Rev. Saude Publica 2013, 47, 881–889. [Google Scholar] [CrossRef]
- Abreu, F.V.S.; Ribeiro, I.P.; Ferreira-de-Brito, A.; Santos, A.; Miranda, R.M.; Bonelly, I.S.; Neves, M.; Bersot, M.I.; Santos, T.P.D.; Gomes, M.Q.; et al. Haemagogus leucocelaenus and haemagogus janthinomys are the primary vectors in the major yellow fever outbreak in brazil, 2016–2018. Emerg. Microbes Infect. 2019, 8, 218–231. [Google Scholar] [CrossRef]
- Eklund, C. Trivittatus virus. In International Catalogue of Arboviruses Including Certain Other Virus of Vertebrates; Karabatsos, N., Ed.; American Society of Tropical Medicine and Hygiene: San Antonio, TX, USA, 1985; pp. 1035–1036. [Google Scholar]
- Groseth, A.; Vine, V.; Weisend, C.; Ebihara, H. Complete genome sequence of trivittatus virus. Arch. Virol. 2015, 160, 2637–2639. [Google Scholar] [CrossRef]
- Possas, C.; Martins, R.M.; Oliveira, R.L.; Homma, A. Urgent call for action: Avoiding spread and re-urbanisation of yellow fever in brazil. Memórias Inst. Oswaldo Cruz 2018, 113, 1–2. [Google Scholar] [CrossRef]
- Malmsten, J.; Dalin, A.; Moutailler, S.; Devillers, E.; Gondard, M.; Felton, A. Vector-borne zoonotic pathogens in eurasian moose (alces alces alces). Vector Borne Zoonotic Dis. 2019, 19, 207–211. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Stage Collected | Collection SITE | GPS Coordinates | Urban Rural Sylvatic | Mosquito Species | Number of Arthropods Screened | Virus Detected through Microfluidic System | Type of Confirmation Performed |
|---|---|---|---|---|---|---|---|
| Adult | Baraboye | 12°41′11.2″ N/12°24′39.2″ W | Rural | Ae. furcifer, Ae. dalzieli, Ae. taylori, Ae. vittatus, An. coustani, An. funestus, Ma. uniformis, sandflies | 97 | - | - |
| Ngari | 12°38′07.3″ N/12°14′59.5″ W | Rural | Ae. furcifer, Ae. dalzieli, Ae. aegypti, Ae. argenteopunctatus, Ae. hirsutus, Ae. mcintoshi, Cx. quinquefasciatus, Ae. vittatus, An. coustani, An. funestus, | 96 | - | - | |
| Silling | 12°32′36.5″ N/12°16′18.7″ W | Rural | Ae. luteocephalus, An. gambiae | 3 | - | - | |
| Tenkoto | 12°40′23.1″ N/12°16′37.1″ W | Rural | Ae. furcifer | 5 | - | - | |
| Velingara | 12°27′33.9″ N/12°03′17.3″ W | Rural | Ae. aegypti, Ae. dalzieli, Ae. furcifer, Ae. luteocephalus, Ae. vittatus, An. coustani | 43 | - | - | |
| Kedougou (C1F) | 12°39′42.1″ N/12°16′05.2″ W | Sylvatic | Ae. aegypti, Ae. furcifer, Ae. luteocephalus, Ae. taylori, Ae. unilineatus, Ae. vittatus, An. coustani, An. funestus, An. nili, Cx. perfuscus, Ma. africana, Ma. uniformis | 151 | - | - | |
| Kedougou (D1F) | 12°36′43.9″ N/12°14′50.7″ W | Sylvatic | Ae. aegypti, Ae. africanus, Ae. dalzieli, Ae. furcifer, Ae. luteocephalus, Ae. taylori, Ae. unilineatus, Ae. vittatus, An. coustani, An. funestus, An. nili, Cx. annulioris, Cx. bitaeniorhynchus, Cx. poicilipes, Cx. perfuscus, Ma. africana, Ma. uniformis | 278 | YFV * | Literature | |
| Kedougou (E2F) | 12°29′21.2″ N/12°06′06.2″ W | Sylvatic | Ae. aegypti, Ae. africanus, Ae. argenteopunctatus, Ae. dalzieli, Ae. furcifer, Ae. luteocephalus, Ae. taylori, Ae. unilineatus, Ae. vittatus, An. coustani, An. funestus, Cx. poicilipes, Ma. uniformis | 261 | - | - | |
| Total | 8 | 934 | |||||
| Larvae | Dalaba | 12°33′25.6″ N/12°10′41.0″ W | Rural | Ae. aegypti | 24 | - | - |
| Ngari | 12°38′07.3″ N/12°14′59.5″ W | Rural | Ae. aegypti, Ae. vittatus | 18 | - | - | |
| Kedougou (D1F) | 12°36′43.9″ N/12°14′50.7″ W | Sylvatic | Ae. aegypti, Ae. bromeliae, Ae. furcifer, Ae. longipalpis, Ae. luteocephalus, Ae. taylori, Ae. unilineatus, Ae. vittatus | 237 | - | - | |
| Kedougou (E2F) | 12°29′21.2″ N/12°06′06.2″ W | Sylvatic | Ae. aegypti, Ae. bromeliae, Ae. longipalpis, Ae. luteocephalus, Ae. neoafricanus, Ae. taylori, Ae. unilineatus, Ae. vittatus, Er. chrysogaster | 123 | - | - | |
| Total | 4 | 402 |
| Collection Site | GPS Coordinates | Urban Rural Sylvatic | Mosquito Species | Number of Mosquitoes Screened | Virus Detected through Microfluidic System | Type of Confirmation Performed |
|---|---|---|---|---|---|---|
| Mondulkiri, Cambodia | 12°10′28.4″/106°53′40.9″ | Sylvatic | Ae. aegypti, Ae. albopictus, Ae. gardnerri imitator, Ae. prominens, An. barbirostris, An. indefinitus, An. jamesii, An. kochi, An. mimulus complex, An. philippinensis, An. roperi, An. umbrosus, An. vegus, Ar. annulipalpis, Ar. dolichocephalus, Ar. flavus, Ar. foliatus/kuchingensis, Ar. moultoni, Ar. subalbatus, Ar. theobaldi, Cx. bitaeniorhynchus, Cx. brevipalpis, Cx. fuscocephala, Cx. perplexus/whitei, Cx. sitiens, Cx. vishnui complex, Hz. catesi, Hz. demeilloni | 492 | - | - |
| Collection Site | GPS Coordinates | Urban Rural Sylvatic | Mosquito Species | Number of Mosquitoes Screened | Virus Detected through Microfluidic System | Type of Confirmation Performed |
|---|---|---|---|---|---|---|
| Belo Horizonte | 19°51′59.29″ S/44° 0′43.51″ W | Urban Forest | Ae. albopictus, Ae. aegypti, Cx. quinquefasciatus, Sa. albiprivus | 17 | - | - |
| Casimiro de Abreu | 22°26′33.31″ S/42°12′30.34″ W | Sylvatic | Ae. scapularis | 24 | - | - |
| Domingos Martins | 20°17′12.48″ S/40°50′14.35″ W | Sylvatic | Ae. albopictus | 7 | - | - |
| Goiânia | 6°40′16.32″ S/49°22′49.93″ W | Urban Forest | Aedeomiya, Ae. aegypti, Ae. albopictus, Aedes sp., Coquillettidia sp., Culex sp., Hg. leucocelaenus, Limatus sp., Mansonia sp., Orthopodomyia sp., Psorophora sp., Sabethes sp., Wyeomyia sp. | 689 | - | - |
| Guapimirim | 22°28′56.31″ S/42°59′26.36″ W | Sylvatic | Ru. frontosa | 10 | - | - |
| Macaé | 22°18′17.54″ S/42° 0′8.80″ W | Sylvatic | Ae. scapularis, Wyeomyia sp. | 25 | - | - |
| Manaus | 3°00′12.78″ S/59°55′37.86″ W | Urban Forest | Ae. aegypti, Ae. albopictus, Aedes sp., Culex sp., Hg. leucocelaenus, Limatus sp., Orthopodomyia sp., Psorophora sp., Sabethes sp., Trichoposopum sp., Uranotenia sp., Wyeomyia sp. | 3939 | CHIKV * | Isolation, conventional and real-time PCR failed to confirm the result |
| Maricá | 22°55′24.44″ S/42°42′27.88″ W | Sylvatic | Ae. aegypti, Ae. albopictus, Ae. scapularis, Ae. taeniorhynchus, Culex sp., Cx. nigripalpus, Hg. janthinomys, Hg. leucocelaenus, Li. durhamii, Ru. humboldti | 198 | YFV § | YFV confirmed by PCR |
| Miguel Pereira | 22°29′3.21″ S/43°18′15.98″ W | Sylvatic | Sh. fluviatilis | 12 | - | _ |
| Nova Friburgo | 22°24′46.35″ S/42°18′58.57″ W | Sylvatic | Ru. humboldti | 2 | CHIKV £ | Isolation, conventional and real-time PCR failed to confirm the result |
| Queluz | 22°41′52.33″ S/44°43′43.91″ W | Sylvatic | Wy. pilicauda, Wy. confusa | 6 | - | - |
| Rio de Janeiro | 22°52′45.7″ S/43°18′10.0″ W | Urban | Ae. aegypti, Ae. albopictus, Cx. quinquefasciatus | 261 | - | - |
| 22°56′6.57″ S/43°26′42.19″ W | Urban Forest | Ae. aegypti, Ae. albopictus, Aedes sp., Coquillettidia sp., Culex sp., Hg. leucocelaenus, Mansonia sp., Psorophora sp., Runchomyia sp., Sabethes sp., Trichoposopum sp., Wyeomyia sp. | 2447 | TVTV $ | Isolation, conventional and real-time PCR failed to confirm the result | |
| Serra | 20° 6′46.89″ S/40°11′12.53″ W | Sylvatic | Ae. albopictus, Cx. quinquefasciatus | 26 | - | - |
| Simonésia | 19°55′12.06″ S/41°54′20.23″ W | Sylvatic | Ae. albopictus, Cq. venezuelensis, Hg. janthinomys, Hg. leucocelaenus, Sa. albiprivus | 41 | - | - |
| Teresópolis | 22°26′58.56″ S/42°59′5.43″ W | Sylvatic | Ru. frontosa | 1 | - | - |
| Total 15 | 7705 |
| Collection Site | GPS Coordinates | Urban RuralSylvatic | Mosquito Species | Number of Mosquitoes Screened | Virus Detected through Microfluidic System | Type of Confirmation Performed |
|---|---|---|---|---|---|---|
| Gosier | 16° 12′ 21.229″ N/61° 29′ 31.438″ W | Urban | Ae. aegypti, Cx. quinquefasciatus, An. albimanus | 399 | - | - |
| Deshaies | 16° 18′ 24.973″ N/61° 47′ 39.556″ W | Urban/Periurban | Ae. aegypti, Cx. quinquefasciatus, An. albimanus, Cx. bisulcatus, De. magnus | 306 | ZIKV * | Confirmed by real-time PCR on head-thorax of individual mosquitoes, isolation of the virus, and full genome sequencing |
| Petit Bourg | 16° 11′ 29.476″ N/61° 35′ 25.753″ W | Urban/Periurban | Ae. aegypti, Cx. quinquefasciatus, Cx. nigripalpus, Culex sp. | 422 | ZIKV * | Confirmed by real-time PCR on head-thorax of individual mosquitoes, isolation of the virus, and full genome sequencing |
| Le Moule | 16° 19′ 52.342″ N/61° 20′ 37.41″ W | Urban/Periurban | Ae. aegypti, Cx. quinquefasciatus, Culex sp. | 202 | ZIKV * | Confirmed by real-time PCR on head-thorax of individual mosquitoes, isolation of the virus, and full genome sequencing |
| Saint François | 16° 15′ 5.141″ N/61° 16′ 26.825″ W | Urban | Ae. aegypti, Cx. quinquefasciatus | 356 | ZIKV * | Confirmed by real-time PCR on head-thorax of individual mosquitoes, isolation of the virus, and full genome sequencing |
| Sainte Anne | 16° 13′ 31.613″ N/61° 23′ 9.377″ W | Urban/Periurban | Ae. aegypti, Cx. quinquefasciatus, Cx. nigripalpus, Culex sp. | 325 | ZIKV * | Confirmed by real-time PCR on head-thorax of individual mosquitoes, isolation of the virus, and full genome sequencing |
| Baie Mahault | 16° 16′ 3.979″ N/61° 35′ 13.337″ W | Urban | Ae. aegypti, Cx. quinquefasciatus | 19 | - | - |
| Le Lamentin | 16° 16′ 17.36″ N/61° 37′ 59.754″ W | Urban/Periurban | Ae. aegypti, Cx. quinquefasciatus | 27 | - | - |
| Goyave | 16° 7′ 26.447″ N/61° 34′ 40.253″ W | Urban/Periurban | Ae. aegypti, Cx. quinquefasciatus, Culex sp. | 86 | - | - |
| Morne-à-L′eau | 16° 19′ 53.832″ N/61° 27′ 25.855″ W | Urban | Ae. aegypti, Cx. quinquefasciatus | 19 | - | - |
| Pointe-à-Pître | 16° 14′ 54.499″ N/61° 32′ 18.888″ W | Urban | Ae. aegypti, Cx. quinquefasciatus | 5 | - | - |
| Saint-Claude | 16° 1′ 36.077″ N/61° 42′ 6.703″ W | Urban/Periurban | Ae. aegypti | 1 | - | - |
| Petit Canal | 16° 22′ 49.03″ N/61° 29′ 14.384″ W | Urban/Periurban | Culex sp. | 6 | - | - |
| Total 13 | 2173 |
| Collection Site | GPS Coordinates | Urban Rural Sylvatic | Mosquito Species | Number of Mosquitoes Screened | Virus Detected through Microfluidic System | Type of Confirmation Performed |
|---|---|---|---|---|---|---|
| Cayenne | 4°55′53.08″ N/52°18′55.99″ W | Urban | Ae. aegypti, Ae. scapularis, Cx. quinquefasciatus, Ma. titillans, Cq. venezualensis, Cq. albicosta | 1928 | ZIKV * | Confirmed by real-time PCR on head-thorax of individual mosquitoes, isolation of the virus, and full genome sequencing |
| Remire-Montjoly | 4°53′34.01″ N/52°16′34.32″ W | Urban/Periurban | Ae. aegypti, Ae. scapularis, Cx. quinquefasciatus, Ma. titillans, Cq. venezualensis | 1078 | _ | - |
| Matoury | 4°50′52.22″ N/52°19′41.58″ W | Urban/Periurban | Ae. aegypti, Ae. taeniorhynchus, Cx. quinquefasciatus, Ma. titillans, Cq. venezualensis, Cq. albicosta | 936 | ZIKV * | Confirmed by real-time PCR on head-thorax of individual mosquitoes, isolation of the virus, and full genome sequencing |
| Total 3 | 3942 |
| Collection Site | GPS | Urban Rural Sylvatic | Mosquito Species | Number of Mosquitoes Screened | Virus Detected through Microfluidic System | Type of Confirmation Performed |
|---|---|---|---|---|---|---|
| Paramaribo | 5°51′54.2″ N/55°11′33.4″ W | Urban (Paramaribo) | Undetermined | 4 | - | - |
| Roti shop | 5°51′59.616″ N/55°6′20.952 W | Urban (Paramaribo) | Ae. aegypti | 68 | - | - |
| Car mechanic | 5°50′35.8″ N 55°06′56.7″ W | Urban (Paramaribo) | Ae. aegypti | 96 | - | - |
| Kwikfit car mechanic | 5°50′38.1″ N 55°07′23.3″ W | Urban (Paramaribo) | Ae. aegypti | 567 | - | - |
| Family home | 5°50′33.3″ N 55°07′17.5″ W | Urban (Paramaribo) | Ae. aegypti | 296 | - | - |
| Outpatient clinic | 5°50′30.3″ N/55°7′8.615″ W | Urban (Paramaribo) | Ae. aegypti | 103 | - | - |
| Chi min restaurant | 5°49′54.408″ N/55°8′24.683 W | Urban (Paramaribo) | Ae. aegypti | 78 | - | - |
| Albertine retirement home | 5°48′49.572″ N/55°11′27.6″ W | Urban (Paramaribo) | Ae. aegypti | 661 | - | - |
| Medisch Opvoedkundig Bureau (MOB) | 5°49′43.212″ N/55°10′41.375″ W | Urban (Paramaribo) | Ae. aegypti | 376 | - | - |
| Brownsweg | 5°0′57.384″ N/55°10′2.172″ W | Rural/Sylvatic | Ae. aegypti, Culex sp., Undetermined | 21 | - | - |
| Brownsberg | 4°56′36.24″ N/55°10′6.6″ W | Rural/Sylvatic | Ae. aegypti, Culex sp., Haemagogus sp., Undetermined | 40 | - | - |
| Total 11 | 2310 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moutailler, S.; Yousfi, L.; Mousson, L.; Devillers, E.; Vazeille, M.; Vega-Rúa, A.; Perrin, Y.; Jourdain, F.; Chandre, F.; Cannet, A.; et al. A New High-Throughput Tool to Screen Mosquito-Borne Viruses in Zika Virus Endemic/Epidemic Areas. Viruses 2019, 11, 904. https://doi.org/10.3390/v11100904
Moutailler S, Yousfi L, Mousson L, Devillers E, Vazeille M, Vega-Rúa A, Perrin Y, Jourdain F, Chandre F, Cannet A, et al. A New High-Throughput Tool to Screen Mosquito-Borne Viruses in Zika Virus Endemic/Epidemic Areas. Viruses. 2019; 11(10):904. https://doi.org/10.3390/v11100904
Chicago/Turabian StyleMoutailler, Sara, Lena Yousfi, Laurence Mousson, Elodie Devillers, Marie Vazeille, Anubis Vega-Rúa, Yvon Perrin, Frédéric Jourdain, Fabrice Chandre, Arnaud Cannet, and et al. 2019. "A New High-Throughput Tool to Screen Mosquito-Borne Viruses in Zika Virus Endemic/Epidemic Areas" Viruses 11, no. 10: 904. https://doi.org/10.3390/v11100904
APA StyleMoutailler, S., Yousfi, L., Mousson, L., Devillers, E., Vazeille, M., Vega-Rúa, A., Perrin, Y., Jourdain, F., Chandre, F., Cannet, A., Chantilly, S., Restrepo, J., Guidez, A., Dusfour, I., Vieira Santos de Abreu, F., Pereira dos Santos, T., Jiolle, D., Visser, T. M., Koenraadt, C. J. M., ... Failloux, A.-B. (2019). A New High-Throughput Tool to Screen Mosquito-Borne Viruses in Zika Virus Endemic/Epidemic Areas. Viruses, 11(10), 904. https://doi.org/10.3390/v11100904