Redox Biology of Respiratory Viral Infections




Abstract
1. Introduction
2. Respiratory Viruses
2.1. Influenza Viruses
2.2. Human Respiratory Syncytial Virus
2.3. Human Rhinovirus
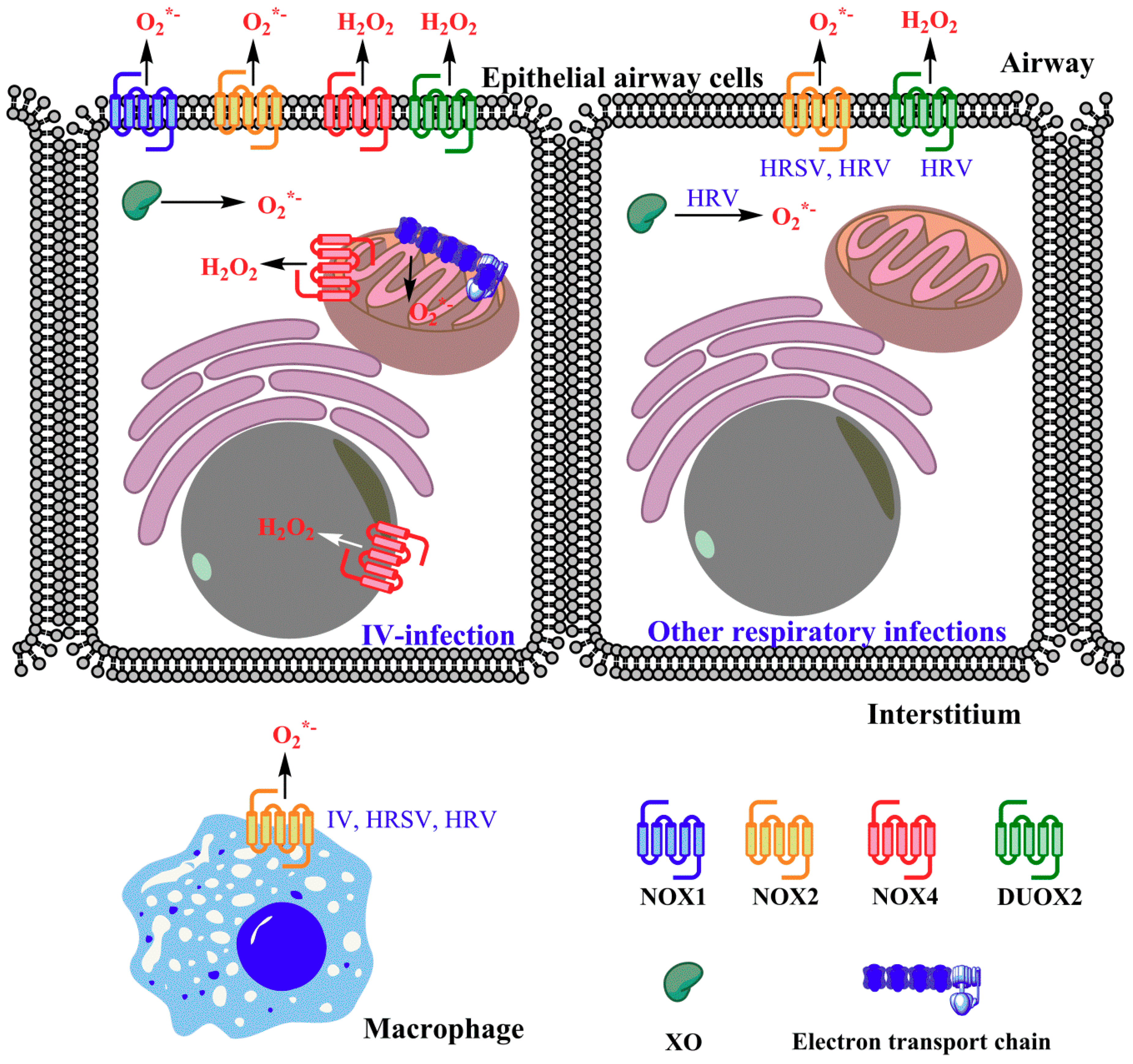
3. Enhanced ROS Production during Viral Respiratory Infections
4. Sources of ROS in the Infected Cells
5. Respiratory Viruses and Antioxidant Defense Pathways
6. Role of ROS in the Life Cycle and Propagation of Respiratory Viruses
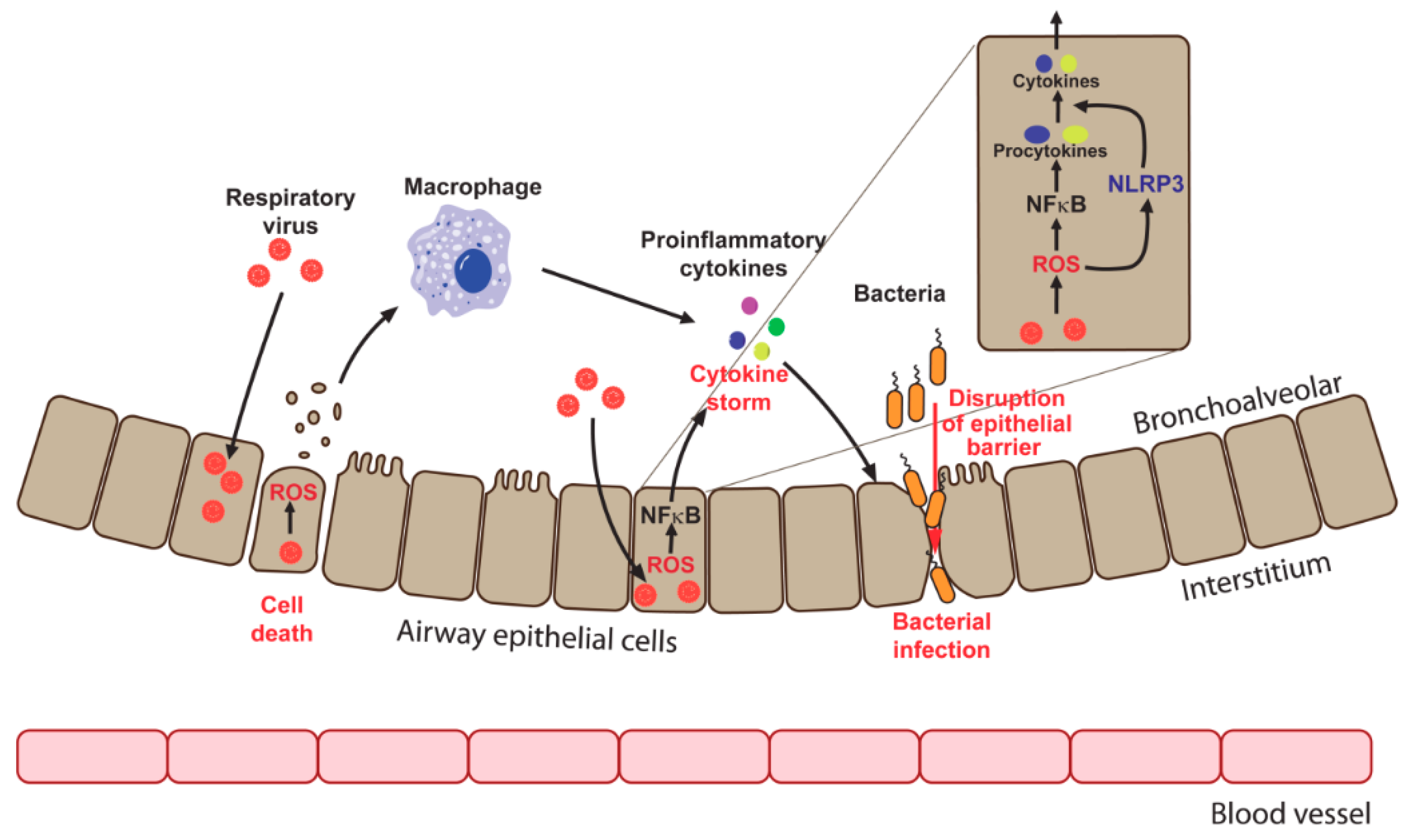
7. ROS in Respiratory Virus Pathology
8. Antioxidant Therapy of Respiratory Viruses
9. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Van der Vliet, A.; Janssen-Heininger, Y.M. Hydrogen peroxide as a damage signal in tissue injury and inflammation: Murderer, mediator, or messenger? J. Cell. Biochem. 2014, 115, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, S.; Nolin, J.; McMillan, D.; Wouters, E.; Janssen-Heininger, Y.; Reynaert, N. Thiol redox chemistry: Role of protein cysteine oxidation and altered redox homeostasis in allergic inflammation and asthma. J. Cell. Biochem. 2015, 116, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2013; 2 volumes. [Google Scholar]
- World Health Organization. Influenza (Seasonal) Fact Sheet No 211; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Colman, P.M. Influenza virus neuraminidase: Structure, antibodies, and inhibitors. Protein Sci. 1994, 3, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Pielak, R.M.; Schnell, J.R.; Chou, J.J. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA 2009, 106, 7379–7384. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.L.; Amarasinghe, G.K.; Banyai, K.; Bao, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.X.; Briese, T.; et al. Taxonomy of the order Mononegavirales: Update 2016. Arch. Virol. 2016, 161, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Walsh, E.E. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 2000, 13, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Bont, L.; Checchia, P.A.; Fauroux, B.; Figueras-Aloy, J.; Manzoni, P.; Paes, B.; Simoes, E.A.; Carbonell-Estrany, X. Defining the Epidemiology and Burden of Severe Respiratory Syncytial Virus Infection Among Infants and Children in Western Countries. Infect. Dis. Ther. 2016, 5, 271–298. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.L.; Kopp, B.T.; Paul, G.; Landgrave, L.C.; Hayes, D., Jr.; Thompson, R. Respiratory syncytial virus: Current and emerging treatment options. Clinicoecon. Outcomes Res. 2014, 6, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Lamson, D.M.; St George, K.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Oh, E.; Kim, Y.; Jung, W.W.; Kim, H.S.; Lee, J.; Sul, D. Enhanced oxidative damage to DNA, lipids, and proteins and levels of some antioxidant enzymes, cytokines, and heat shock proteins in patients infected with influenza H1N1 virus. Acta Virol. 2014, 58, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.P.; Lee, J.C.; Loke, W.M.; Yeo, L.L.; Quek, A.M.; Lim, E.C.; Halliwell, B.; Seet, R.C. Does influenza A infection increase oxidative damage? Antioxid. Redox Signal. 2014, 21, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Erkekoglu, P.; Asci, A.; Ceyhan, M.; Kizilgun, M.; Schweizer, U.; Atas, C.; Kara, A.; Kocer Giray, B. Selenium levels, selenoenzyme activities and oxidant/antioxidant parameters in H1N1-infected children. Turk. J. Pediatr. 2013, 55, 271–282. [Google Scholar] [PubMed]
- Nin, N.; Sanchez-Rodriguez, C.; Ver, L.S.; Cardinal, P.; Ferruelo, A.; Soto, L.; Deicas, A.; Campos, N.; Rocha, O.; Ceraso, D.H.; et al. Lung histopathological findings in fatal pandemic influenza A (H1N1). Med. Intensiv. 2012, 36, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Buffinton, G.D.; Christen, S.; Peterhans, E.; Stocker, R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic. Res. Commun. 1992, 16, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Hennet, T.; Peterhans, E.; Stocker, R. Alterations in antioxidant defences in lung and liver of mice infected with influenza A virus. J. Gen. Virol. 1992, 73 Pt 1, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell. Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Lowther, S.; Stambas, J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J. Virol. 2015, 89, 2672–2683. [Google Scholar] [CrossRef] [PubMed]
- Casola, A.; Burger, N.; Liu, T.; Jamaluddin, M.; Brasier, A.R.; Garofalo, R.P. Oxidant tone regulates RANTES gene expression in airway epithelial cells infected with respiratory syncytial virus. Role in viral-induced interferon regulatory factor activation. J. Biol. Chem. 2001, 276, 19715–19722. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Garcia-Carpizo, V.; Guijarro, T.; Garcia-Gomez, A.; Navarro, D.; Aranda, A.; Zambrano, A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence 2016, 7, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Li, L.Y.; Zhang, M.; Zhang, Q. Inactivated Sendai virus induces apoptosis mediated by reactive oxygen species in murine melanoma cells. Biomed. Environ. Sci. 2016, 29, 877–884. [Google Scholar] [PubMed]
- Qian, M.; Tan, H.M.; Yu, N.; Wang, T.; Zhang, Q. Inactivated Sendai Virus Induces ROS-dependent Apoptosis and Autophagy in Human Prostate Cancer Cells. Biomed. Environ. Sci. 2018, 31, 280–289. [Google Scholar] [PubMed]
- Moreno-Solis, G.; de la Torre-Aguilar, M.J.; Torres-Borrego, J.; Llorente-Cantarero, F.J.; Fernandez-Gutierrez, F.; Gil-Campos, M.; Tunez-Finana, I.; Perez-Navero, J.L. Oxidative Stress and Inflamatory Plasma Biomarkers in Respiratory Syncytial Virus Bronchiolitis. Clin. Respir. J. 2015, 11, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Okamoto, S.; Sawa, T.; Yoshitake, J.; Tamura, F.; Ichimori, K.; Miyazaki, K.; Sasamoto, K.; Maeda, H. 8-nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Jantzi, P.D.; Esham, D.L.; Spratt, H.; Kurosky, A.; Casola, A.; Garofalo, R.P. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 2011, 183, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell. Mol. Biol. 2009, 41, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.; Tian, B.; Boldogh, I.; Garofalo, R.P.; Brasier, A.R. Respiratory syncytial virus infection induces a reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required for cytokine expression. J. Virol. 2009, 83, 10605–10615. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Todokoro, M.; Arakawa, H. RS virus-induced inflammation and the intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation 2009, 32, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Sinha, M.; Liu, T.; Hong, C.; Luxon, B.A.; Garofalo, R.P.; Casola, A. Identification of human metapneumovirus-induced gene networks in airway epithelial cells by microarray analysis. Virology 2008, 374, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.C.; Kaul, P.; Singh, I.; Turner, R.B. The role of oxidative stress in rhinovirus induced elaboration of IL-8 by respiratory epithelial cells. Free Radic. Biol. Med. 1999, 26, 454–462. [Google Scholar] [CrossRef]
- Papi, A.; Contoli, M.; Gasparini, P.; Bristot, L.; Edwards, M.R.; Chicca, M.; Leis, M.; Ciaccia, A.; Caramori, G.; Johnston, S.L.; et al. Role of xanthine oxidase activation and reduced glutathione depletion in rhinovirus induction of inflammation in respiratory epithelial cells. J. Biol. Chem. 2008, 283, 28595–28606. [Google Scholar] [CrossRef] [PubMed]
- Kaul, P.; Biagioli, M.C.; Singh, I.; Turner, R.B. Rhinovirus-induced oxidative stress and interleukin-8 elaboration involves p47-phox but is independent of attachment to intercellular adhesion molecule-1 and viral replication. J. Infect. Dis. 2000, 181, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.; Porto, B.N. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef] [PubMed]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.; Bose, S. TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Stambas, J.; Bozinovski, S.; Broughton, B.R.; Drummond, G.R.; Selemidis, S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011, 7, e1001271. [Google Scholar] [CrossRef] [PubMed]
- To, E.E.; Broughton, B.R.; Hendricks, K.S.; Vlahos, R.; Selemidis, S. Influenza A virus and TLR7 activation potentiate NOX2 oxidase-dependent ROS production in macrophages. Free Radic. Res. 2014, 48, 940–947. [Google Scholar] [CrossRef] [PubMed]
- To, E.E.; Vlahos, R.; Luong, R.; Halls, M.L.; Reading, P.C.; King, P.T.; Chan, C.; Drummond, G.R.; Sobey, C.G.; Broughton, B.R.S.; et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat. Commun. 2017, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Duval, A.; Martel, A.; Soucy-Faulkner, A.; Grandvaux, N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-κB in airway epithelial cells. J. Immunol. 2008, 180, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Snelgrove, R.J.; Edwards, L.; Rae, A.J.; Hussell, T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur. J. Immunol. 2006, 36, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Trocme, C.; Deffert, C.; Cachat, J.; Donati, Y.; Tissot, C.; Papacatzis, S.; Braunersreuther, V.; Pache, J.C.; Krause, K.H.; Holmdahl, R.; et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J. Pathol. 2015, 235, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Soucy-Faulkner, A.; Mukawera, E.; Fink, K.; Martel, A.; Jouan, L.; Nzengue, Y.; Lamarre, D.; Vande Velde, C.; Grandvaux, N. Requirement of NOX2 and reactive oxygen species for efficient RIG-I-mediated antiviral response through regulation of MAVS expression. PLoS Pathog. 2010, 6, e1000930. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Mariani, M.; Fink, K. Lung epithelial NOX/DUOX and respiratory virus infections. Clin. Sci. (Lond.) 2015, 128, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Van den Brand, J.M.; Stittelaar, K.J.; van Amerongen, G.; Reperant, L.; de Waal, L.; Osterhaus, A.D.; Kuiken, T. Comparison of temporal and spatial dynamics of seasonal H3N2, pandemic H1N1 and highly pathogenic avian influenza H5N1 virus infections in ferrets. PLoS ONE 2012, 7, e42343. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.V.; Bagci, U.; Chu, Y.K.; Squier, B.; Fraig, M.; Uriarte, S.M.; Guo, H.; Mollura, D.J.; Jonsson, C.B. Lower Respiratory Tract Infection of the Ferret by 2009 H1N1 Pandemic Influenza A Virus Triggers Biphasic, Systemic, and Local Recruitment of Neutrophils. J. Virol. 2015, 89, 8733–8748. [Google Scholar] [CrossRef] [PubMed]
- Geerdink, R.J.; Pillay, J.; Meyaard, L.; Bont, L. Neutrophils in respiratory syncytial virus infection: A target for asthma prevention. J. Allergy Clin. Immunol. 2015, 136, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, C.H.; Ryu, J.H.; Kim, M.J.; Park, C.Y.; Lee, J.M.; Holtzman, M.J.; Yoon, J.H. Reactive oxygen species induce antiviral innate immune response through IFN-λ regulation in human nasal epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2013, 49, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Selemidis, S.; Seow, H.J.; Broughton, B.R.; Vinh, A.; Bozinovski, S.; Sobey, C.G.; Drummond, G.R.; Vlahos, R. NOX1 oxidase suppresses influenza a virus-induced lung inflammation and oxidative stress. PLoS ONE 2013, 8, e60792. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, A.R.; De La Cruz, J.A.; Cao, W.; Patel, J.; Belser, J.A.; McCoy, J.; Liepkalns, J.S.; Amoah, S.; Cheng, G.; Ranjan, P.; et al. NADPH Oxidase 1 Is Associated with Altered Host Survival and T Cell Phenotypes after Influenza A Virus Infection in Mice. PLoS ONE 2016, 11, e0149864. [Google Scholar] [CrossRef] [PubMed]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef] [PubMed]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.N.; Hershenson, M.B.; Sajjan, U.S. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [PubMed]
- Tattoli, I.; Carneiro, L.A.; Jehanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J.; Kundu, J.K.; Na, H.K.; Lee, J.S. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J. Nutr. 2005, 135, S2993–S3001. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, C.H.; Kim, M.J.; Ryu, J.H.; Seong, S.Y.; Kim, S.; Lim, S.J.; Holtzman, M.J.; Yoon, J.H. The Induction of Pattern-Recognition Receptor Expression against Influenza A Virus through Duox2-Derived Reactive Oxygen Species in Nasal Mucosa. Am. J. Respir. Cell Mol. Biol. 2015, 53, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Strengert, M.; Jennings, R.; Davanture, S.; Hayes, P.; Gabriel, G.; Knaus, U.G. Mucosal reactive oxygen species are required for antiviral response: Role of Duox in influenza a virus infection. Antioxid. Redox Signal. 2014, 20, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.N.; Kim, J.Y.; Kim, H.; Kim, D.Y.; Won, T.B.; Han, D.H.; Rhee, C.S.; Kim, H.J. Duox2 is required for the transcription of pattern recognition receptors in acute viral lung infection: An interferon-independent regulatory mechanism. Antivir. Res. 2016, 134, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Harper, R.W.; Xu, C.; Eiserich, J.P.; Chen, Y.; Kao, C.Y.; Thai, P.; Setiadi, H.; Wu, R. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005, 579, 4911–4917. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Ganesan, S.; Comstock, A.T.; Meldrum, C.A.; Mahidhara, R.; Goldsmith, A.M.; Curtis, J.L.; Martinez, F.J.; Hershenson, M.B.; Sajjan, U. Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ando, M.; Oda, T.; Doi, T.; Ijiri, S.; Araki, S.; Maeda, H. Dependence on O2− generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Investig. 1990, 85, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Akaike, T.; Hamamoto, T.; Suzuki, F.; Hirano, T.; Maeda, H. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 1989, 244, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, M.J.; Park, D.Y.; Chung, H.J.; Kim, C.H.; Yoon, J.H.; Kim, H.J. Mitochondrial reactive oxygen species modulate innate immune response to influenza A virus in human nasal epithelium. Antivir. Res. 2015, 119, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Armeanu, S.; Prinz, F.; Ungerechts, G.; Wybranietz, W.; Spiegel, M.; Bernlohr, C.; Cecconi, F.; Gregor, M.; Neubert, W.J.; et al. Caspase-8 and Apaf-1-independent caspase-9 activation in Sendai virus-infected cells. J. Biol. Chem. 2002, 277, 29817–29824. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Akaike, T.; Ichimori, K.; Akuta, T.; Kaneko, K.; Nakayama, H.; Stuehr, D.J.; Maeda, H. Superoxide generation mediated by 8-nitroguanosine, a highly redox-active nucleic acid derivative. Biochem. Biophys. Res. Commun. 2003, 311, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, A.; Iles, K.E.; Liu, G.; Noah, D.L.; Noah, J.W.; Matalon, S. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J. 2009, 23, 3829–3842. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.; Pyo, C.W.; Jung, K.I.; Choi, S.Y. Influenza A virus PB1-F2 is involved in regulation of cellular redox state in alveolar epithelial cells. Biochem. Biophys. Res. Commun. 2015, 459, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Flory, E.; Kunz, M.; Scheller, C.; Jassoy, C.; Stauber, R.; Rapp, U.R.; Ludwig, S. Influenza virus-induced NF-κB-dependent gene expression is mediated by overexpression of viral proteins and involves oxidative radicals and activation of IκB kinase. J. Biol. Chem. 2000, 275, 8307–8314. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Lin, K.H.; Hsieh, T.H.; Shiu, S.Y.; Li, J.Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 2006, 46, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Shuvaeva, T.; Dodia, C.; Kazi, A.; Feinstein, S.I.; Fisher, A.B. Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase A(2) activities. Arch. Biochem. Biophys. 2009, 485, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, B.; Messier, E.M.; Janssen, W.J.; Nahreini, P.; Wang, J.; Hartshorn, K.L.; Mason, R.J. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir. Res. 2012, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Limmon, G.V.; Zheng, D.; Li, N.; Li, L.; Yin, L.; Chow, V.T.; Chen, J.; Engelward, B.P. Major shifts in the spatio-temporal distribution of lung antioxidant enzymes during influenza pneumonia. PLoS ONE 2012, 7, e31494. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.F.; McCorrister, S.; Hu, P.; Chong, P.; Silaghi, A.; Westmacott, G.; Coombs, K.M.; Kobasa, D. Highly Pathogenic H5N1 and Novel H7N9 Influenza A Viruses Induce More Profound Proteomic Host Responses than Seasonal and Pandemic H1N1 Strains. J. Proteome Res. 2015, 14, 4511–4523. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, D.B.; Choi, A.M. Influenza virus induces expression of antioxidant genes in human epithelial cells. Free Radic. Biol. Med. 1994, 16, 821–824. [Google Scholar] [CrossRef]
- Pyo, C.W.; Shin, N.; Jung, K.I.; Choi, J.H.; Choi, S.Y. Alteration of copper-zinc superoxide dismutase 1 expression by influenza A virus is correlated with virus replication. Biochem. Biophys. Res. Commun. 2014, 450, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, R.; Zou, W.; Sun, X.; Liu, X.; Zhao, L.; Wang, S.; Jin, M. The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression. Viruses 2016, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zaas, A.K.; Rao, A.; Dobigeon, N.; Woolf, P.J.; Veldman, T.; Oien, N.C.; McClain, M.T.; Varkey, J.B.; Nicholson, B.; et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet. 2011, 7, e1002234. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.; Mediavilla-Varela, M.; Antonia, S. Indoleamine 2,3-dioxygenase: Is it an immune suppressor? Cancer J. 2010, 16, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Knobil, K.; Otterbein, S.L.; Eastman, D.A.; Jacoby, D.B. Oxidant stress responses in influenza virus pneumonia: Gene expression and transcription factor activation. Am. J. Physiol. 1996, 271, L383–L391. [Google Scholar] [CrossRef] [PubMed]
- Komaravelli, N.; Tian, B.; Ivanciuc, T.; Mautemps, N.; Brasier, A.R.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of Nrf2. Free Radic. Biol. Med. 2015, 88, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Imani, F.; Miller-DeGraff, L.; Walters, D.; Melendi, G.A.; Yamamoto, M.; Polack, F.P.; Kleeberger, S.R. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 2009, 179, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.P.; Kolli, D.; Casola, A. Respiratory syncytial virus infection: Mechanisms of redox control and novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 186–217. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.; Wiktorowicz, J.E.; Soman, K.V.; Boldogh, I.; Forbus, J.D.; Spratt, H.; Garofalo, R.P.; Brasier, A.R. Role of peroxiredoxin 1 and peroxiredoxin 4 in protection of respiratory syncytial virus-induced cysteinyl oxidation of nuclear cytoskeletal proteins. J. Virol. 2010, 84, 9533–9545. [Google Scholar] [CrossRef] [PubMed]
- Hastie, M.L.; Headlam, M.J.; Patel, N.B.; Bukreyev, A.A.; Buchholz, U.J.; Dave, K.A.; Norris, E.L.; Wright, C.L.; Spann, K.M.; Collins, P.L.; et al. The human respiratory syncytial virus nonstructural protein 1 regulates type I and type II interferon pathways. Mol. Cell. Proteom. 2012, 11, 108–127. [Google Scholar] [CrossRef] [PubMed]
- Darville, M.I.; Ho, Y.S.; Eizirik, D.L. NF-κB is required for cytokine-induced manganese superoxide dismutase expression in insulin-producing cells. Endocrinology 2000, 141, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kaul, P.; Singh, I.; Turner, R.B. Effect of rhinovirus challenge on antioxidant enzymes in respiratory epithelial cells. Free Radic. Res. 2002, 36, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Ciriolo, M.R.; Palamara, A.T.; Incerpi, S.; Lafavia, E.; Bue, M.C.; De Vito, P.; Garaci, E.; Rotilio, G. Loss of GSH, oxidative stress, and decrease of intracellular pH as sequential steps in viral infection. J. Biol. Chem. 1997, 272, 2700–2708. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.; Chan, J.Y. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 2010, 244, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Chen, Y.; Seth, S.; Furukawa, S.; Compans, R.W.; Jones, D.P. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 2003, 34, 928–936. [Google Scholar] [CrossRef]
- Nencioni, L.; Iuvara, A.; Aquilano, K.; Ciriolo, M.R.; Cozzolino, F.; Rotilio, G.; Garaci, E.; Palamara, A.T. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003, 17, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Kesic, M.J.; Simmons, S.O.; Bauer, R.; Jaspers, I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic. Biol. Med. 2011, 51, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Pauli, E.K.; Schmolke, M.; Wolff, T.; Viemann, D.; Roth, J.; Bode, J.G.; Ludwig, S. Influenza A virus inhibits type I IFN signaling via NF-κB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008, 4, e1000196. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; Herold, S.; Cakarova, L.; Hoegner, K.; Lohmeyer, J.; Planz, O.; Pleschka, S. Inhibition of influenza virus-induced NF-κB and Raf/MEK/ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo. Antivir. Res. 2011, 92, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Hillesheim, A.; Nordhoff, C.; Boergeling, Y.; Ludwig, S.; Wixler, V. β-catenin promotes the type I IFN synthesis and the IFN-dependent signaling response but is suppressed by influenza A virus-induced RIG-I/NF-κB signaling. Cell Commun. Signal. 2014, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell. Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Martin, L.; Mukawera, E.; Chartier, S.; De Deken, X.; Brochiero, E.; Miot, F.; Grandvaux, N. IFNβ/TNFalpha synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH oxidase-mediated airway antiviral response. Cell. Res. 2013, 23, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Castro, S.; Brasier, A.R.; Jamaluddin, M.; Garofalo, R.P.; Casola, A. Reactive oxygen species mediate virus-induced STAT activation: Role of tyrosine phosphatases. J. Biol. Chem. 2004, 279, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory syncytial virus-mediated NF-κB p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKKβ. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Guan, X.; Yoboua, F.; Zucchini, N.; Fink, K.; Doyon, P.; Martin, L.; Servant, M.J.; Chartier, S. Sustained activation of interferon regulatory factor 3 during infection by paramyxoviruses requires MDA5. J. Innate Immun. 2014, 6, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-κB, and proinflammatory cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.P.; Casola, A.; Bao, X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Barik, S. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr. Top. Microbiol. Immunol. 2013, 372, 173–191. [Google Scholar] [PubMed]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; San Juan-Vergara, H.; Mohapatra, S.; Mohapatra, S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE 2012, 7, e29386. [Google Scholar] [CrossRef] [PubMed]
- Swedan, S.; Musiyenko, A.; Barik, S. Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J. Virol. 2009, 83, 9682–9693. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Papadopoulos, N.G.; Stanciu, L.A.; Bellettato, C.M.; Pinamonti, S.; Degitz, K.; Holgate, S.T.; Johnston, S.L. Reducing agents inhibit rhinovirus-induced up-regulation of the rhinovirus receptor intercellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells. FASEB J. 2002, 16, 1934–1936. [Google Scholar] [CrossRef] [PubMed]
- Palamara, A.T.; Di Francesco, P.; Ciriolo, M.R.; Bue, C.; Lafavia, E.; Rotilio, G.; Garaci, E. Cocaine increases Sendai virus replication in cultured epithelial cells: Critical role of the intracellular redox status. Biochem. Biophys. Res. Commun. 1996, 228, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Dosal, R.; Horan, K.A.; Paludan, S.R. Mitochondria-derived reactive oxygen species negatively regulates immune innate signaling pathways triggered by a DNA virus, but not by an RNA virus. Biochem Biophys. Res. Commun. 2012, 418, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Vendemiale, G.; Grattagliano, I.; Portincasa, P.; Serviddio, G.; Palasciamo, G.; Altomare, E. Oxidative stress in symptom-free HCV carriers: Relation with ALT flare-up. Eur. J. Clin. Investig. 2001, 31, 54–63. [Google Scholar] [CrossRef]
- Jain, S.K.; Pemberton, P.W.; Smith, A.; McMahon, R.F.; Burrows, P.C.; Aboutwerat, A.; Warnes, T.W. Oxidative stress in chronic hepatitis C: Not just a feature of late stage disease. J. Hepatol. 2002, 36, 805–811. [Google Scholar] [CrossRef]
- Bolukbas, C.; Bolukbas, F.F.; Horoz, M.; Aslan, M.; Celik, H.; Erel, O. Increased oxidative stress associated with the severity of the liver disease in various forms of hepatitis B virus infection. BMC Infect. Dis. 2005, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Parrish, M.; Chan, T.K.; Yin, L.; Rai, P.; Yoshiyuki, Y.; Abolhassani, N.; Tan, K.B.; Kiraly, O.; Chow, V.T.; et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell. Mol. Life Sci. 2015, 72, 2973–2988. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Xiao, Y.; Davis, A.S.; Walters, K.A.; Chertow, D.S.; Easterbrook, J.D.; Dunfee, R.L.; Sandouk, A.; Jagger, B.W.; Schwartzman, L.M.; et al. Treatment with the reactive oxygen species scavenger EUK-207 reduces lung damage and increases survival during 1918 influenza virus infection in mice. Free Radic. Biol. Med. 2014, 67, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Uchide, N.; Ohyama, K. Antiviral function of pyrrolidine dithiocarbamate against influenza virus: The inhibition of viral gene replication and transcription. J. Antimicrob. Chemother. 2003, 52, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Yatmaz, S.; Seow, H.J.; Gualano, R.C.; Wong, Z.X.; Stambas, J.; Selemidis, S.; Crack, P.J.; Bozinovski, S.; Anderson, G.P.; Vlahos, R. Glutathione peroxidase-1 reduces influenza A virus-induced lung inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 48, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, I.; Zhang, W.; Brighton, L.E.; Carson, J.L.; Styblo, M.; Beck, M.A. Selenium deficiency alters epithelial cell morphology and responses to influenza. Free Radic. Biol. Med. 2007, 42, 1826–1837. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wei, L.; Jiang, D.; Wang, J.; Cong, X.; Fei, R. SARS-CoV nucleocapsid protein induced apoptosis of COS-1 mediated by the mitochondrial pathway. Artif. Cells Blood Substit Immobil. Biotechnol. 2007, 35, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.W.; Dybdahl-Sissoko, N.; Hinshaw, V.S. The influence of calcium and reactive oxygen species on influenza virus-induced apoptosis. Cell Death Differ. 1996, 3, 191–197. [Google Scholar] [PubMed]
- Blume, C.; Reale, R.; Held, M.; Loxham, M.; Millar, T.M.; Collins, J.E.; Swindle, E.J.; Morgan, H.; Davies, D.E. Cellular crosstalk between airway epithelial and endothelial cells regulates barrier functions during exposure to double-stranded RNA. Immun. Inflamm. Dis. 2017, 5, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar] [PubMed]
- Go, Y.M.; Kang, S.M.; Roede, J.R.; Orr, M.; Jones, D.P. Increased inflammatory signaling and lethality of influenza H1N1 by nuclear thioredoxin-1. PLoS ONE 2011, 6, e18918. [Google Scholar] [CrossRef] [PubMed]
- Mastronarde, J.G.; Monick, M.M.; Hunninghake, G.W. Oxidant tone regulates IL-8 production in epithelium infected with respiratory syncytial virus. Am. J. Respir. Cell. Mol. Biol. 1995, 13, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Tergaonkar, V. Roles of NF-κB in health and disease: Mechanisms and therapeutic potential. Clin. Sci. (Lond.) 2009, 116, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Morcillo, E.; Gimeno, C.; Cortijo, J. N-acetyl-l-cysteine (NAC) inhibit mucin synthesis and pro-inflammatory mediators in alveolar type II epithelial cells infected with influenza virus A and B and with respiratory syncytial virus (RSV). Biochem. Pharmacol. 2011, 82, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Mastronarde, J.G.; Monick, M.M.; Mukaida, N.; Matsushima, K.; Hunninghake, G.W. Activator protein-1 is the preferred transcription factor for cooperative interaction with nuclear factor-κB in respiratory syncytial virus-induced interleukin-8 gene expression in airway epithelium. J. Infect. Dis. 1998, 177, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sanders, S.P.; Siekierski, E.S.; Casolaro, V.; Proud, D. Role of NF-κB in cytokine production induced from human airway epithelial cells by rhinovirus infection. J. Immunol. 2000, 165, 3384–3392. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Kolli, D.; Liu, T.; Shan, Y.; Garofalo, R.P.; Casola, A. Human metapneumovirus small hydrophobic protein inhibits NF-κB transcriptional activity. J. Virol. 2008, 82, 8224–8229. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.R.; Moy, J.N.; Roebuck, K.A. Respiratory syncytial virus and TNF alpha induction of chemokine gene expression involves differential activation of Rel A and NF-κB1. BMC Infect. Dis. 2002, 2, 5. [Google Scholar] [CrossRef]
- Indukuri, H.; Castro, S.M.; Liao, S.M.; Feeney, L.A.; Dorsch, M.; Coyle, A.J.; Garofalo, R.P.; Brasier, A.R.; Casola, A. Ikkepsilon regulates viral-induced interferon regulatory factor-3 activation via a redox-sensitive pathway. Virology 2006, 353, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Brasier, A.R.; Casola, A.; Garofalo, R.P.; Kurosky, A. Respiratory Syncytial Virus Infection Triggers Epithelial HMGB1 Release as a Damage-Associated Molecular Pattern Promoting a Monocytic Inflammatory Response. J. Virol. 2016, 90, 9618–9631. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Lv, Y.; Zhuo, Y.; Chen, C.; Shi, H.; Guo, L.; Yang, G.; Hou, Y.; Tan, R.X.; Li, E. Suppression of IRG-1 Reduces Inflammatory Cell Infiltration and Lung Injury in Respiratory Syncytial Virus Infection by Reducing Production of Reactive Oxygen Species. J. Virol. 2016, 90, 7313–7322. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, X.P.; Zhu, X.L.; Li, T.; Liu, Y. IRG1 increases MHC class I level in macrophages through STAT-TAP1 axis depending on NADPH oxidase mediated reactive oxygen species. Int. Immunopharmacol. 2017, 48, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Kar, S.; van Kuppeveld, F.J.; Triantafilou, M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.C.; Tully, J.E.; Guala, A.S.; Reiss, J.N.; Godburn, K.E.; Pociask, D.A.; Alcorn, J.F.; Riches, D.W.; Dienz, O.; Janssen-Heininger, Y.M.; et al. Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-β release in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Sajjan, U.; Wang, Q.; Zhao, Y.; Gruenert, D.C.; Hershenson, M.B. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 2008, 178, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, F.; DeSando, S.A.; Ivanov, A.I.; Chapman, T.J.; Knowlden, S.A.; Beck, L.A.; Georas, S.N. Sustained protein kinase D activation mediates respiratory syncytial virus-induced airway barrier disruption. J. Virol. 2013, 87, 11088–11095. [Google Scholar] [CrossRef] [PubMed]
- Waldron, R.T.; Rey, O.; Zhukova, E.; Rozengurt, E. Oxidative stress induces protein kinase C-mediated activation loop phosphorylation and nuclear redistribution of protein kinase D. J. Biol. Chem. 2004, 279, 27482–27493. [Google Scholar] [CrossRef] [PubMed]
- Waldron, R.T.; Rozengurt, E. Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J. Biol. Chem. 2000, 275, 17114–17121. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.H.; Yang, W.; Beineke, A.; Dijkman, R.; Matrosovich, M.; Baumgartner, W.; Thiel, V.; Valentin-Weigand, P.; Meng, F.; Herrler, G. The differentiated airway epithelium infected by influenza viruses maintains the barrier function despite a dramatic loss of ciliated cells. Sci. Rep. 2016, 6, 39668. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Kasper, J.; van der Aa, S.; Andeweg, A.C.; Zaaraoui-Boutahar, F.; Goeijenbier, M.; Richard, M.; Herold, S.; Becker, C.; Scott, D.P.; et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur. Respir. J. 2016, 47, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Gotts, J.E.; Abbott, J.; Matthay, M.A. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L395–L406. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Yajjala, V.K.; Bauer, C.; Talmon, G.A.; Fischer, K.J.; Kielian, T.; Metzger, D.W. Nox2-derived oxidative stress results in inefficacy of antibiotics against post-influenza S. aureus pneumonia. J. Exp. Med. 2016, 213, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, R.V.; Harrison, K.; Oyston, P.C.; Lukaszewski, R.A.; Clark, G.C. Targeting the “cytokine storm” for therapeutic benefit. Clin. Vaccine Immunol. 2013, 20, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Komaravelli, N.; Casola, A. Respiratory Viral Infections and Subversion of Cellular Antioxidant Defenses. J. Pharmacogenomics Pharmacoproteomics 2014, 5, 1000141. [Google Scholar] [PubMed]
- Uchide, N.; Toyoda, H. Antioxidant therapy as a potential approach to severe influenza-associated complications. Molecules 2011, 16, 2032–2052. [Google Scholar] [CrossRef] [PubMed]
- Sgarbanti, R.; Amatore, D.; Celestino, I.; Marcocci, M.E.; Fraternale, A.; Ciriolo, M.R.; Magnani, M.; Saladino, R.; Garaci, E.; Palamara, A.T.; et al. Intracellular redox state as target for anti-influenza therapy: Are antioxidants always effective? Curr. Top. Med. Chem. 2014, 14, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Ali-Ahmad, D.; Bonville, C.A.; Rosenberg, H.F.; Domachowske, J.B. Replication of respiratory syncytial virus is inhibited in target cells generating nitric oxide in situ. Front. Biosci. 2003, 8, a48–a53. [Google Scholar] [PubMed]
- Stark, J.M.; Khan, A.M.; Chiappetta, C.L.; Xue, H.; Alcorn, J.L.; Colasurdo, G.N. Immune and functional role of nitric oxide in a mouse model of respiratory syncytial virus infection. J. Infect. Dis. 2005, 191, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Bazhanov, N.; Ansar, M.; Ivanciuc, T.; Garofalo, R.P.; Casola, A. Hydrogen Sulfide: A Novel Player in Airway Development, Pathophysiology of Respiratory Diseases, and Antiviral Defenses. Am. J. Respir. Cell. Mol. Biol. 2017, 57, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ma, Y.; Escaffre, O.; Ivanciuc, T.; Komaravelli, N.; Kelley, J.P.; Coletta, C.; Szabo, C.; Rockx, B.; Garofalo, R.P.; et al. Role of hydrogen sulfide in paramyxovirus infections. J. Virol. 2015, 89, 5557–5568. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Selemidis, S. NADPH oxidases as novel pharmacologic targets against influenza A virus infection. Mol. Pharmacol. 2014, 86, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Stambas, J.; Selemidis, S. Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy. Trends Pharmacol. Sci. 2012, 33, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Geiler, J.; Michaelis, M.; Naczk, P.; Leutz, A.; Langer, K.; Doerr, H.W.; Cinatl, J., Jr. N-acetyl-l-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem. Pharmacol. 2010, 79, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P.; Ungheri, D. Synergistic combination of N-acetylcysteine and ribavirin to protect from lethal influenza viral infection in a mouse model. Int. J. Immunopathol. Pharmacol. 2004, 17, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Garozzo, A.; Tempera, G.; Ungheri, D.; Timpanaro, R.; Castro, A. N-acetylcysteine synergizes with oseltamivir in protecting mice from lethal influenza infection. Int. J. Immunopathol. Pharmacol. 2007, 20, 349–354. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Grassi, C.; Carati, L. Attenuation of influenza-like symptomatology and improvement of cell-mediated immunity with long-term N-acetylcysteine treatment. Eur. Respir. J. 1997, 10, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Sarrion, I.; Armengot, M.; Carda, C.; Martinez, I.; Melero, J.A.; Cortijo, J. Respiratory syncytial virus inhibits ciliagenesis in differentiated normal human bronchial epithelial cells: Effectiveness of N-acetylcysteine. PLoS ONE 2012, 7, e48037. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, N.; Zimmer, C.; Jarasch-Althof, N.; Wutzler, P.; Henke, A. Therapy of experimental influenza virus infection with pyrrolidine dithiocarbamate. Med. Microbiol. Immunol. 2011, 200, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.M.; Guerrero-Plata, A.; Suarez-Real, G.; Adegboyega, P.A.; Colasurdo, G.N.; Khan, A.M.; Garofalo, R.P.; Casola, A. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am. J. Respir. Crit. Care Med. 2006, 174, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Serkedjieva, J.; Roeva, I.; Angelova, M.; Dolashka, P.; Voelter, W.G. Combined protective effect of a fungal Cu/Zn-containing superoxide dismutase and rimantadine hydrochloride in experimental murine influenza a virus infection. Acta Virol. 2003, 47, 53–56. [Google Scholar] [PubMed]
- Wyde, P.R.; Moore, D.K.; Pimentel, D.M.; Gilbert, B.E.; Nimrod, R.; Panet, A. Recombinant superoxide dismutase (SOD) administered by aerosol inhibits respiratory syncytial virus infection in cotton rats. Antivir. Res. 1996, 31, 173–184. [Google Scholar] [CrossRef]
- Sidwell, R.W.; Huffman, J.H.; Bailey, K.W.; Wong, M.H.; Nimrod, A.; Panet, A. Inhibitory effects of recombinant manganese superoxide dismutase on influenza virus infections in mice. Antimicrob. Agents Chemother. 1996, 40, 2626–2631. [Google Scholar] [PubMed]
- Suliman, H.B.; Ryan, L.K.; Bishop, L.; Folz, R.J. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L69–L78. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Martinez, I.; Melero, J.A.; Tenor, H.; Cortijo, J. Roflumilast inhibits respiratory syncytial virus infection in human differentiated bronchial epithelial cells. PLoS ONE 2013, 8, e69670. [Google Scholar] [CrossRef] [PubMed]
- Yamaya, M.; Nishimura, H.; Shinya, K.; Hatachi, Y.; Sasaki, T.; Yasuda, H.; Yoshida, M.; Asada, M.; Fujino, N.; Suzuki, T.; et al. Inhibitory effects of carbocisteine on type A seasonal influenza virus infection in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L160–168. [Google Scholar] [CrossRef] [PubMed]
- Asada, M.; Yoshida, M.; Hatachi, Y.; Sasaki, T.; Yasuda, H.; Deng, X.; Nishimura, H.; Kubo, H.; Nagatomi, R.; Yamaya, M. l-carbocisteine inhibits respiratory syncytial virus infection in human tracheal epithelial cells. Respir. Physiol. Neurobiol. 2012, 180, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Furuya, A.; Uozaki, M.; Yamasaki, H.; Arakawa, T.; Arita, M.; Koyama, A.H. Antiviral effects of ascorbic and dehydroascorbic acids in vitro. Int. J. Mol. Med. 2008, 22, 541–545. [Google Scholar] [PubMed]
- Kim, Y.; Kim, H.; Bae, S.; Choi, J.; Lim, S.Y.; Lee, N.; Kong, J.M.; Hwang, Y.I.; Kang, J.S.; Lee, W.J. Vitamin C Is an Essential Factor on the Anti-viral Immune Responses through the Production of Interferon-α/β at the Initial Stage of Influenza A Virus (H3N2) Infection. Immune Netw. 2013, 13, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Li, Y.F.; Tang, L.P.; Tsoi, B.; Chen, M.; Chen, H.; Chen, X.M.; Tan, R.R.; Kurihara, H.; He, R.R. A new mechanism of vitamin C effects on A/FM/1/47(H1N1) virus-induced pneumonia in restraint-stressed mice. Biomed. Res. Int. 2015, 2015, 675149. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yao, D.F.; Ohuchi, M.; Ide, M.; Yano, M.; Okumura, Y.; Kido, H. Ambroxol suppresses influenza-virus proliferation in the mouse airway by increasing antiviral factor levels. Eur. Respir. J. 2002, 19, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Palamara, A.T.; Nencioni, L.; Aquilano, K.; De Chiara, G.; Hernandez, L.; Cozzolino, F.; Ciriolo, M.R.; Garaci, E. Inhibition of influenza A virus replication by resveratrol. J. Infect. Dis. 2005, 191, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.H.; Zang, N.; Li, S.M.; Wang, L.J.; Deng, Y.; He, Y.; Yang, X.Q.; Liu, E.M. Resveratrol Inhibits respiratory syncytial virus-induced IL-6 production, decreases viral replication, and downregulates TRIF expression in airway epithelial cells. Inflammation 2012, 35, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Zang, N.; Xie, X.; Deng, Y.; Wu, S.; Wang, L.; Peng, C.; Li, S.; Ni, K.; Luo, Y.; Liu, E. Resveratrol-mediated gamma interferon reduction prevents airway inflammation and airway hyperresponsiveness in respiratory syncytial virus-infected immunocompromised mice. J. Virol. 2011, 85, 13061–13068. [Google Scholar] [CrossRef] [PubMed]
- Uchide, N.; Ohyama, K.; Bessho, T.; Yuan, B.; Yamakawa, T. Effect of antioxidants on apoptosis induced by influenza virus infection: Inhibition of viral gene replication and transcription with pyrrolidine dithiocarbamate. Antivir. Res. 2002, 56, 207–217. [Google Scholar] [CrossRef]
- Mileva, M.; Bakalova, R.; Tancheva, L.; Galabov, A.; Ribarov, S. Effect of vitamin E supplementation on lipid peroxidation in blood and lung of influenza virus infected mice. Comp. Immunol. Microbiol. Infect. Dis. 2002, 25, 1–11. [Google Scholar] [CrossRef]
- Hayek, M.G.; Taylor, S.F.; Bender, B.S.; Han, S.N.; Meydani, M.; Smith, D.E.; Eghtesada, S.; Meydani, S.N. Vitamin E supplementation decreases lung virus titers in mice infected with influenza. J. Infect. Dis. 1997, 176, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Shi, Z.; Huang, H.; Zhu, H.; Zhou, P.; Zhu, H.; Ju, D. Ability of recombinant human catalase to suppress inflammation of the murine lung induced by influenza A. Inflammation 2014, 37, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.L.; Shi, Z.H.; Huang, H.; Zhu, H.G.; Zhou, P.; Ju, D. Therapeutic effect of recombinant human catalase on H1N1 influenza-induced pneumonia in mice. Inflammation 2010, 33, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Gaudernak, E.; Seipelt, J.; Triendl, A.; Grassauer, A.; Kuechler, E. Antiviral effects of pyrrolidine dithiocarbamate on human rhinoviruses. J. Virol. 2002, 76, 6004–6015. [Google Scholar] [CrossRef] [PubMed]
- Krenn, B.M.; Holzer, B.; Gaudernak, E.; Triendl, A.; van Kuppeveld, F.J.; Seipelt, J. Inhibition of polyprotein processing and RNA replication of human rhinovirus by pyrrolidine dithiocarbamate involves metal ions. J. Virol. 2005, 79, 13892–13899. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Yamaya, M.; Sasaki, T.; Inoue, D.; Nakayama, K.; Yamada, M.; Asada, M.; Yoshida, M.; Suzuki, T.; Nishimura, H.; et al. Carbocisteine inhibits rhinovirus infection in human tracheal epithelial cells. Eur. Respir. J. 2006, 28, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Yamaya, M.; Nishimura, H.; Nadine, L.K.; Ota, C.; Kubo, H.; Nagatomi, R. Ambroxol inhibits rhinovirus infection in primary cultures of human tracheal epithelial cells. Arch. Pharm. Res. 2014, 37, 520–529. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Antioxidant | Influenza Virus | Human Respiratory Syncytial Virus | ||||||
|---|---|---|---|---|---|---|---|---|
| Model System | Antiviral Activity | Synergism with | Ref | Model System | Antiviral Activity | Synergism with | Ref | |
| N-acetylcysteine (NAC) | Cells Mice Human | Yes | Ribavirin, oseltamivir | [185,186,187,188] | Cells | ? * | NA | [31,32,143,189] |
| Pyrrolidine dithiocarbamate (PDTC) | Cells Mice | Yes | ND ** | [132,190] | ND | ND | ND | ND |
| Glutathione | Cells Mice | Yes | ND | [108,109] | Cells | Yes | ND | [32] |
| Butylated hydroxyanisole (BHA) | - | - | - | - | Cells Mice | No | ND | [31,191] |
| SOD1 | Cells Mice | No | Rimantadine | [74,95,192] | Rats | Yes | ND | [193] |
| SOD2 | Mice | No | Ribavirin | [194] | Rats | Yes | ND | [193] |
| SOD3 | Mice | No | ND | [195] | ND | ND | ND | ND |
| Roflumilast | - | - | - | - | Cells | Yes | NA | [196] |
| Carbocisteine | Cells | Yes | ND | [197] | Cells | Yes | NA | [198] |
| Ascorbic acid | Cells Mice | Yes | ND | [199,200,201] | ND | ND | ND | ND |
| Ambroxol | Mice | Yes | ND | [202] | ND | ND | ND | ND |
| Resveratrol | Cells Mice | Yes | ND | [203] | Cells Mice | Yes | ND | [204,205] |
| Vitamin E or Trolox | Cells Mice | ? *** | ND | [206,207,208] | ND | ND | ND | ND |
| Catalase | Mice | Yes | ND | [209,210] | Cells | No | ND | [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. https://doi.org/10.3390/v10080392
Khomich OA, Kochetkov SN, Bartosch B, Ivanov AV. Redox Biology of Respiratory Viral Infections. Viruses. 2018; 10(8):392. https://doi.org/10.3390/v10080392
Chicago/Turabian StyleKhomich, Olga A., Sergey N. Kochetkov, Birke Bartosch, and Alexander V. Ivanov. 2018. "Redox Biology of Respiratory Viral Infections" Viruses 10, no. 8: 392. https://doi.org/10.3390/v10080392
APA StyleKhomich, O. A., Kochetkov, S. N., Bartosch, B., & Ivanov, A. V. (2018). Redox Biology of Respiratory Viral Infections. Viruses, 10(8), 392. https://doi.org/10.3390/v10080392