The Diverse Roles of microRNAs at the Host–Virus Interface


Abstract
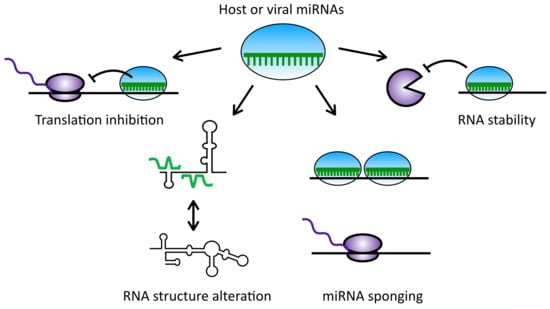
1. Introduction
2. Herpesviruses
2.1. Cellular miRNAs in Herpesvirus Infection
2.1.1. MiRNAs Implicated in Latency Maintenance
2.1.2. MiRNAs Implicated in Immune Evasion
2.1.3. MiRNAs Implicated in Cell Cycle Control and Tumorigenesis
2.2. Herpesvirus-Encoded miRNAs
2.2.1. Viral miRNAs with Cellular Targets
2.2.2. Viral miRNAs Regulating Viral Transcripts
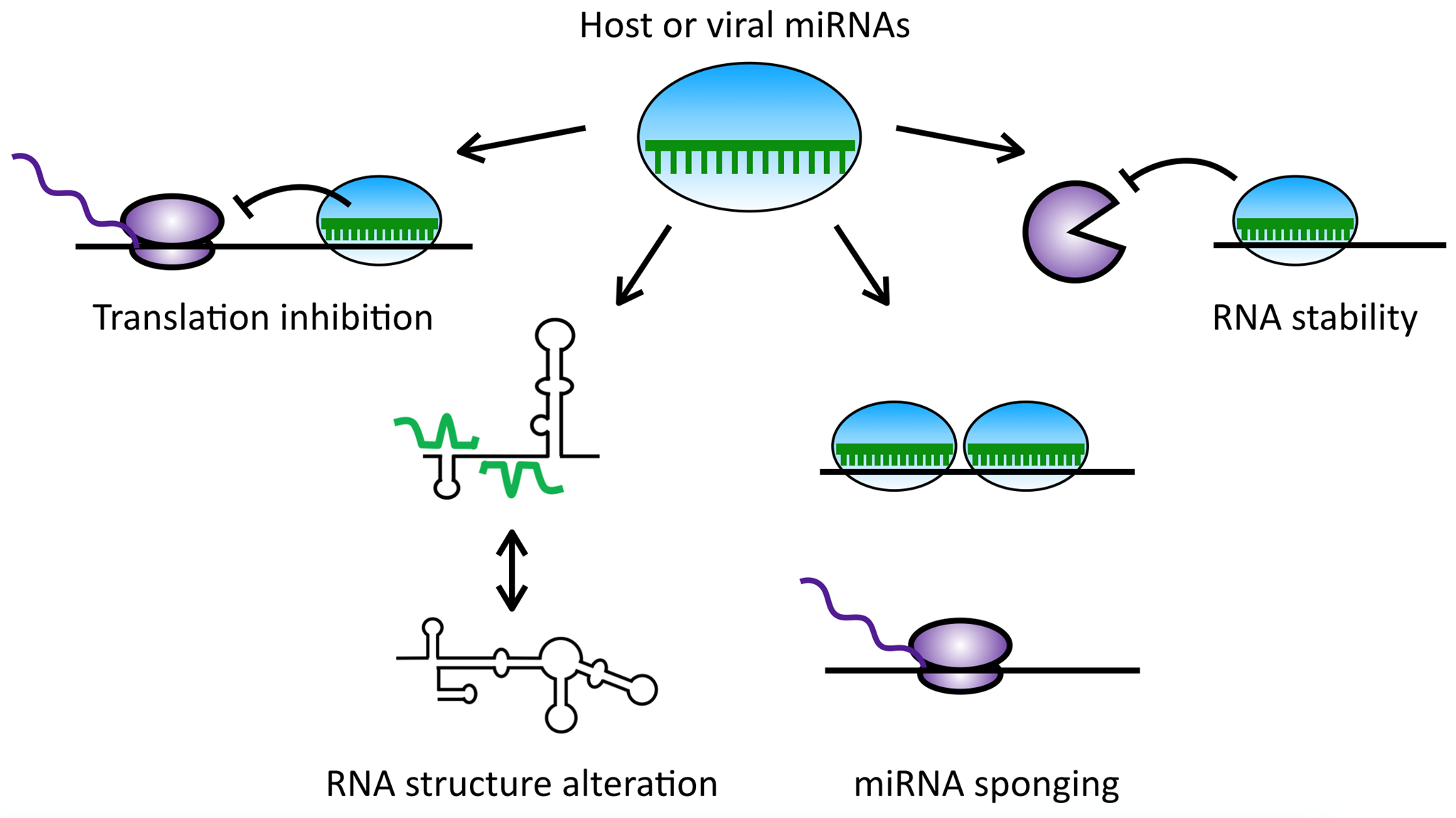
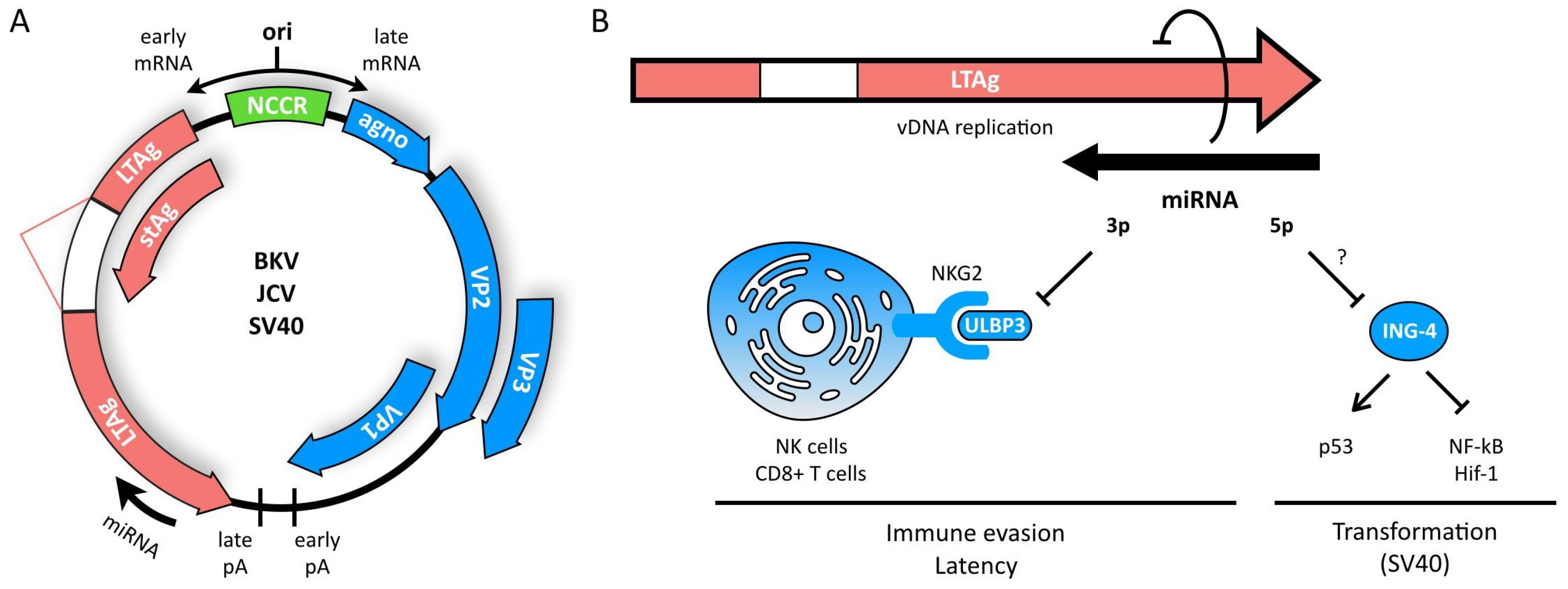
3. Polyomaviruses
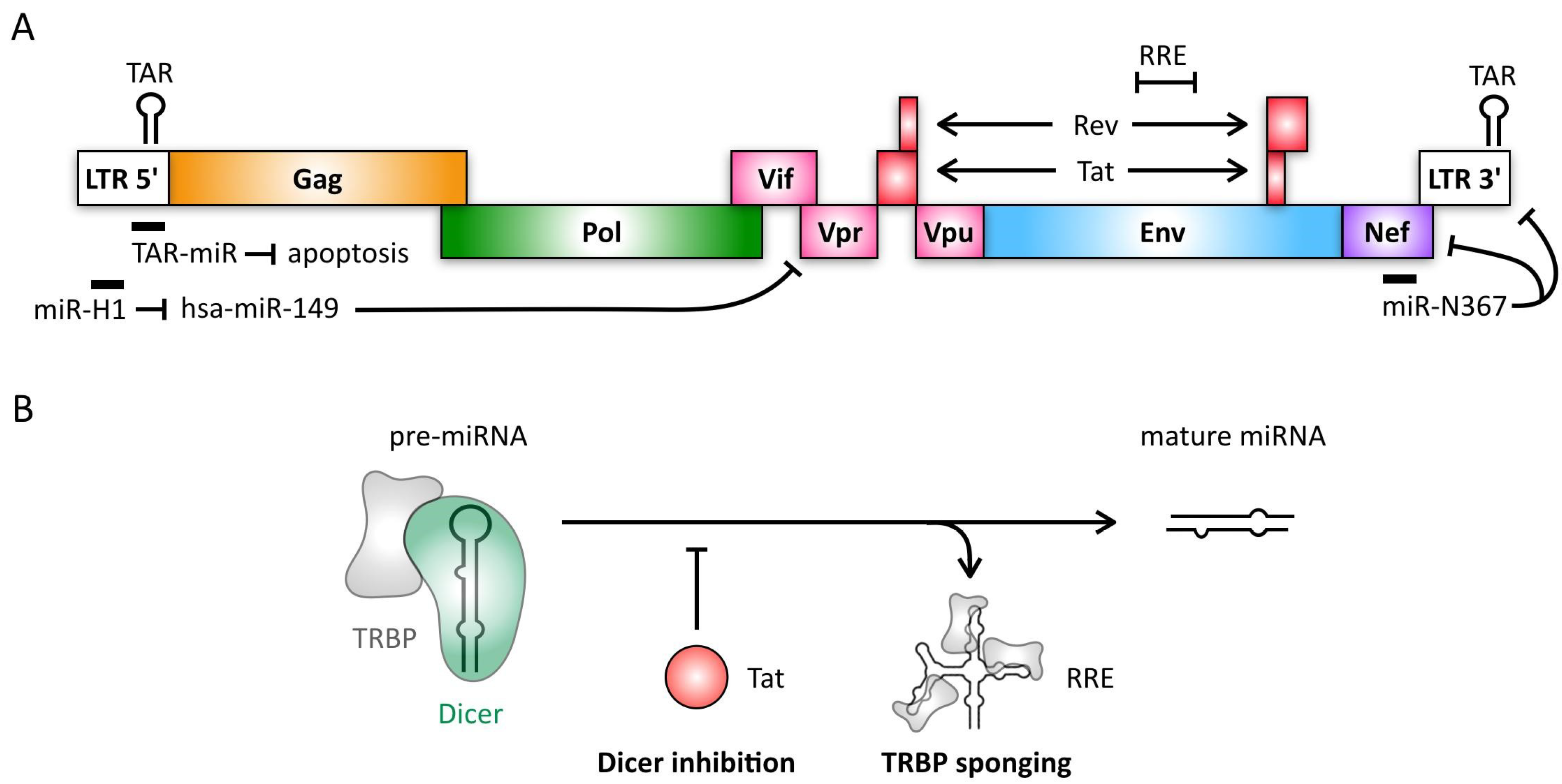
4. Retroviruses
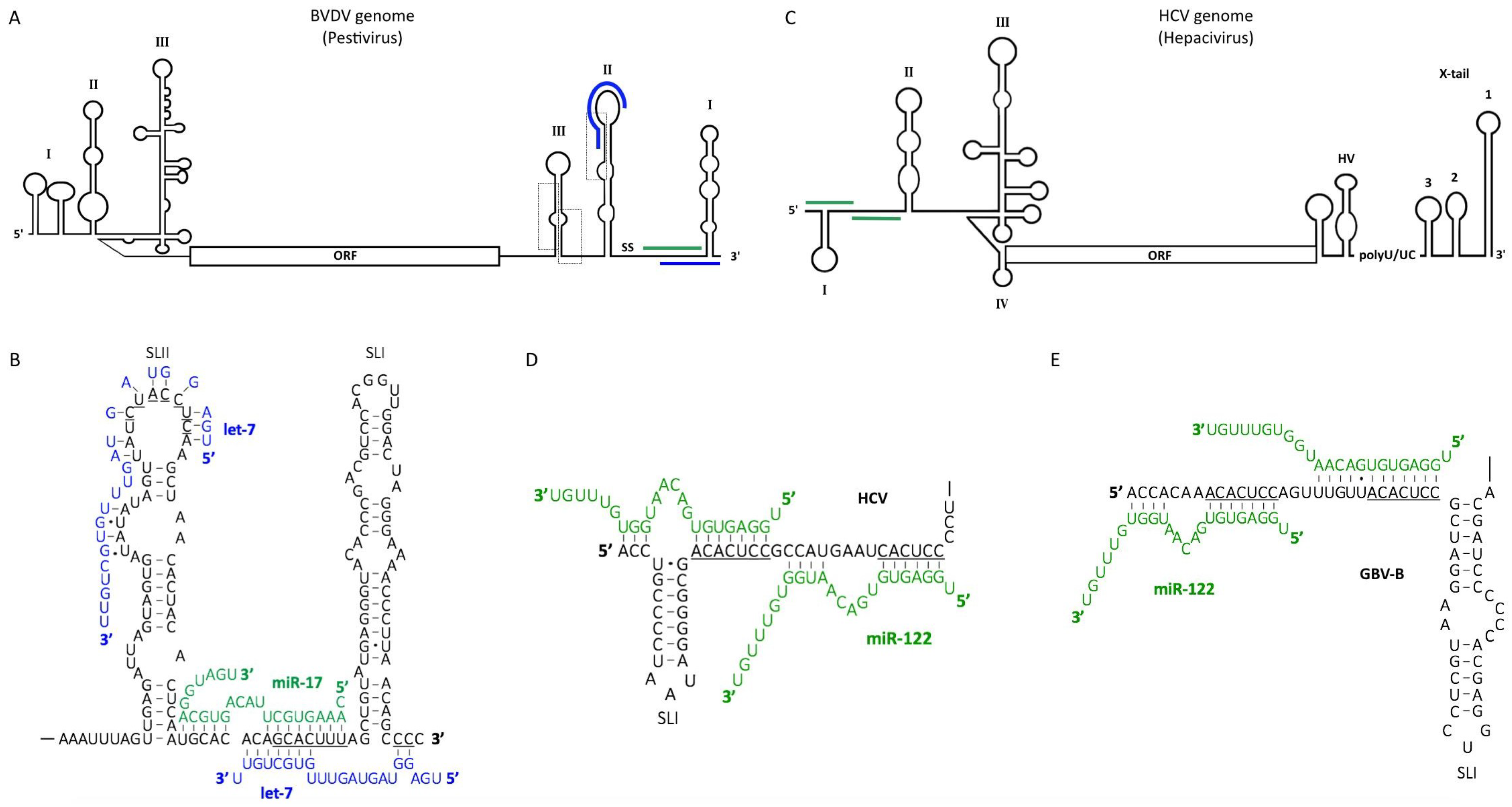
5. Pestiviruses
6. Hepaciviruses
6.1. The Role of miR-122 in the Hepatitis C Virus (HCV) Life Cycle
6.2. MiR-122 Protects the HCV Genome from Cellular Pyrophosphatase and 5′ Exonuclease Activities
6.3. MiR-122 Binding to the 5′ UTR Alters the Structure of the HCV Genome
6.4. Dysregulation of miR-122 May Contribute to Viral Pathogenesis
6.5. MiR-122 Binding May Be a Common Strategy for Viral RNA Accumulation among Hepaciviruses
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Knight, S.W.; Bass, B.L. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 2001, 293, 2269–2271. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; McLachlan, J.; Pasquinelli, A.E.; Balint, E.; Tuschl, T.; Zamore, P.D. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Zhou, K.; Smith, A.M.; Noland, C.L.; Doudna, J.A. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013, 41, 6568–6576. [Google Scholar] [CrossRef] [PubMed]
- Mourelatos, Z.; Dostie, J.; Paushkin, S.; Sharma, A.; Charroux, B.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. miRNPs: A novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev. 2002, 16, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Rajewsky, N. The impact of miRNA target sites in coding sequences and in 3′ UTRs. PLoS ONE 2011, 6, e18067. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Syed, A.P.; Bilen, B.; Zavolan, M. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res. 2013, 23, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Meijer, H.A.; Smith, E.M.; Bushell, M. Regulation of miRNA strand selection: Follow the leader? Biochem. Soc. Trans. 2014, 42, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.Y.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Zhou, Y.H.; Chen, W.; Khaitovich, P. Sequence features associated with microRNA strand selection in humans and flies. BMC Genom. 2009, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Shin, C.; Nam, J.W.; Farh, K.K.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code: Functional sites with centered pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Westholm, J.O.; Lai, E.C. Vive la difference: Biogenesis and evolution of microRNAs in plants and animals. Genome Biol. 2011, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, N. microRNA target predictions in animals. Nat. Genet. 2006, 38, S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, N.; Socci, N.D. Computational identification of microRNA targets. Dev. Biol. 2004, 267, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Vienberg, S.; Geiger, J.; Madsen, S.; Dalgaard, L.T. MicroRNAs in metabolism. Acta Physiol. 2017, 219, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Cochella, L. A framework for understanding the roles of miRNAs in animal development. Development 2017, 144, 2548–2559. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, P.E.; Nimonkar, A.V. Herpes virus replication. IUBMB Life 2003, 55, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J. Overview of classification. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Cullen, B.R. Herpesvirus microRNAs: Phenotypes and functions. Curr. Opin. Virol. 2011, 1, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Pinnoji, R.C.; Bedadala, G.R.; George, B.; Holland, T.C.; Hill, J.M.; Hsia, S.C. Repressor element-1 silencing transcription factor/neuronal restrictive silencer factor (REST/NRSF) can regulate HSV-1 immediate-early transcription via histone modification. Virol. J. 2007, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Diao, C.; Yang, X.; Yang, Z.; Liu, M.; Li, X.; Tang, H. ICP4-induced miR-101 attenuates HSV-1 replication. Sci. Rep. 2016, 6, 23205. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.Q.; Li, Y.X.; Zhang, Y.; Li, X.; Tang, H. MiR-101 regulates HSV-1 replication by targeting ATP5B. Antivir. Res. 2011, 89, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Wilson, D.W. ATP depletion blocks herpes simplex virus DNA packaging and capsid maturation. J. Virol. 1999, 73, 2006–2015. [Google Scholar] [PubMed]
- Pan, D.; Flores, O.; Umbach, J.L.; Pesola, J.M.; Bentley, P.; Rosato, P.C.; Leib, D.A.; Cullen, B.R.; Coen, D.M. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 2014, 15, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Varani, S.; Landini, M.P. Cytomegalovirus-induced immunopathology and its clinical consequences. Herpesviridae 2011, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Britt, W. Manifestations of human cytomegalovirus infection: Proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 2008, 325, 417–470. [Google Scholar] [PubMed]
- O’Connor, C.M.; Vanicek, J.; Murphy, E.A. Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency. J. Virol. 2014, 88, 5524–5532. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A.; Hawkins, J.B.; Tracy, S.I.; Shapiro, M. The pathogenesis of Epstein-Barr virus persistent infection. Curr. Opin. Virol. 2013, 3, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Ellis-Connell, A.L.; Iempridee, T.; Xu, I.; Mertz, J.E. Cellular microRNAs 200b and 429 regulate the Epstein-Barr virus switch between latency and lytic replication. J. Virol. 2010, 84, 10329–10343. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.L.; Wang, Z.; Yu, X.; Mertz, J.E. Either ZEB1 or ZEB2/SIP1 can play a central role in regulating the Epstein-Barr virus latent-lytic switch in a cell-type-specific manner. J. Virol. 2010, 84, 6139–6152. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, S.; Pan, Q.; Blencowe, B.J.; Claycomb, J.M.; Frappier, L. Epstein-Barr virus EBNA1 protein regulates viral latency through effects on let-7 microRNA and dicer. J. Virol. 2014, 88, 11166–11177. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 1985, 313, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Frappier, L. EBNA1 and host factors in Epstein-Barr virus latent DNA replication. Curr. Opin. Virol. 2012, 2, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Li, W.; Tang, Q.; Yao, S.; Lv, Z.; Feng, N.; Ma, X.; Bai, Z.; Zeng, Y.; Qin, D.; et al. Cellular microRNAs 498 and 320d regulate herpes simplex virus 1 induction of Kaposi’s sarcoma-associated herpesvirus lytic replication by targeting RTA. PLoS ONE 2013, 8, e55832. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Ma, X.; Shen, C.; Cao, X.; Feng, N.; Qin, D.; Zeng, Y.; Zhu, J.; Gao, S.J.; Lu, C. Inhibition of Kaposi’s sarcoma-associated herpesvirus lytic replication by HIV-1 Nef and cellular microRNA hsa-miR-1258. J. Virol. 2014, 88, 4987–5000. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Suemasa, F.; Sagara, H.; Nakamura, S.; Ino, Y.; Kobayashi, K.; Hiramatsu, H.; Haraguchi, T.; Kurokawa, K.; Todo, T.; et al. MiR-199a Inhibits Secondary Envelopment of Herpes Simplex Virus-1 Through the Downregulation of Cdc42-specific GTPase Activating Protein Localized in Golgi Apparatus. Sci. Rep. 2017, 7, 6650. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zhao, Y.; Clement, C.; Neumann, D.M.; Lukiw, W.J. HSV-1 infection of human brain cells induces miRNA-146a and Alzheimer-type inflammatory signaling. Neuroreport 2009, 20, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: A nontargeted metabolomic study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef] [PubMed]
- Ru, J.; Sun, H.; Fan, H.; Wang, C.; Li, Y.; Liu, M.; Tang, H. MiR-23a facilitates the replication of HSV-1 through the suppression of interferon regulatory factor 1. PLoS ONE 2014, 9, e114021. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ru, J.; Zhang, J.; Zhu, L.H.; Liu, M.; Li, X.; Tang, H. miR-23a targets interferon regulatory factor 1 and modulates cellular proliferation and paclitaxel-induced apoptosis in gastric adenocarcinoma cells. PLoS ONE 2013, 8, e64707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, J.; Tang, J.; Zhou, L.; Zhou, M. MicroRNA-649 promotes HSV-1 replication by directly targeting MALT1. J. Med. Virol. 2017, 89, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Hoffman, S.; Beal, A.M.; Dykon, A.; Ringenberg, M.A.; Hughes, A.C.; Dare, L.; Anderson, A.D.; Finger, J.; Kasparcova, V.; et al. MALT1 Protease Activity Is Required for Innate and Adaptive Immune Responses. PLoS ONE 2015, 10, e0127083. [Google Scholar] [CrossRef] [PubMed]
- Lagos, D.; Pollara, G.; Henderson, S.; Gratrix, F.; Fabani, M.; Milne, R.S.; Gotch, F.; Boshoff, C. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 2010, 12, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.R.; Liu, X.J.; Li, X.J.; Shen, Z.Z.; Yang, B.; Wu, C.C.; Li, J.F.; Miao, L.F.; Ye, H.Q.; Qiao, G.H.; et al. MicroRNA miR-21 attenuates human cytomegalovirus replication in neural cells by targeting Cdc25a. J. Virol. 2015, 89, 1070–1082. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, M.; Liao, S.; Liu, W.; Dai, G.; Wu, G.; Chen, L. Hsa-miR-27b is up-regulated in cytomegalovirus-infected human glioma cells, targets engrailed-2 and inhibits its expression. Exp. Biol. Med. 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Cramer, E.M.; Shao, Y.; Wang, Y.; Yuan, Y. miR-190 is upregulated in Epstein-Barr Virus type I latency and modulates cellular mRNAs involved in cell survival and viral reactivation. Virology 2014, 464–465, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.; Motsch, N.; Zhu, J.Y.; Barth, S.; Okoniewski, M.; Reineke, T.; Tinguely, M.; Faggioni, A.; Trivedi, P.; Meister, G.; et al. microRNA profiling in Epstein-Barr virus-associated B-cell lymphoma. Nucleic Acids Res. 2011, 39, 1880–1893. [Google Scholar] [CrossRef] [PubMed]
- Onnis, A.; Navari, M.; Antonicelli, G.; Morettini, F.; Mannucci, S.; De Falco, G.; Vigorito, E.; Leoncini, L. Epstein-Barr nuclear antigen 1 induces expression of the cellular microRNA hsa-miR-127 and impairing B-cell differentiation in EBV-infected memory B cells. New insights into the pathogenesis of Burkitt lymphoma. Blood Cancer J. 2012, 2, e84. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.H.; Wu, M.F.; Wu, Y.H.; Chang, S.J.; Lin, S.F.; Sharp, T.V.; Wang, H.W. The M type K15 protein of Kaposi’s sarcoma-associated herpesvirus regulates microRNA expression via its SH2-binding motif to induce cell migration and invasion. J. Virol. 2009, 83, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Hu, T.F.; Chen, Y.C.; Tsai, Y.N.; Tsai, Y.H.; Cheng, C.C.; Wang, H.W. The manipulation of miRNA-gene regulatory networks by KSHV induces endothelial cell motility. Blood 2011, 118, 2896–2905. [Google Scholar] [CrossRef] [PubMed]
- Punj, V.; Matta, H.; Schamus, S.; Tamewitz, A.; Anyang, B.; Chaudhary, P.M. Kaposi’s sarcoma-associated herpesvirus-encoded viral FLICE inhibitory protein (vFLIP) K13 suppresses CXCR4 expression by upregulating miR-146a. Oncogene 2010, 29, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Bridge, G.; Monteiro, R.; Henderson, S.; Emuss, V.; Lagos, D.; Georgopoulou, D.; Patient, R.; Boshoff, C. The microRNA-30 family targets DLL4 to modulate endothelial cell behavior during angiogenesis. Blood 2012, 120, 5063–5072. [Google Scholar] [CrossRef] [PubMed]
- Redpath, S.; Angulo, A.; Gascoigne, N.R.; Ghazal, P. Immune checkpoints in viral latency. Annu. Rev. Microbiol. 2001, 55, 531–560. [Google Scholar] [CrossRef] [PubMed]
- Leucci, E.; Onnis, A.; Cocco, M.; De Falco, G.; Imperatore, F.; Giuseppina, A.; Costanzo, V.; Cerino, G.; Mannucci, S.; Cantisani, R.; et al. B-cell differentiation in EBV-positive Burkitt lymphoma is impaired at posttranscriptional level by miRNA-altered expression. Int. J. Cancer 2010, 126, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Trotter, M.W.; Lagos, D.; Bourboulia, D.; Henderson, S.; Makinen, T.; Elliman, S.; Flanagan, A.M.; Alitalo, K.; Boshoff, C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 2004, 36, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Jakymiw, A.; Findlay, V.; Parsons, C. KSHV-Encoded MicroRNAs: Lessons for Viral Cancer Pathogenesis and Emerging Concepts. Int. J. Cell Biol. 2012, 2012, 603961. [Google Scholar] [CrossRef] [PubMed]
- Rosean, T.R.; Holman, C.J.; Tompkins, V.S.; Jing, X.; Krasowski, M.D.; Rose-John, S.; Janz, S. KSHV-encoded vIL-6 collaborates with deregulated c-Myc to drive plasmablastic neoplasms in mice. Blood Cancer J. 2016, 6, e398. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.G.; Majerciak, V.; Uldrick, T.S.; Wang, X.; Kruhlak, M.; Yarchoan, R.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesviral IL-6 and human IL-6 open reading frames contain miRNA binding sites and are subject to cellular miRNA regulation. J. Pathol. 2011, 225, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.G.; Pripuzova, N.; Majerciak, V.; Kruhlak, M.; Le, S.Y.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus ORF57 promotes escape of viral and human interleukin-6 from microRNA-mediated suppression. J. Virol. 2011, 85, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Liang, D.; Lan, K. MicroRNAs and unusual small RNAs discovered in Kaposi’s sarcoma-associated herpesvirus virions. J. Virol. 2012, 86, 12717–12730. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.A.; Costa, H.; Landazuri, N.; Lui, W.O.; Hultenby, K.; Rahbar, A.; Yaiw, K.C.; Soderberg-Naucler, C. Human cytomegalovirus microRNAs are carried by virions and dense bodies and are delivered to target cells. J. Gen. Virol. 2017, 98, 1058–1072. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Kennedy, E.M.; Whisnant, A.W.; Cullen, B.R. Induced Packaging of Cellular MicroRNAs into HIV-1 Virions Can Inhibit Infectivity. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Sullivan, C.S. Virus-encoded microRNAs. Virology 2011, 411, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Sullivan, C.S. Virus-encoded microRNAs: An overview and a look to the future. PLoS Pathog. 2012, 8, e1003018. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Iizasa, H.; Kanehiro, Y.; Fekadu, S.; Yoshiyama, H. Herpesviral microRNAs in Cellular Metabolism and Immune Responses. Front. Microbiol. 2017, 8, 1318. [Google Scholar] [CrossRef] [PubMed]
- Cardin, S.; Borchert, G.M. Viral MicroRNAs, Host MicroRNAs Regulating Viruses, and Bacterial MicroRNA-Like RNAs. In Bioinformatics in MicroRNA Research; Al, H.J.E., Ed.; Humana Press: New York, NY, USA, 2017; pp. 39–56. [Google Scholar]
- Markus, A.; Golani, L.; Ojha, N.K.; Borodiansky-Shteinberg, T.; Kinchington, P.R.; Goldstein, R.S. Varicella-Zoster Virus Expresses Multiple Small Noncoding RNAs. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Guo, Z.; Zhang, X.; Guo, L.; Liu, L.; Liao, Y.; Wang, J.; Wang, L.; Li, Q. A microRNA encoded by HSV-1 inhibits a cellular transcriptional repressor of viral immediate early and early genes. Sci. China Life Sci. 2013, 56, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Enk, J.; Levi, A.; Weisblum, Y.; Yamin, R.; Charpak-Amikam, Y.; Wolf, D.G.; Mandelboim, O. HSV1 MicroRNA Modulation of GPI Anchoring and Downstream Immune Evasion. Cell Rep. 2016, 17, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Liu, Q.; Wang, S.; Ren, Z.; Kitazato, K.; Yang, D.; Wang, Y. HSV-1-encoded microRNA miR-H1 targets Ubr1 to promote accumulation of neurodegeneration-associated protein. Virus Genes 2018, 54, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gerlier, D. Viral hijacking of cellular ubiquitination pathways as an anti-innate immunity strategy. Viral Immunol. 2006, 19, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host immune system gene targeting by a viral miRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Esteso, G.; Luzon, E.; Sarmiento, E.; Gomez-Caro, R.; Steinle, A.; Murphy, G.; Carbone, J.; Vales-Gomez, M.; Reyburn, H.T. Altered microRNA expression after infection with human cytomegalovirus leads to TIMP3 downregulation and increased shedding of metalloprotease substrates, including MICA. J. Immunol. 2014, 193, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S.; Kim, S.; Kim, D.; Ahn, J.H.; Ahn, K. Human cytomegalovirus clinical strain-specific microRNA miR-UL148D targets the human chemokine RANTES during infection. PLoS Pathog. 2012, 8, e1002577. [Google Scholar] [CrossRef] [PubMed]
- Maghazachi, A.A.; Al-Aoukaty, A.; Schall, T.J. CC chemokines induce the generation of killer cells from CD56+ cells. Eur. J. Immunol. 1996, 26, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Hook, L.M.; Grey, F.; Grabski, R.; Tirabassi, R.; Doyle, T.; Hancock, M.; Landais, I.; Jeng, S.; McWeeney, S.; Britt, W.; et al. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 2014, 15, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Albanese, M.; Tagawa, T.; Buschle, A.; Hammerschmidt, W. MicroRNAs of Epstein-Barr Virus Control Innate and Adaptive Antiviral Immunity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yu, F.; Wu, W.; Wang, Y.; Ding, H.; Qian, L. Epstein-Barr virus-encoded microRNAs as regulators in host immune responses. Int. J. Biol. Sci. 2018, 14, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Wulff, B.E.; Alla, N.R.; Maragkakis, M.; Megraw, M.; Hatzigeorgiou, A.; Iwakiri, D.; Takada, K.; Wiedmer, A.; Showe, L.; et al. Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J. Biol. Chem. 2010, 285, 33358–33370. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; O’Hara, A.; Araujo, I.; Barreto, J.; Carvalho, E.; Sapucaia, J.B.; Ramos, J.C.; Luz, E.; Pedroso, C.; Manrique, M.; et al. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1-3. Cancer Res. 2008, 68, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Qin, Z.; Wang, J.; Zheng, X.; Lu, J.; Zhang, X.; Wei, L.; Peng, Q.; Zheng, Y.; Ou, C.; et al. Epstein-Barr Virus miR-BART6-3p Inhibits the RIG-I Pathway. J. Innate Immun. 2017, 9, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.Y.; Siu, K.L.; Kok, K.H.; Lung, R.W.; Tsang, C.M.; To, K.F.; Kwong, D.L.; Tsao, S.W.; Jin, D.Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Navari, M.; Etebari, M.; Ibrahimi, M.; Leoncini, L.; Piccaluga, P. Pathobiologic Roles of Epstein-Barr Virus-Encoded MicroRNAs in Human Lymphomas. Int. J. Mol. Sci. 2018, 19, 1168. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Taylor, G.S.; Sauce, D.; Rickinson, A.B. Cellular responses to viral infection in humans: Lessons from Epstein-Barr virus. Annu. Rev. Immunol. 2007, 25, 587–617. [Google Scholar] [CrossRef] [PubMed]
- Pagano, J.S.; Blaser, M.; Buendia, M.A.; Damania, B.; Khalili, K.; Raab-Traub, N.; Roizman, B. Infectious agents and cancer: Criteria for a causal relation. Semin. Cancer Biol. 2004, 14, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Happel, C.; Ramalingam, D.; Ziegelbauer, J.M. Virus-Mediated Alterations in miRNA Factors and Degradation of Viral miRNAs by MCPIP1. PLoS Biol. 2016, 14, e2000998. [Google Scholar] [CrossRef] [PubMed]
- Mizgalska, D.; Wegrzyn, P.; Murzyn, K.; Kasza, A.; Koj, A.; Jura, J.; Jarzab, B.; Jura, J. Interleukin-1-inducible MCPIP protein has structural and functional properties of RNase and participates in degradation of IL-1beta mRNA. FEBS J. 2009, 276, 7386–7399. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Kearney, P.; Plaisance, K.; Parsons, C.H. Pivotal advance: Kaposi’s sarcoma-associated herpesvirus (KSHV)-encoded microRNA specifically induce IL-6 and IL-10 secretion by macrophages and monocytes. J. Leukoc. Biol. 2010, 87, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Samols, M.A.; Plaisance, K.B.; Boss, I.W.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Kaposi’s sarcoma-associated herpesvirus encodes an ortholog of miR-155. J. Virol. 2007, 81, 12836–12845. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature 2007, 450, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Corcoran, D.L.; Mukherjee, N.; Skalsky, R.L.; Hafner, M.; Nusbaum, J.D.; Shamulailatpam, P.; Love, C.L.; Dave, S.S.; Tuschl, T.; et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 2011, 10, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Shamulailatpam, P.; Raja, A.N.; Gottwein, E. Kaposi’s sarcoma-associated herpesvirus encodes a mimic of cellular miR-23. J. Virol. 2013, 87, 11821–11830. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Zhu, Y.; Jones, T.; Bai, Z.; Huang, Y.; Gao, S.J. A Kaposi’s sarcoma-associated herpesvirus microRNA and its variants target the transforming growth factor beta pathway to promote cell survival. J. Virol. 2012, 86, 11698–11711. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Freitas, E.; Sullivan, R.; Mohan, S.; Bacelieri, R.; Branch, D.; Romano, M.; Kearney, P.; Oates, J.; Plaisance, K.; et al. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog. 2010, 6, e1000742. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Gao, Y.; Lin, X.; He, Z.; Zhao, Q.; Deng, Q.; Lan, K. A human herpesvirus miRNA attenuates interferon signaling and contributes to maintenance of viral latency by targeting IKKepsilon. Cell Res. 2011, 21, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Boss, I.W.; Nadeau, P.E.; Abbott, J.R.; Yang, Y.; Mergia, A.; Renne, R. A Kaposi’s sarcoma-associated herpesvirus-encoded ortholog of microRNA miR-155 induces human splenic B-cell expansion in NOD/LtSz-scid IL2Rgammanull mice. J. Virol. 2011, 85, 9877–9886. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, R.; Lin, X.; Liang, D.; Deng, Q.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded microRNA miR-K12-11 attenuates transforming growth factor beta signaling through suppression of SMAD5. J. Virol. 2012, 86, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R. Physiological roles of miR-155. Immunology 2015, 145, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Liao, J.; Huang, Q.; Nie, Y.; Wu, K. HSV-1 miR-H6 inhibits HSV-1 replication and IL-6 expression in human corneal epithelial cells in vitro. Clin. Dev. Immunol. 2012, 2012, 192791. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Patel, A.; Krause, P.R. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J. Virol. 2009, 83, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Samaniego, L.A.; Wu, N.; DeLuca, N.A. The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J. Virol. 1997, 71, 4614–4625. [Google Scholar] [PubMed]
- Tang, S.; Bertke, A.S.; Patel, A.; Wang, K.; Cohen, J.I.; Krause, P.R. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc. Natl. Acad. Sci. USA 2008, 105, 10931–10936. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Han, Z.; Zhou, G.; Roizman, B. Patterns of accumulation of miRNAs encoded by herpes simplex virus during productive infection, latency, and on reactivation. Proc. Natl. Acad. Sci. USA 2015, 112, E49–55. [Google Scholar] [CrossRef] [PubMed]
- Grey, F.; Meyers, H.; White, E.A.; Spector, D.H.; Nelson, J. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog. 2007, 3, e163. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; To, K.F.; Lo, K.W.; Lung, R.W.; Hui, J.W.; Liao, G.; Hayward, S.D. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc. Natl. Acad. Sci. USA 2007, 104, 16164–16169. [Google Scholar] [CrossRef] [PubMed]
- Lung, R.W.; Tong, J.H.; Sung, Y.M.; Leung, P.S.; Ng, D.C.; Chau, S.L.; Chan, A.W.; Ng, E.K.; Lo, K.W.; To, K.F. Modulation of LMP2A expression by a newly identified Epstein-Barr virus-encoded microRNA miR-BART22. Neoplasia 2009, 11, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Wang, Z.; Ke, Q.; Hong, M.; Qian, Y.; Zhao, X.; Liu, Y.; Kim, H.J.; Ritz, J.; Cantor, H.; et al. Signaling by the Epstein-Barr virus LMP1 protein induces potent cytotoxic CD4(+) and CD8(+) T cell responses. Proc. Natl. Acad. Sci. USA 2018, 115, E686–E695. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, R.J.; Tong, S.; Zhang, G.; Zong, J.; Chen, Y.; Jin, D.Y.; Chen, M.R.; Pan, J.; Chen, H. NF-kappaB Signaling Regulates Expression of Epstein-Barr Virus BART MicroRNAs and Long Noncoding RNAs in Nasopharyngeal Carcinoma. J. Virol. 2016, 90, 6475–6488. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, W.; Gao, S.J.; Lu, C. KSHV microRNAs: Tricks of the Devil. Trends Microbiol. 2017, 25, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Liang, D.; He, Z.; Deng, Q.; Robertson, E.S.; Lan, K. miR-K12-7-5p encoded by Kaposi’s sarcoma-associated herpesvirus stabilizes the latent state by targeting viral ORF50/RTA. PLoS ONE 2011, 6, e16224. [Google Scholar] [CrossRef] [PubMed]
- Bellare, P.; Ganem, D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: An evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 2009, 6, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Li, Z.; Chu, C.Y.; Feng, J.; Feng, J.; Sun, R.; Rana, T.M. MicroRNAs encoded by Kaposi’s sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 2010, 11, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Stedman, W.; Yousef, M.; Renne, R.; Lieberman, P.M. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 2010, 84, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Meshesha, M.K.; Bentwich, Z.; Solomon, S.A.; Avni, Y.S. In vivo expression of human cytomegalovirus (HCMV) microRNAs during latency. Gene 2016, 575, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, M.J. The Human Polyomaviruses: An Overview. In Human Polyomaviruses; Khalili, K., Stoner, G.L., Eds.; Wiley-Liss, Inc.: New York, NY, USA, 2002; pp. 53–71. [Google Scholar]
- Lagatie, O.; Tritsmans, L.; Stuyver, L.J. The miRNA world of polyomaviruses. Virol. J. 2013, 10, 268. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B.; Consortium, I.R. ICTV Virus Taxonomy Profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Khalili, K. Polyomaviruses and human cancer: Molecular mechanisms underlying patterns of tumorigenesis. Virology 2004, 324, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Zhang, Z.; DeCerbo, J.N.; Carmichael, G.G. Gene regulation by sense-antisense overlap of polyadenylation signals. RNA 2009, 15, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Seo, G.J.; Fink, L.H.; O’Hara, B.; Atwood, W.J.; Sullivan, C.S. Evolutionarily conserved function of a viral microRNA. J. Virol. 2008, 82, 9823–9828. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Grundhoff, A.T.; Tevethia, S.; Pipas, J.M.; Ganem, D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 2005, 435, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Sung, C.K.; Pack, C.D.; Grundhoff, A.; Lukacher, A.E.; Benjamin, T.L.; Ganem, D. Murine Polyomavirus encodes a microRNA that cleaves early RNA transcripts but is not essential for experimental infection. Virology 2009, 387, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Broekema, N.M.; Imperiale, M.J. miRNA regulation of BK polyomavirus replication during early infection. Proc. Natl. Acad. Sci. USA 2013, 110, 8200–8205. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.C.; Li, Y.J.; Chen, H.C.; Wu, H.H.; Weng, C.H.; Chen, Y.C.; Lee, C.C.; Chang, M.Y.; Hsu, H.H.; Yen, T.H.; et al. Polyomavirus BK-encoded microRNA suppresses autoregulation of viral replication. Biochem. Biophys. Res. Commun. 2014, 447, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Bauman, Y.; Mandelboim, O. MicroRNA based immunoevasion mechanism of human polyomaviruses. RNA Biol. 2011, 8, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Bauman, Y.; Nachmani, D.; Vitenshtein, A.; Tsukerman, P.; Drayman, N.; Stern-Ginossar, N.; Lankry, D.; Gruda, R.; Mandelboim, O. An identical miRNA of the human JC and BK polyoma viruses targets the stress-induced ligand ULBP3 to escape immune elimination. Cell Host Microbe 2011, 9, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.M.; Bass, C.R.; Kincaid, R.P.; Ulug, E.T.; Sullivan, C.S. The Murine Polyomavirus MicroRNA Locus Is Required To Promote Viruria during the Acute Phase of Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Zhang, Z.; Fan, J.; Cui, Z.; Zhang, X.E. Functionally orthologous viral and cellular microRNAs studied by a novel dual-fluorescent reporter system. PLoS ONE 2012, 7, e36157. [Google Scholar] [CrossRef] [PubMed]
- Garkavtsev, I.; Kozin, S.V.; Chernova, O.; Xu, L.; Winkler, F.; Brown, E.; Barnett, G.H.; Jain, R.K. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature 2004, 428, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.H.; Jones, S.N. The ING gene family in the regulation of cell growth and tumorigenesis. J. Cell. Physiol. 2009, 218, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Shiseki, M.; Nagashima, M.; Pedeux, R.M.; Kitahama-Shiseki, M.; Miura, K.; Okamura, S.; Onogi, H.; Higashimoto, Y.; Appella, E.; Yokota, J.; et al. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003, 63, 2373–2378. [Google Scholar] [PubMed]
- Li, S.; Zeng, A.; Hu, Q.; Yan, W.; Liu, Y.; You, Y. miR-423-5p contributes to a malignant phenotype and temozolomide chemoresistance in glioblastomas. Neuro Oncol. 2017, 19, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Micolucci, L.; Akhtar, M.M.; Olivieri, F.; Rippo, M.R.; Procopio, A.D. Diagnostic value of microRNAs in asbestos exposure and malignant mesothelioma: Systematic review and qualitative meta-analysis. Oncotarget 2016, 7, 58606–58637. [Google Scholar] [CrossRef] [PubMed]
- Sevinc, E.D.; Egeli, U.; Cecener, G.; Tezcan, G.; Tunca, B.; Gokgoz, S.; Tasdelen, I.; Tolunay, S.; Evrensel, T. Association of miR-1266 with recurrence/metastasis potential in estrogen receptor positive breast cancer patients. Asian Pac. J. Cancer Prev. 2015, 16, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lu, M.H.; Zhang, D.; Hao, N.B.; Fan, Y.H.; Wu, Y.Y.; Wang, S.M.; Xie, R.; Fang, D.C.; Zhang, H.; et al. miR-1207-5p and miR-1266 suppress gastric cancer growth and invasion by targeting telomerase reverse transcriptase. Cell Death Dis. 2014, 5, e1034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ren, D.; Wu, X.; Lin, X.; Ye, L.; Lin, C.; Wu, S.; Zhu, J.; Peng, X.; Song, L. miR-1266 Contributes to Pancreatic Cancer Progression and Chemoresistance by the STAT3 and NF-κB Signaling Pathways. Mol. Ther. Nucleic Acids 2018, 11, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, D.C.; Li, Z.F.; Liu, C.X.; Xiao, Y.M.; Zhang, B.; Li, X.D.; Zhao, J.; Chen, L.P.; Xing, X.M.; et al. Upregulation of miR-27a contributes to the malignant transformation of human bronchial epithelial cells induced by SV40 small T antigen. Oncogene 2011, 30, 3875–3886. [Google Scholar] [CrossRef] [PubMed]
- Lerner, M.; Lundgren, J.; Akhoondi, S.; Jahn, A.; Ng, H.F.; Akbari Moqadam, F.; Oude Vrielink, J.A.; Agami, R.; Den Boer, M.L.; Grander, D.; et al. MiRNA-27a controls FBW7/hCDC4-dependent cyclin E degradation and cell cycle progression. Cell Cycle 2011, 10, 2172–2183. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Cui, H.; Banerjee, S.; Tan, Z.; Salomao, R.; Fu, M.; Abraham, E.; Thannickal, V.J.; Liu, G. miR-27a regulates inflammatory response of macrophages by targeting IL-10. J. Immunol. 2014, 193, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Park, J.L.; Kim, M.; Song, K.S.; Kim, S.Y.; Kim, Y.S. Cell-Free miR-27a, a Potential Diagnostic and Prognostic Biomarker for Gastric Cancer. Genom. Inform. 2015, 13, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Saib, A. Early steps of retrovirus replicative cycle. Retrovirology 2004, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.C.; Peterlin, B.M. Charting HIV’s remarkable voyage through the cell: Basic science as a passport to future therapy. Nat. Med. 2002, 8, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ye, L.; Hou, W.; Zhou, Y.; Wang, Y.J.; Metzger, D.S.; Ho, W.Z. Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood 2009, 113, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, M.; Pandhare, J.; Dash, C. Are microRNAs Important Players in HIV-1 Infection? An Update. Viruses 2018, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Liu, H.; Rice, A.P. miR-132 enhances HIV-1 replication. Virology 2013, 438, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Lioy, D.T.; Ma, L.; Impey, S.; Mandel, G.; Goodman, R.H. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat. Neurosci. 2007, 10, 1513–1514. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Saavedra, M.; Antoun, G.; Yanagiya, A.; Oliva-Hernandez, R.; Cornejo-Palma, D.; Perez-Iratxeta, C.; Sonenberg, N.; Cheng, H.Y. miRNA-132 orchestrates chromatin remodeling and translational control of the circadian clock. Hum. Mol. Genet. 2011, 20, 731–751. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Ito, M.; Tsutsumi, Y.; Ichikawa, Y.; Okuyama, H.; Brisibe, E.A.; Saksena, N.K.; Fujii, Y.R. HIV-1 nef suppression by virally encoded microRNA. Retrovirology 2004, 1, 44. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Fujii, Y.R. Regulation of human immunodeficiency virus 1 transcription by nef microRNA. J. Gen. Virol. 2005, 86, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, D.L.; Plante, I.; Landry, P.; Barat, C.; Janelle, M.E.; Flamand, L.; Tremblay, M.J.; Provost, P. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008, 36, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Klase, Z.; Winograd, R.; Davis, J.; Carpio, L.; Hildreth, R.; Heydarian, M.; Fu, S.; McCaffrey, T.; Meiri, E.; Ayash-Rashkovsky, M.; et al. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology 2009, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Kaul, D.; Ahlawat, A.; Gupta, S.D. HIV-1 genome-encoded hiv1-mir-H1 impairs cellular responses to infection. Mol. Cell Biochem. 2009, 323, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Bennasser, Y.; Le, S.Y.; Benkirane, M.; Jeang, K.T. Evidence that HIV-1 encodes an siRNA and a suppressor of RNA silencing. Immunity 2005, 22, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Bennasser, Y.; Jeang, K.T. HIV-1 Tat interaction with Dicer: Requirement for RNA. Retrovirology 2006, 3, 95. [Google Scholar] [CrossRef] [PubMed]
- Sardo, L.; Vakil, P.R.; Elbezanti, W.; El-Sayed, A.; Klase, Z. The inhibition of microRNAs by HIV-1 Tat suppresses beta catenin activity in astrocytes. Retrovirology 2016, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.D.; Jaskiewicz, L.; Zhang, H.; Laine, S.; Sack, R.; Gatignol, A.; Filipowicz, W. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005, 6, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Daniels, S.M.; Sinck, L.; Ward, N.J.; Melendez-Pena, C.E.; Scarborough, R.J.; Azar, I.; Rance, E.; Daher, A.; Pang, K.M.; Rossi, J.J.; et al. HIV-1 RRE RNA acts as an RNA silencing suppressor by competing with TRBP-bound siRNAs. RNA Biol. 2015, 12, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.W.; Le Grice, S.F. HIV Rev Assembly on the Rev Response Element (RRE): A Structural Perspective. Viruses 2015, 7, 3053–3075. [Google Scholar] [CrossRef] [PubMed]
- Lecellier, C.H.; Dunoyer, P.; Arar, K.; Lehmann-Che, J.; Eyquem, S.; Himber, C.; Saib, A.; Voinnet, O. A cellular microRNA mediates antiviral defense in human cells. Science 2005, 308, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Khodakaram-Tafti, A.; Farjanikish, G.H. Persistent bovine viral diarrhea virus (BVDV) infection in cattle herds. Iran J. Vet. Res. 2017, 18, 154–163. [Google Scholar] [PubMed]
- Scheel, T.K.; Luna, J.M.; Liniger, M.; Nishiuchi, E.; Rozen-Gagnon, K.; Shlomai, A.; Auray, G.; Gerber, M.; Fak, J.; Keller, I.; et al. A Broad RNA Virus Survey Reveals Both miRNA Dependence and Functional Sequestration. Cell Host Microbe 2016, 19, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Moes, L.; Wirth, M. The internal initiation of translation in bovine viral diarrhea virus RNA depends on the presence of an RNA pseudoknot upstream of the initiation codon. Virol. J. 2007, 4, 124. [Google Scholar] [CrossRef] [PubMed]
- Austermann-Busch, S.; Becher, P. RNA structural elements determine frequency and sites of nonhomologous recombination in an animal plus-strand RNA virus. J. Virol. 2012, 86, 7393–7402. [Google Scholar] [CrossRef] [PubMed]
- Schult, P.; Roth, H.; Adams, R.L.; Mas, C.; Imbert, L.; Orlik, C.; Ruggieri, A.; Pyle, A.M.; Lohmann, V. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 2018, 9, 2613. [Google Scholar] [CrossRef] [PubMed]
- Isken, O.; Grassmann, C.W.; Sarisky, R.T.; Kann, M.; Zhang, S.; Grosse, F.; Kao, P.N.; Behrens, S.E. Members of the NF90/NFAR protein group are involved in the life cycle of a positive-strand RNA virus. EMBO J. 2003, 22, 5655–5665. [Google Scholar] [CrossRef] [PubMed]
- Pankraz, A.; Thiel, H.J.; Becher, P. Essential and nonessential elements in the 3′ nontranslated region of Bovine viral diarrhea virus. J. Virol. 2005, 79, 9119–9127. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.H.; Moore, M.J.; Luna, J.M.; Nishiuchi, E.; Fak, J.; Darnell, R.B.; Rice, C.M. Global mapping of miRNA-target interactions in cattle (Bos taurus). Sci. Rep. 2017, 7, 8190. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Lozoya, T.; Dominguez, F.; Romero-Ruiz, A.; Steffani, L.; Martinez, S.; Monterde, M.; Ferri, B.; Nunez, M.J.; AinhoaRomero, E.; Zamora, O.; et al. The Lin28/Let-7 system in early human embryonic tissue and ectopic pregnancy. PLoS ONE 2014, 9, e87698. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, G.C.; Tscherner, A.; Nalpathamkalam, T.; Merico, D.; LaMarre, J. MicroRNA Expression during Bovine Oocyte Maturation and Fertilization. Int. J. Mol. Sci. 2016, 17, 396. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.T. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008, 133, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Chase, C.C. The impact of BVDV infection on adaptive immunity. Biologicals 2013, 41, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Jopling, C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol. 2012, 9, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.P.; Jopling, C.L. Regulation and biological function of the liver-specific miR-122. Biochem. Soc. Trans. 2010, 38, 1553–1557. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.L.; Sarnow, P. Modulation of hepatitis C virus RNA abundance and the isoprenoid biosynthesis pathway by microRNA miR-122 involves distinct mechanisms. J. Virol. 2010, 84, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huys, A.; Thibault, P.A.; Wilson, J.A. Requirements for human Dicer and TRBP in microRNA-122 regulation of HCV translation and RNA abundance. Virology 2012, 433, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Randall, G.; Panis, M.; Cooper, J.D.; Tellinghuisen, T.L.; Sukhodolets, K.E.; Pfeffer, S.; Landthaler, M.; Landgraf, P.; Kan, S.; Lindenbach, B.D.; et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. USA 2007, 104, 12884–12889. [Google Scholar] [CrossRef] [PubMed]
- Machlin, E.S.; Sarnow, P.; Sagan, S.M. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. USA 2011, 108, 3193–3198. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.L.; Pirakitikulr, N.; Pyle, A.M. Functional RNA structures throughout the Hepatitis C Virus genome. Curr. Opin. Virol. 2017, 24, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Amador-Canizares, Y.; Bernier, A.; Wilson, J.A.; Sagan, S.M. miR-122 does not impact recognition of the HCV genome by innate sensors of RNA but rather protects the 5′ end from the cellular pyrophosphatases, DOM3Z and DUSP11. Nucleic Acids Res. 2018, 46, 5139–5158. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Lam, V.L.; Chirayil, R.P.; Randall, G.; Sullivan, C.S. RNA triphosphatase DUSP11 enables exonuclease XRN-mediated restriction of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2018, 115, 8197–8202. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Coyle, S.M.; Doudna, J.A. Coupled 5′ nucleotide recognition and processivity in Xrn1-mediated mRNA decay. Mol. Cell 2011, 41, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.; Poole, T.L. 5′-exonuclease-2 of Saccharomyces cerevisiae. Purification and features of ribonuclease activity with comparison to 5′-exonuclease-1. J. Biol. Chem. 1995, 270, 16063–16069. [Google Scholar] [CrossRef] [PubMed]
- Sedano, C.D.; Sarnow, P. Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell Host Microbe 2014, 16, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Michal, J.J.; Zhang, L.; Ding, B.; Lunney, J.K.; Liu, B.; Jiang, Z. Interferon induced IFIT family genes in host antiviral defense. Int. J. Biol. Sci. 2013, 9, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yamane, D.; Lemon, S.M. Dissecting the roles of the 5′ exoribonucleases Xrn1 and Xrn2 in restricting hepatitis C virus replication. J. Virol. 2015, 89, 4857–4865. [Google Scholar] [CrossRef] [PubMed]
- Locker, N.; Easton, L.E.; Lukavsky, P.J. HCV and CSFV IRES domain II mediate eIF2 release during 80S ribosome assembly. EMBO J. 2007, 26, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Toledano, R.; Ariza-Mateos, A.; Birk, A.; Martinez-Garcia, B.; Gomez, J. In vitro characterization of a miR-122-sensitive double-helical switch element in the 5′ region of hepatitis C virus RNA. Nucleic Acids Res. 2009, 37, 5498–5510. [Google Scholar] [CrossRef] [PubMed]
- Meijer, H.A.; Kong, Y.W.; Lu, W.T.; Wilczynska, A.; Spriggs, R.V.; Robinson, S.W.; Godfrey, J.D.; Willis, A.E.; Bushell, M. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 2013, 340, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, C.S.; Winlow, P.L.; Parsons, A.L.; Jopling, C.L. Eukaryotic translation initiation factor 4AII contributes to microRNA-122 regulation of hepatitis C virus replication. Nucleic Acids Res. 2018, 46, 6330–6343. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.M.; Walton, C.M.; Wu, C.H.; Wu, G.Y. Secondary structure and hybridization accessibility of hepatitis C virus 3′-terminal sequences. J. Virol. 2002, 76, 9563–9574. [Google Scholar] [CrossRef] [PubMed]
- Friebe, P.; Bartenschlager, R. Role of RNA structures in genome terminal sequences of the hepatitis C virus for replication and assembly. J. Virol. 2009, 83, 11989–11995. [Google Scholar] [CrossRef] [PubMed]
- Hopcraft, S.E.; Azarm, K.D.; Israelow, B.; Leveque, N.; Schwarz, M.C.; Hsu, T.H.; Chambers, M.T.; Sourisseau, M.; Semler, B.L.; Evans, M.J. Viral Determinants of miR-122-Independent Hepatitis C Virus Replication. mSphere 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Ono, C.; Fukuhara, T.; Motooka, D.; Nakamura, S.; Okuzaki, D.; Yamamoto, S.; Tamura, T.; Mori, H.; Sato, A.; Uemura, K.; et al. Characterization of miR-122-independent propagation of HCV. PLoS Pathog. 2017, 13, e1006374. [Google Scholar] [CrossRef] [PubMed]
- Amador-Cañizares, Y.; Panigrahi, M.; Huys, A.; Kunden, R.D.; Adams, H.M.; Schinold, M.J.; Wilson, J.A. miR-122, small RNA annealing and sequence mutations alter the predicted structure of the Hepatitis C virus 5′ UTR RNA to stabilize and promote viral RNA accumulation. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Scheel, T.K.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Yan, Y.; Chai, H.; Chen, S.; Xiong, X.; Sun, D.; Yu, Y.; Deng, L.; Cheng, F. Pyruvate kinase M2 affects liver cancer cell behavior through up-regulation of HIF-1alpha and Bcl-xL in culture. Biomed. Pharmacother. 2015, 69, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ghazwani, M.; Zhang, Y.; Lu, J.; Li, J.; Fan, J.; Gandhi, C.R.; Li, S. miR-122 regulates collagen production via targeting hepatic stellate cells and suppressing P4HA1 expression. J. Hepatol. 2013, 58, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.J.; Iyengar, S.; Blahnik, K.R.; Ajuha, T.P.; Jiang, J.X.; Farnham, P.J.; Zern, M. Epigenetic modulation of miR-122 facilitates human embryonic stem cell self-renewal and hepatocellular carcinoma proliferation. PLoS ONE 2011, 6, e27740. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Investig. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Pfeffer, S.; Baumert, T.F. miR-122 acts as a tumor suppressor in hepatocarcinogenesis in vivo. J. Hepatol. 2013, 58, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Sagan, S.M.; Sarnow, P.; Wilson, J.A. Modulation of GB Virus B RNA Abundance by MicroRNA-122: Dependence on and Escape from MicroRNA-122 Restriction. J. Virol. 2013, 87, 7338–7347. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Simmonds, P.; Gerold, G.; Qaisar, N.; Jain, K.; Henriquez, J.A.; Firth, C.; Hirschberg, D.L.; Rice, C.M.; Shields, S.; et al. Characterization of a canine homolog of hepatitis C. virus. Proc. Natl. Acad. Sci. USA 2011, 108, 11608–11613. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Simmonds, P.; Scheel, T.K.; Hjelle, B.; Cullen, J.M.; Burbelo, P.D.; Chauhan, L.V.; Duraisamy, R.; Sanchez Leon, M.; Jain, K.; et al. Identification of rodent homologs of hepatitis C virus and pegiviruses. MBio 2013, 4, e00216–e00213. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.L.; Schoeniger, A.; Becher, P.; Baechlein, C. Mutational Analysis of the Bovine Hepacivirus Internal Ribosome Entry Site. J. Virol. 2018, 92, 1974-17. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Scheel, T.K.H.; Luna, J.M.; Chung, H.; Nishiuchi, E.; Scull, M.A.; Echeverria, N.; Ricardo-Lax, I.; Kapoor, A.; Lipkin, I.W.; et al. miRNA independent hepacivirus variants suggest a strong evolutionary pressure to maintain miR-122 dependence. PLoS Pathog. 2017, 13, e1006694. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Klimstra, W.B. MicroRNA Regulation of RNA Virus Replication and Pathogenesis. Trends Mol. Med. 2017, 23, 80–93. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | miRNA | Targets 1 | Predicted Roles | References |
|---|---|---|---|---|
| HSV-1 | miR-101 | ATP5B | Blocks DNA packaging and capsid maturation | [34] |
| GRSF1 | Attenuates viral replication | [33] | ||
| miR-138 | ICPO (v) | Inhibits lytic cycle gene expression | [36] | |
| miR-199a | ARHGAP21 | Alters Golgi function disturbing viral envelopment | [48] | |
| miR-146a | Complement factor H | Immune evasion | [49] | |
| Arachidonic cascade | Alzheimer-type neurological changes | [49,50] | ||
| miR-23a | IRF1 | Innate immune evasion | [51,52] | |
| miR-649 | MALT1 | Innate and adaptive immune evasion | [53,54] | |
| miR-132 | p300 | Innate immune evasion | [55] | |
| HCMV | miR-200 family | UL112 (v) | Inhibits viral reactivation | [39] |
| miR-21-5p | Cdc25a | Inhibits viral replication | [56] | |
| miR-27b | EN2 | Alters glioma cell morphology, neurological disorders | [57] | |
| miR-132 | p300 | Innate immune evasion | [55] | |
| EBV | miR-200b, -429 | ZEB1, ZEB2 | Reactivation of lytic cycle, viral replication | [41,42] |
| let-7 | Dicer | Promotes latency | [43] | |
| miR-190 | NR4A3 | Inhibits lytic cycle | [58] | |
| TP53INP1 | Enhances cell survival | [58] | ||
| miR-424 | SIAH1 | Inhibits apoptosis | [59] | |
| miR-127 | BLIMP1, XBP-1 | Lymphomagenesis, blocks B-cell differentiation | [60] | |
| KSHV | miR-320d, -498, -1258 | RTA (v) | Inhibition of reactivation | [46,47] |
| miR-132 | p300 | Innate immune evasion | [55] | |
| miR-21 | Pdcd4, PTEN | Cell migration, invasion, angiogenesis | [61,62,63] | |
| miR-31 | FAT4 | Cell migration | [61,64] | |
| miR-146a | CXCR4 | Cell migration | [65] | |
| miR-221/222 cluster | ETS1, ETS2 | Cell migration | [64] | |
| miR-30b/c | DLL4 | Angiogenesis | [66] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernier, A.; Sagan, S.M. The Diverse Roles of microRNAs at the Host–Virus Interface. Viruses 2018, 10, 440. https://doi.org/10.3390/v10080440
Bernier A, Sagan SM. The Diverse Roles of microRNAs at the Host–Virus Interface. Viruses. 2018; 10(8):440. https://doi.org/10.3390/v10080440
Chicago/Turabian StyleBernier, Annie, and Selena M. Sagan. 2018. "The Diverse Roles of microRNAs at the Host–Virus Interface" Viruses 10, no. 8: 440. https://doi.org/10.3390/v10080440
APA StyleBernier, A., & Sagan, S. M. (2018). The Diverse Roles of microRNAs at the Host–Virus Interface. Viruses, 10(8), 440. https://doi.org/10.3390/v10080440