Co-Infection Patterns in Individual Ixodes scapularis Ticks Reveal Associations between Viral, Eukaryotic and Bacterial Microorganisms


Abstract
1. Introduction
2. Materials and Methods
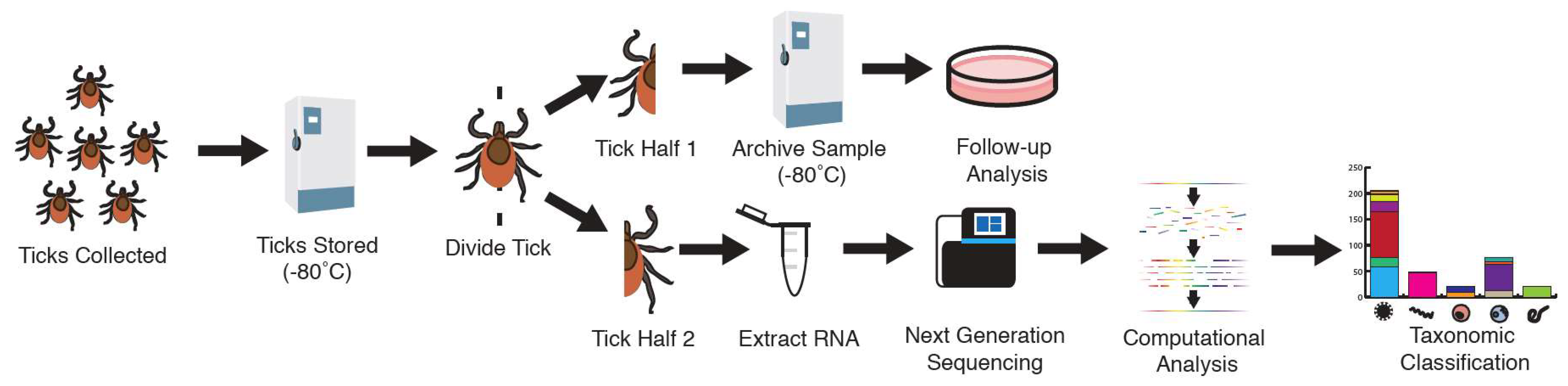
2.1. Sample Collection
2.2. RNA Extraction
2.3. Shotgun Metagenomic Library Preparation
2.4. Sequence Analysis
2.5. Validation of Sequencing by PCR
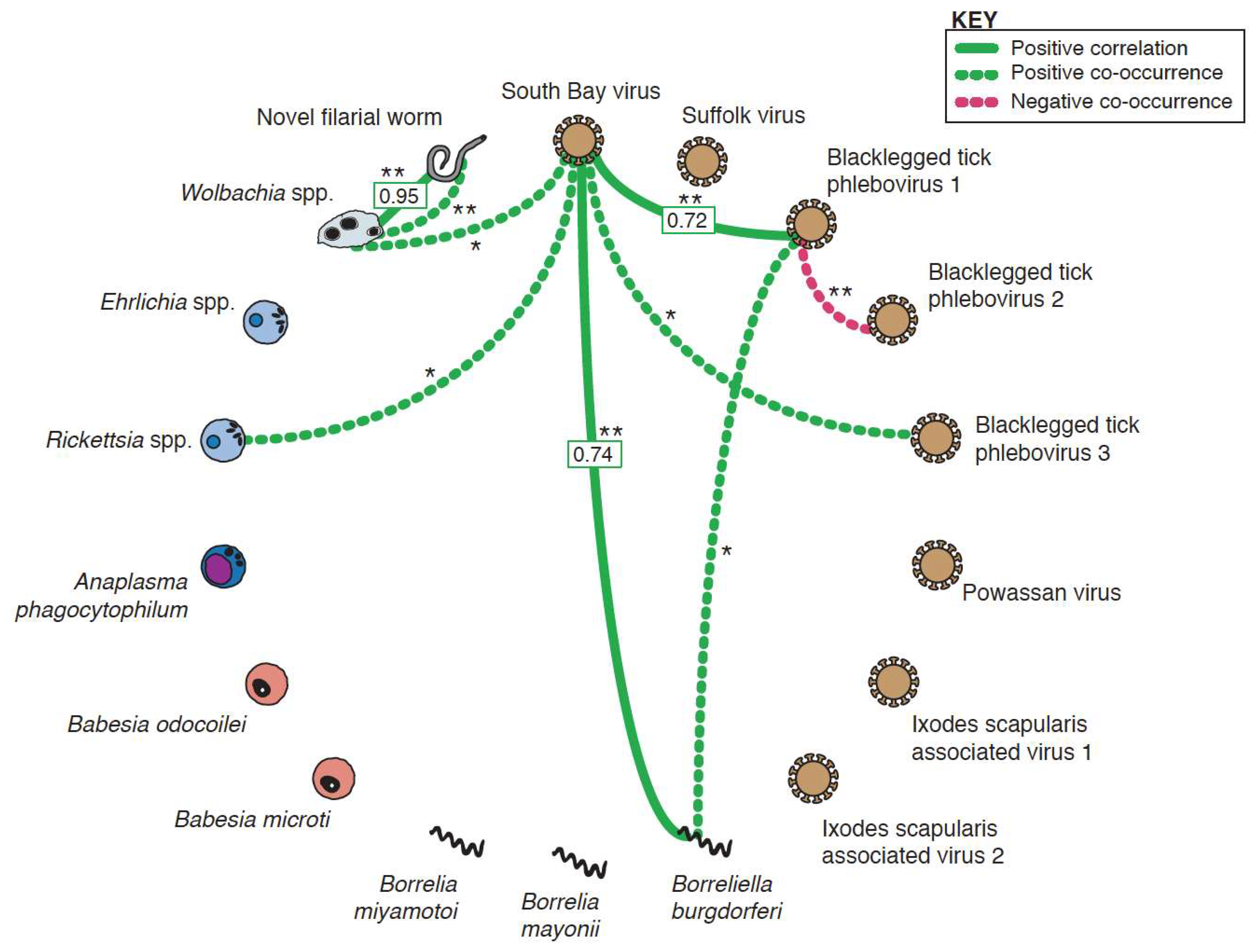
2.6. Statistical Analysis of Microbial Relationships
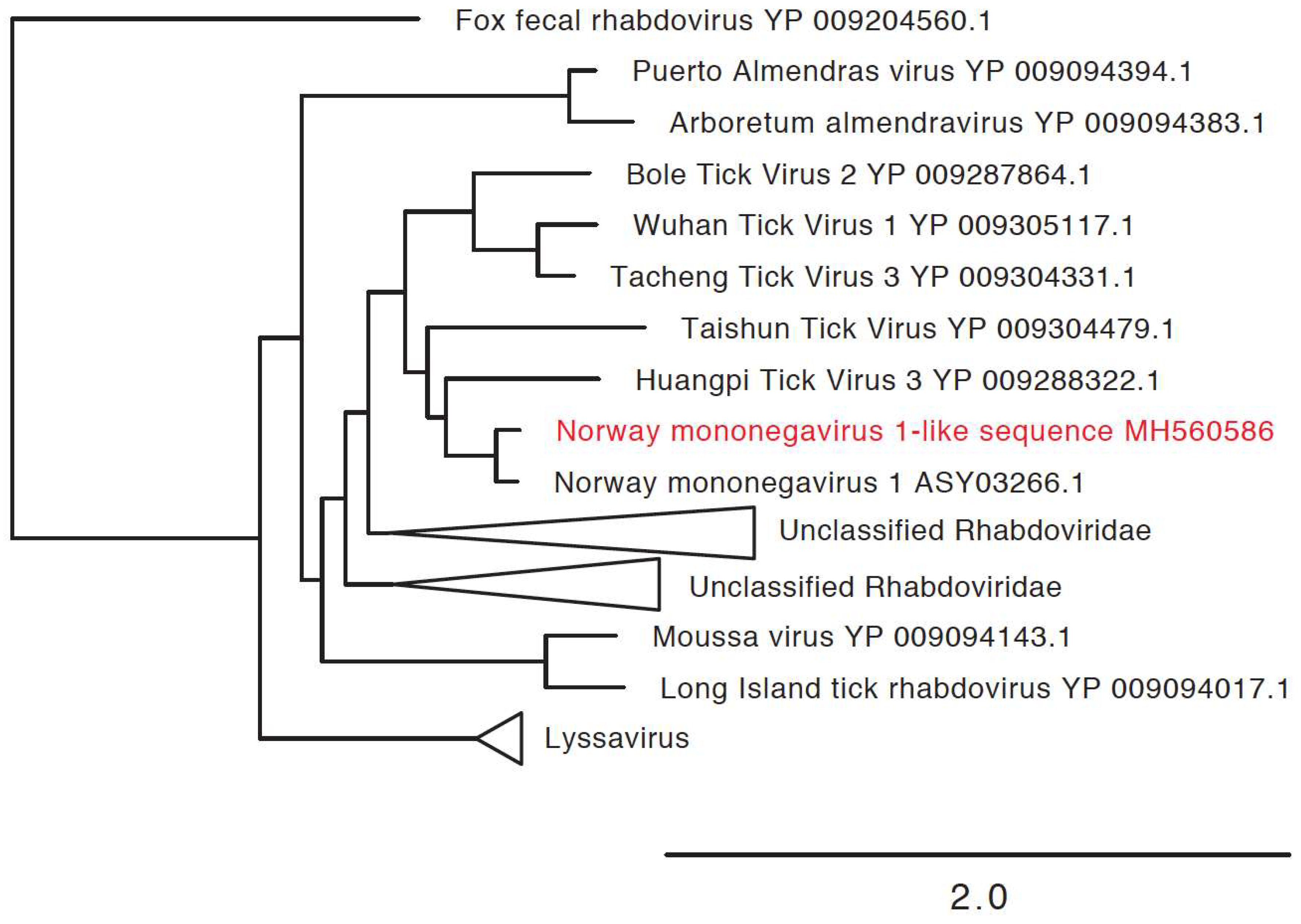
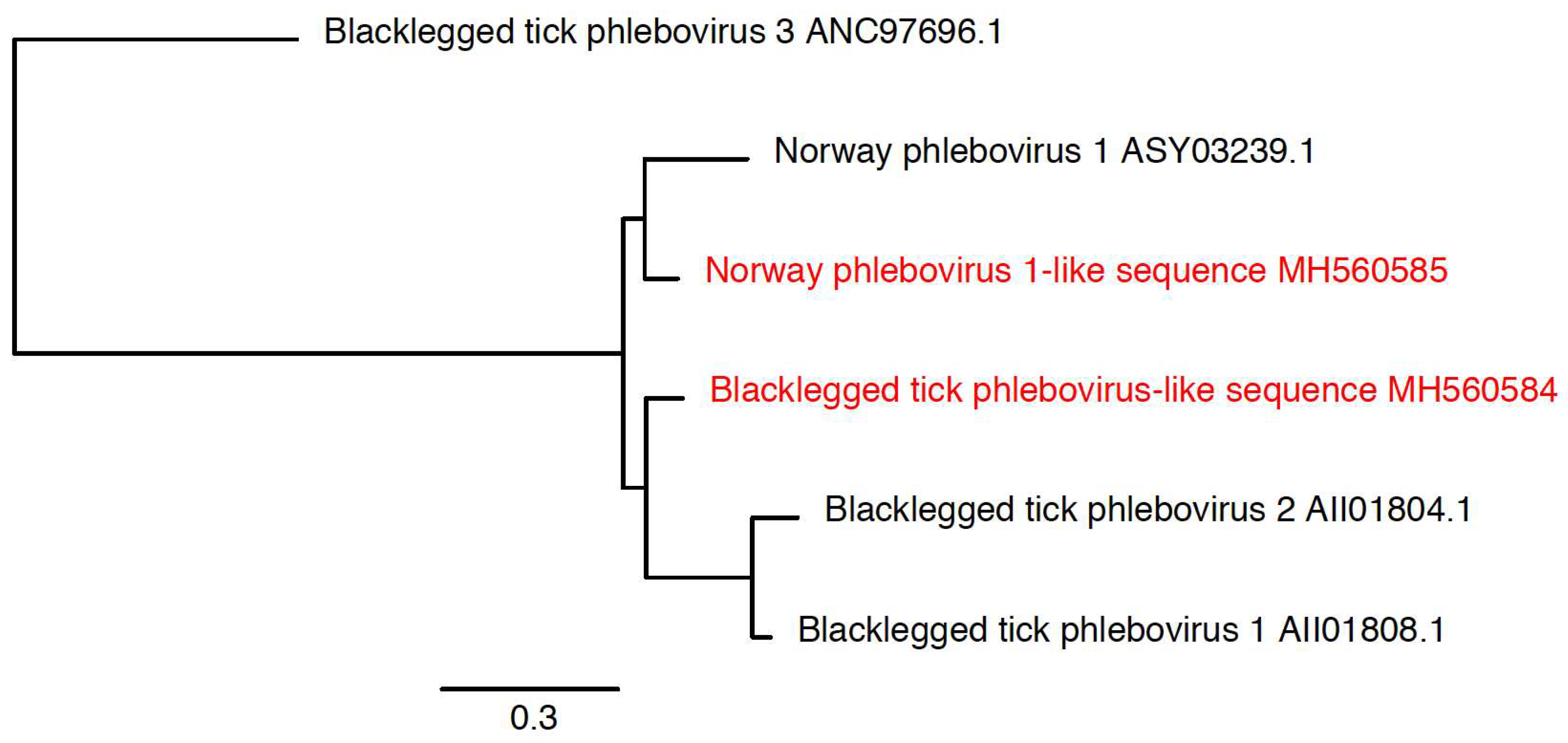
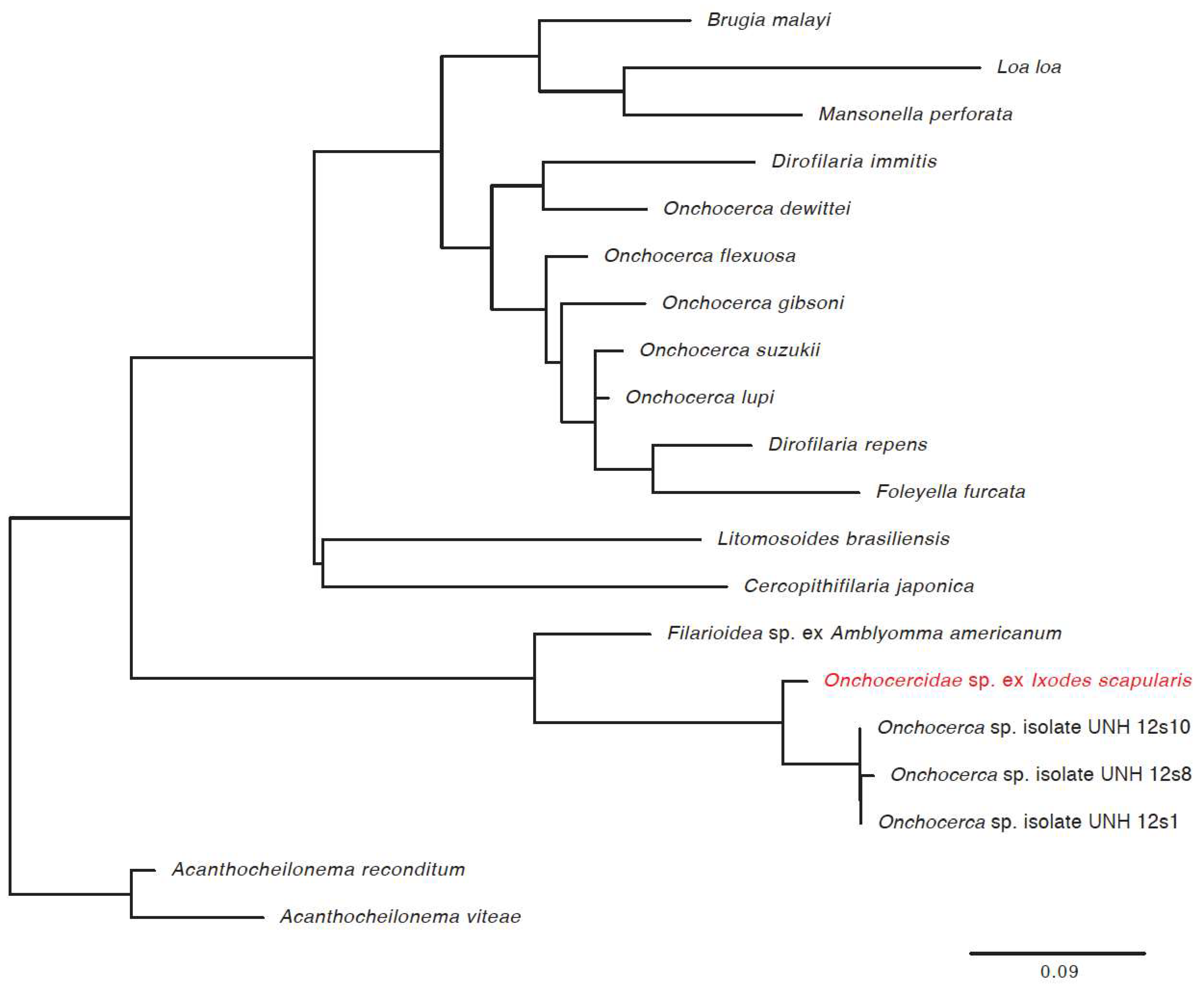
2.7. Phylogenetic Analysis of Novel Microorganisms
3. Results
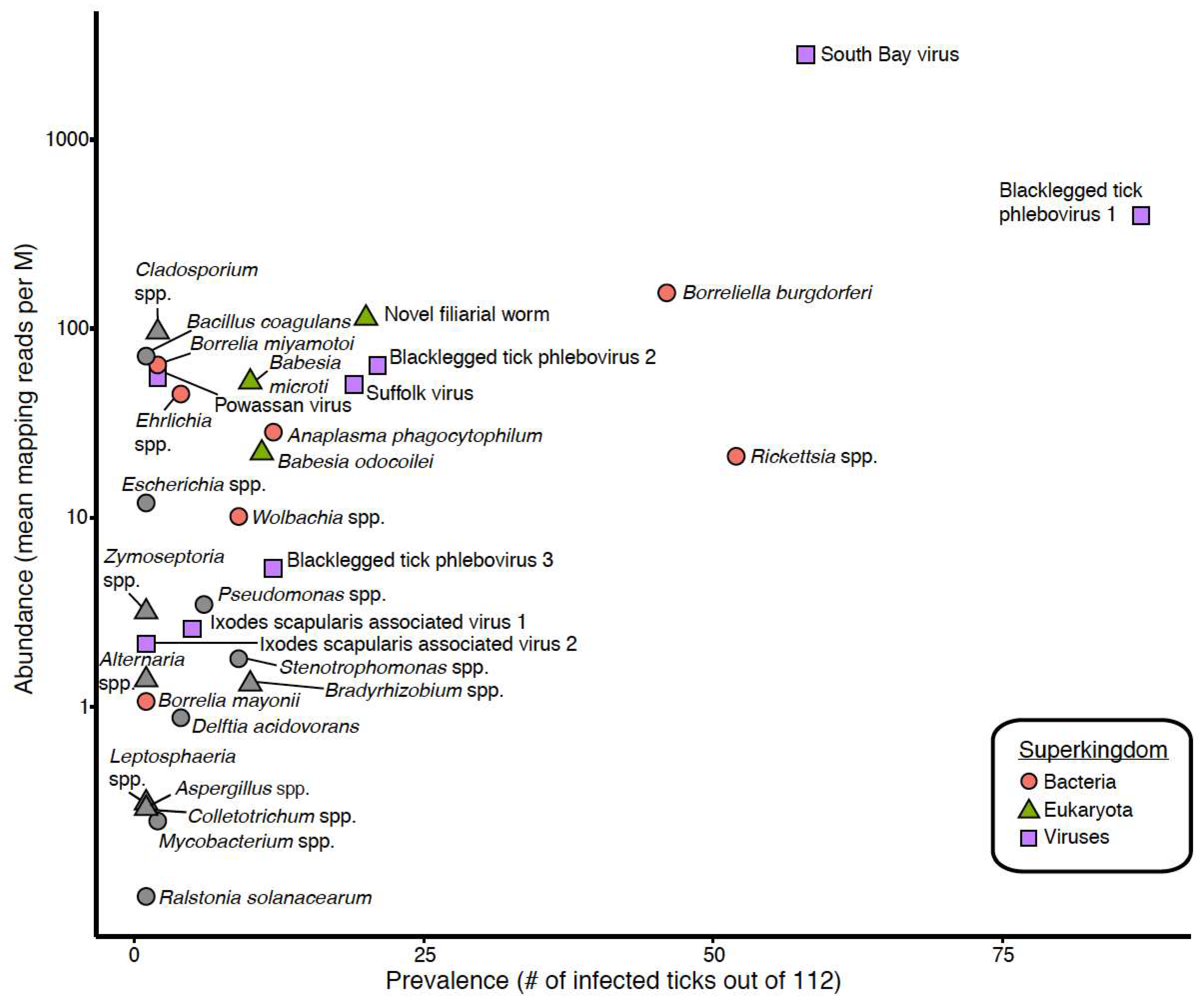
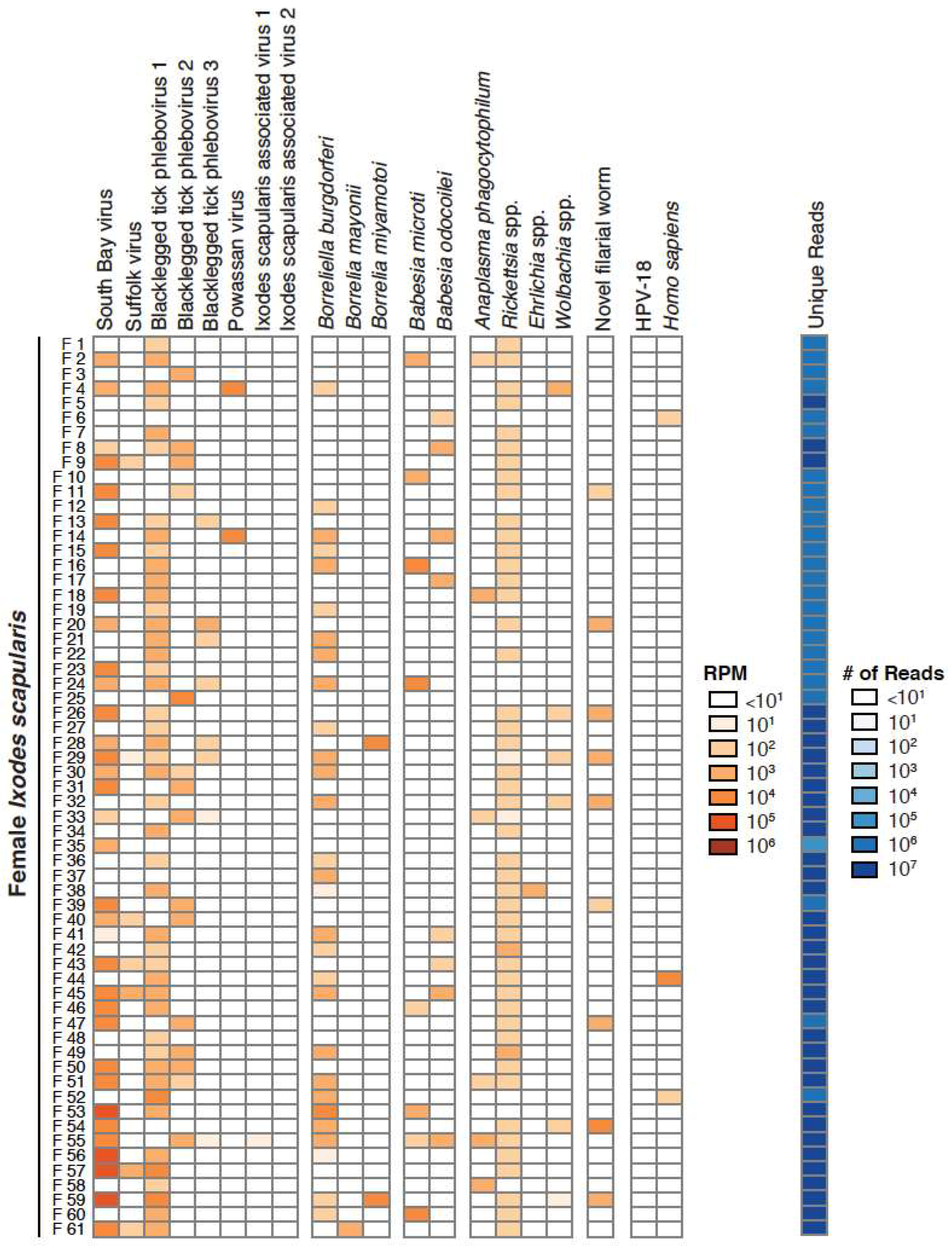
3.1. Taxonomic Assessment of the Ixodes Scapularis Microbiome
3.2. Validation of Metagenomic Sequencing Results
3.3. Detection of New Microorganisms
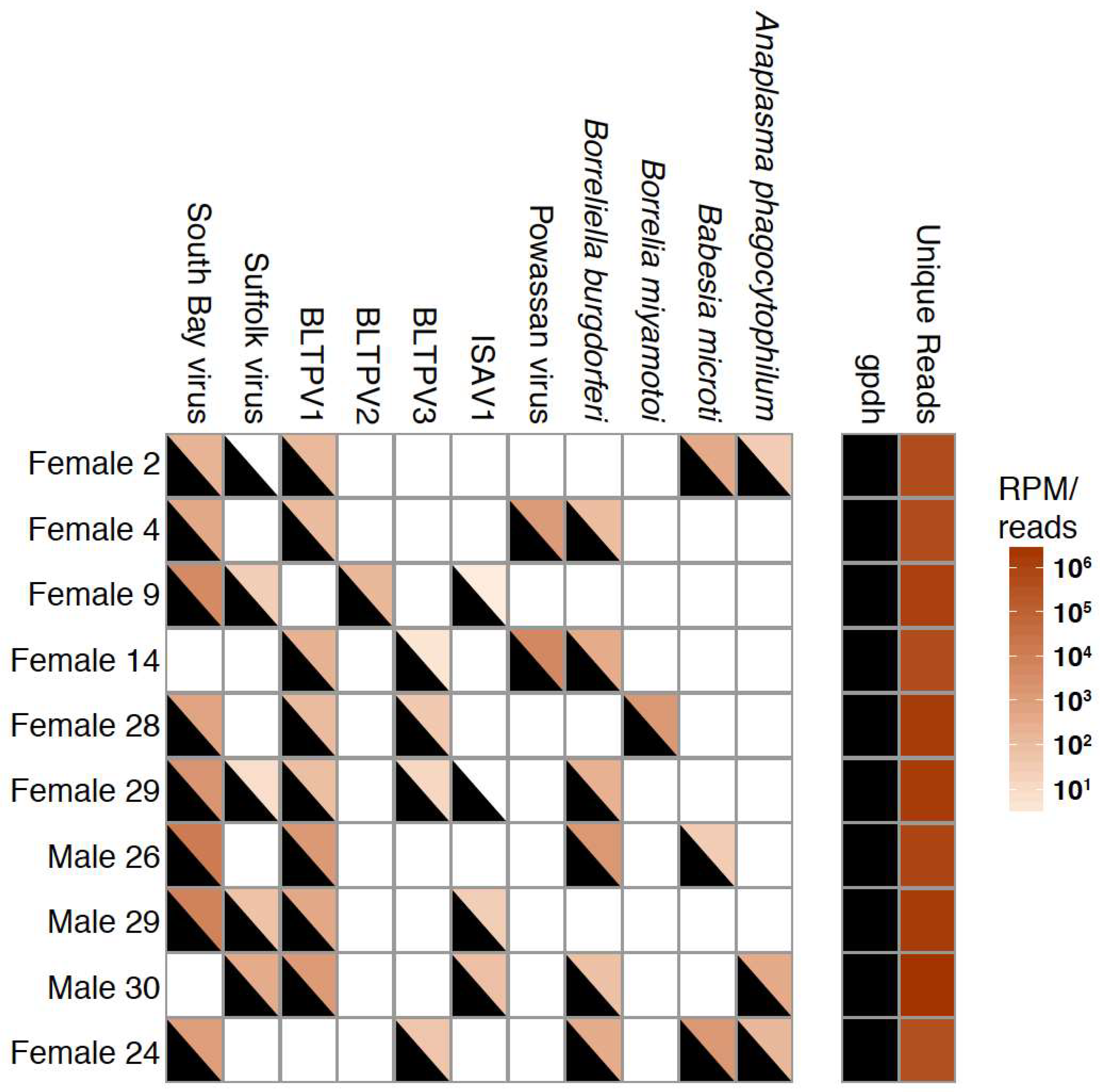
3.4. Co-Occurrence and Correlation Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Christina, A.N.; Shubhayu, S.; Kiersten, J.K.; Mark, J.D.; Manjunath, B.S.; Alison, H.; Paul, S.M. Incidence of Clinician-Diagnosed Lyme Disease, United States, 2005–2010. Emerg. Infect. Dis. 2015, 21, 1625–1631. [Google Scholar] [CrossRef]
- Hinckley, A.F.; Connally, N.P.; Meek, J.I.; Johnson, B.J.; Kemperman, M.M.; Feldman, K.A.; White, J.L.; Mead, P.S. Lyme Disease Testing by Large Commercial Laboratories in the United States. Clin. Infect. Dis. 2014, 59, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Eisen, R.J.; Eisen, L. The Blacklegged Tick, Ixodes scapularis: An Increasing Public Health Concern. Trends Parasitol. 2018, 34, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Nelder, M.P.; Russell, C.B.; Sheehan, N.J.; Sander, B.; Moore, S.; Li, Y.; Johnson, S.; Patel, S.N.; Sider, D. Human pathogens associated with the blacklegged tick Ixodes scapularis: A systematic review. Parasites Vectors 2016, 9, 265. [Google Scholar] [CrossRef] [PubMed]
- Sonenshine, D.E.; Roe, R.M. Biology of Ticks, 2nd ed.; Oxford University Press: Oxford, UK, 2013; Volume 2, ISBN 978-0-19-974406-0. [Google Scholar]
- Scoles, G.A.; Papero, M.; Beati, L.; Fish, D. A Relapsing Fever Group Spirochete Transmitted by Ixodes scapularis Ticks. Vector-Borne Zoonotic Dis. 2001, 1, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.C.; Hojgaard, A.; Hoxmeier, J.C.; Replogle, A.J.; Respicio-Kingry, L.B.; Sexton, C.; Williams, M.A.; Pritt, B.S.; Schriefer, M.E.; Eisen, L. Vector competence of the blacklegged tick, Ixodes scapularis, for the recently recognized Lyme borreliosis spirochete Candidatus Borrelia mayonii. Ticks Tick-Borne Dis. 2016, 7, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Ebel, G.D. Update on Powassan Virus: Emergence of a North American Tick-Borne Flavivirus. Annu. Rev. Entomol. 2009, 55, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Swanson, S.J.; Neitzel, D.; Reed, K.D.; Belongia, E.A. Coinfections Acquired from Ixodes Ticks. Clin. Microbiol. Rev. 2006, 19, 708–727. [Google Scholar] [CrossRef] [PubMed]
- Moutailler, S.; Valiente Moro, C.; Vaumourin, E.; Michelet, L.; Tran, F.H.; Devillers, E.; Cosson, J.-F.; Gasqui, P.; Van, V.T.; Mavingui, P.; et al. Co-infection of Ticks: The Rule Rather Than the Exception. PLoS Negl. Trop. Dis. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Diuk-Wasser, M.A.; Vannier, E.; Krause, P.J. Coinfection by the tick-borne pathogens Babesia microti and Borrelia burgdorferi: Ecological, epidemiological and clinical consequences. Trends Parasitol. 2016, 32, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Cowdry, E.V. A Group of Microorganisms Transmitted Hereditarily in Ticks and Apparently Unassociated with Disease. J. Exp. Med. 1925, 41, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.L.; Fish, D. Acquisition of coinfection and simultaneous transmission of Borrelia burgdorferi and Ehrlichia phagocytophila by Ixodes scapularis ticks. Infect. Immun. 2000, 68, 2183–2186. [Google Scholar] [CrossRef] [PubMed]
- Greay, T.L.; Gofton, A.W.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Recent insights into the tick microbiome gained through next-generation sequencing. Parasites Vectors 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.M.; Duron, O. The Tick Microbiome: Why Non-pathogenic Microorganisms Matter in Tick Biology and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Fikrig, E. Tick microbiome: The force within. Trends Parasitol. 2015, 31, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Telford Sam, R. Status of the “East Side Hypothesis” (Transovarial Interference) 25 Years Later. Ann. N. Y. Acad. Sci. 2009, 1166, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Gurfield, N.; Grewal, S.; Cua, L.S.; Torres, P.J.; Kelley, S.T. Endosymbiont interference and microbial diversity of the Pacific coast tick, Dermacentor occidentalis, in San Diego County, California. PeerJ 2017, 5, e3202. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.M.; Dark, M.J.; Reif, K.E.; Ueti, M.W.; Kappmeyer, L.S.; Scoles, G.A.; Palmer, G.H.; Brayton, K.A. Superinfection Exclusion of the Ruminant Pathogen Anaplasma marginale in Its Tick Vector Is Dependent on the Time between Exposures to the Strains. Appl. Environ. Microbiol. 2016, 82, 3217–3224. [Google Scholar] [CrossRef] [PubMed]
- Gall, C.A.; Reif, K.E.; Scoles, G.A.; Mason, K.L.; Mousel, M.; Noh, S.M.; Brayton, K.A. The bacterial microbiome of Dermacentor andersoni ticks influences pathogen susceptibility. ISME J. 2016, 10, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Jasinskas, A.; Barbour, A.G. Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS ONE 2007, 2, e405. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; DePonte, K.; Fish, D.; Fikrig, E. Gut Microbiota of the Tick Vector Ixodes scapularis Modulate Colonization of the Lyme Disease Spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, R.; Williams, S.H.; Sameroff, S.; Sanchez Leon, M.; Jain, K.; Lipkin, W.I. Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J. Virol. 2014, 88, 11480–11492. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Hu, C.; Zhang, D.; Tang, S.; Zhang, Z.; Kou, Z.; Fan, Z.; Bente, D.; Zeng, C.; Li, T. Metagenomic Profile of the Viral Communities in Rhipicephalus spp. Ticks from Yunnan, China. PLoS ONE 2015, 10, e0121609. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, R.; Sameroff, S.; Tagliafierro, T.; Jain, K.; Williams, S.H.; Cucura, D.M.; Rochlin, I.; Monzon, J.; Carpi, G.; Tufts, D.; et al. Identification of Novel Viruses in Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis Ticks. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Vanmechelen, B.; Laenen, L.; Vergote, V.; Maes, P. Grotenhout Virus, a Novel Nairovirus Found in Ixodes ricinus in Belgium. Genome Announc. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Moutailler, S.; Popovici, I.; Devillers, E.; Vayssier-Taussat, M.; Eloit, M. Diversity of viruses in Ixodes ricinus, and characterization of a neurotropic strain of Eyach virus. New Microbes New Infect 2016, 11, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.M.; Ng, T.F.F.; Suzuki, Y.; Tsujimoto, H.; Deng, X.; Delwart, E.; Rasgon, J.L. Bunyaviruses are common in male and female Ixodes scapularis ticks in central Pennsylvania. PeerJ 2016, 4, e2324. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.A.; Gall, C.A.; Mason, K.L.; Scoles, G.A.; Brayton, K.A. The characterization and manipulation of the bacterial microbiome of the Rocky Mountain wood tick, Dermacentor andersoni. Parasites Vectors 2015, 8, 632. [Google Scholar] [CrossRef] [PubMed]
- Trout Fryxell, R.T.; DeBruyn, J.M. The Microbiome of Ehrlichia-Infected and Uninfected Lone Star Ticks (Amblyomma americanum). PLoS ONE 2016, 11, e0146651. [Google Scholar] [CrossRef]
- Zhang, X.-C.; Yang, Z.-N.; Lu, B.; Ma, X.-F.; Zhang, C.-X.; Xu, H.-J. The composition and transmission of microbiome in hard tick, Ixodes persulcatus, during blood meal. Ticks Tick-borne Dis. 2014, 5, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Lyme Disease Maps | Lyme Disease | CDC. Available online: http://www.cdc.gov/lyme/stats/maps.html (accessed on 14 October 2016).
- Hoon-Hanks, L.L.; Layton, M.L.; Ossiboff, R.J.; Parker, J.S.L.; Dubovi, E.J.; Stenglein, M.D. Respiratory disease in ball pythons (Python regius) experimentally infected with ball python nidovirus. Virology 2018, 517, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Stenglein, M.D.; Jacobson, E.R.; Chang, L.-W.; Sanders, C.; Hawkins, M.G.; Guzman, D.S.-M.; Drazenovich, T.; Dunker, F.; Kamaka, E.K.; Fisher, D.; et al. Widespread Recombination, Reassortment, and Transmission of Unbalanced Compound Viral Genotypes in Natural Arenavirus Infections. PLOS Pathog. 2015, 11, e1004900. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Gulia-Nuss, M.; Nuss, A.B.; Meyer, J.M.; Sonenshine, D.E.; Roe, R.M.; Waterhouse, R.M.; Sattelle, D.B.; de la Fuente, J.; Ribeiro, J.M.; Megy, K.; et al. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat. Commun. 2016, 7, 10507. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information (US) BLAST® Command Line Applications User Manual. Available online: https://www.ncbi.nlm.nih.gov/books/NBK279690/ (accessed on 1 May 2018).
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M. The Sequence Read Archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Hojgaard, A.; Lukacik, G.; Piesman, J. Detection of Borrelia burgdorferi, Anaplasma phagocytophilum and Babesia microti, with two different multiplex PCR assays. Ticks Tick-Borne Dis. 2014, 5, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Koči, J.; Šimo, L.; Park, Y. Validation of Internal Reference Genes for Real-Time Quantitative Polymerase Chain Reaction Studies in the Tick, Ixodes scapularis (Acari: Ixodidae). J. Med. Entomol. 2013, 50, 79–84. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2016. [Google Scholar]
- Griffith, D.M.; Veech, J.A.; Marsh, C.J. Cooccur: Probabilistic Species Co-Occurrence Analysis in R. J. Stat. Softw. 2016, 69, 1–17. [Google Scholar] [CrossRef]
- Revelle, W. Procedures for Psychological, Psychometric, and Personality Research; Northwestern University: Evanston, IL, USA, 2017. [Google Scholar]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Namrata, P.; Miller, M.J.; Shilpa, M.; Reddy, R.P.; Bandoski, C.; Rossi, J.M.; Sapi, E. Filarial Nematode Infection in Ixodes scapularis Ticks Collected from Southern Connecticut. Vet. Sci. 2014, 1. [Google Scholar] [CrossRef]
- Van Treuren, W.; Ponnusamy, L.; Brinkerhoff, R.J.; Gonzalez, A.; Parobek, C.M.; Juliano, J.J.; Andreadis, T.G.; Falco, R.C.; Ziegler, L.B.; Hathaway, N.; et al. Variation in the Microbiota of Ixodes Ticks with Regard to Geography, Species, and Sex. Appl. Environ. Microbiol. 2015, 81, 6200–6209. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Committee on Lyme Disease and Other Tick-Borne Diseases: The State of the Science. Critical Needs and Gaps in Understanding Prevention, Amelioration, and Resolution of Lyme and Other Tick-Borne Diseases; The Short-Term and Long-Term Outcomes: Workshop Report; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Ponnusamy, L.; Gonzalez, A.; Van Treuren, W.; Weiss, S.; Parobek, C.M.; Juliano, J.J.; Knight, R.; Roe, R.M.; Apperson, C.S.; Meshnick, S.R. Diversity of Rickettsiales in the Microbiome of the Lone Star Tick, Amblyomma americanum. Appl. Environ. Microbiol. 2014, 80, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Prachayangprecha, S.; Schapendonk, C.M.E.; Koopmans, M.P.; Osterhaus, A.D.M.E.; Schürch, A.C.; Pas, S.D.; van der Eijk, A.A.; Poovorawan, Y.; Haagmans, B.L.; Smits, S.L. Exploring the Potential of Next-Generation Sequencing in Detection of Respiratory Viruses. J. Clin. Microbiol. 2014, 52, 3722–3730. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, J.H.-O.; Shi, M.; Bohlin, J.; Eldholm, V.; Brynildsrud, O.B.; Paulsen, K.M.; Andreassen, Å.; Holmes, E.C. Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 2017, 7, 10870. [Google Scholar] [CrossRef] [PubMed]
- Ebel, G.D.; Foppa, I.; Spielman, A.; Telford, S.R. A focus of deer tick virus transmission in the northcentral United States. Emerg. Infect. Dis. 1999, 5, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Brackney, D.E.; Nofchissey, R.A.; Fitzpatrick, K.A.; Brown, I.K.; Ebel, G.D. Stable prevalence of Powassan virus in Ixodes scapularis in a northern Wisconsin focus. Am. J. Trop. Med. Hyg. 2008, 79, 971–973. [Google Scholar] [PubMed]
- Steiner, F.E.; Pinger, R.R.; Vann, C.N.; Grindle, N.; Civitello, D.; Clay, K.; Fuqua, C. Infection and Co-infection Rates of Anaplasma phagocytophilum Variants, Babesia spp., Borrelia burgdorferi, and the Rickettsial Endosymbiont in Ixodes scapularis (Acari: Ixodidae) from Sites in Indiana, Maine, Pennsylvania, and Wisconsin. J. Med. Entomol. 2008, 45, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Telford Iii, S.R.; Goethert, H.K.; Cunningham, J.A. Prevalence of Ehrlichia muris in Wisconsin Deer Ticks Collected During the Mid 1990s. Open Microbiol. J. 2011, 5, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Michalski, M.; Rosenfield, C.; Erickson, M.; Selle, R.; Bates, K.; Essar, D.; Massung, R. Anaplasma phagocytophilum in central and western Wisconsin: A molecular survey. Parasitol. Res. 2006, 99, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Lee, X.; Coyle, D.R.; Johnson, D.K.H.; Murphy, M.W.; McGeehin, M.A.; Murphy, R.J.; Raffa, K.F.; Paskewitz, S.M. Prevalence of Borrelia burgdorferi and Anaplasma phagocytophilum in Ixodes scapularis (Acari: Ixodidae) nymphs collected in managed red pine forests in Wisconsin. J. Med. Entomol. 2014, 51, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Nieto, N.C.; Porter, W.T.; Wachara, J.C.; Lowrey, T.J.; Martin, L.; Motyka, P.J.; Salkeld, D.J. Using citizen science to describe the prevalence and distribution of tick bite and exposure to tick-borne diseases in the United States. PLoS ONE 2018, 13, e0199644. [Google Scholar] [CrossRef] [PubMed]
- Halbach, R.; Junglen, S.; van Rij, R.P. Mosquito-specific and mosquito-borne viruses: Evolution, infection, and host defense. Curr. Opin. Insect Sci. 2017, 22, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Bolling, B.G.; Weaver, S.C.; Tesh, R.B.; Vasilakis, N. Insect-Specific Virus Discovery: Significance for the Arbovirus Community. Viruses 2015, 7, 4911–4928. [Google Scholar] [CrossRef] [PubMed]
- Abrao, E.P.; da Fonseca, B.A.L. Infection of Mosquito Cells (C6/36) by Dengue-2 Virus Interferes with Subsequent Infection by Yellow Fever Virus. Vector-Borne Zoonotic Dis. 2016, 16, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Potiwat, R.; Komalamisra, N.; Thavara, U.; Tawatsin, A.; Siriyasatien, P. Competitive Suppression between Chikungunya and Dengue Virus in Aedes Albopictus C6/36 Cell Line. Southeast Asian J. Trop. Med. Public Health 2011, 42, 1388–1394. [Google Scholar] [PubMed]
- Karpf, A.R.; Lenches, E.; Strauss, E.G.; Strauss, J.H.; Brown, D.T. Superinfection exclusion of alphaviruses in three mosquito cell lines persistently infected with Sindbis virus. J. Virol. 1997, 71, 7119–7123. [Google Scholar] [PubMed]
- Puck, T.T.; Lee, H.H. Mechanism of cell wall penetration by viruses. II. Demonstration of cyclic permeability change accompanying virus infection of Escherichia coli B cells. J. Exp. Med. 1955, 101, 151–175. [Google Scholar] [CrossRef] [PubMed]
- Beaty, B.; Sundin, D.; Chandler, L.; Bishop, D. Evolution of bunyaviruses by genome reassortment in dually infected mosquitoes (Aedes triseriatus). Science 1985, 230, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Plantard, O.; Bouju-Albert, A.; Malard, M.-A.; Hermouet, A.; Capron, G.; Verheyden, H. Detection of Wolbachia in the Tick Ixodes ricinus is Due to the Presence of the Hymenoptera Endoparasitoid Ixodiphagus hookeri. PLoS ONE 2012, 7, e30692. [Google Scholar] [CrossRef] [PubMed]
- Bouchery, T.; Lefoulon, E.; Karadjian, G.; Nieguitsila, A.; Martin, C. The symbiotic role of Wolbachia in Onchocercidae and its impact on filariasis. Clin. Microbiol. Infect. 2013, 19, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Evaluating the Information Content of Shallow Shotgun Metagenomics | Biorxiv. Available online: https://www.biorxiv.org/content/early/2018/05/12/320986 (accessed on 14 June 2018).
- Roossinck, M.J.; Bazán, E.R. Symbiosis: Viruses as Intimate Partners. Annu. Rev. Virol. 2017, 4, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.A.; McCutcheon, J.P.; Nakabachi, A. Genomics and Evolution of Heritable Bacterial Symbionts. Annu. Rev. Genet. 2008, 42, 165–190. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Move over, bacteria! Viruses make their mark as mutualistic microbial symbionts. J. Virol. 2015, 89, 6532–6535. [Google Scholar] [CrossRef] [PubMed]
- Engelstädter, J.; Hurst, G.D.D. The Ecology and Evolution of Microbes that Manipulate Host Reproduction. Annu. Rev. Ecolol. Evol. Syst. 2009, 40, 127–149. [Google Scholar] [CrossRef]
- McCutcheon, J.P.; Moran, N.A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 2011, 10, 13–26. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tick Sex | All Ticks | ||||
|---|---|---|---|---|---|
| Taxon | Male | Female | (n = 112) | Nucleotide Identity 1 | Average Mapping Reads Per Million Unique Reads (RPM) |
| (n = 51) | (n = 61) | ||||
| Viruses | |||||
| Blacklegged tick phlebovirus 1 | 82.4% | 73.8% | 77.7% | 97.6% | 395 |
| South Bay virus | 47.1% | 55.7% | 51.8% | 98.0% | 2796 |
| Blacklegged tick phlebovirus 2 | 11.8% | 24.6% | 18.8% | 97.1% | 63 |
| Suffolk virus | 23.5% | 11.5% | 17.0% | 98.1% | 50 |
| Blacklegged tick phlebovirus 3 | 7.8% | 13.1% | 10.7% | 98.1% | 5 |
| Ixodes scapularis associated virus 1 | 7.8% | 1.6% | 4.5% | 98.3% | 3 |
| Powassan virus | 0% | 3.3% | 1.8% | 100.0% | 55 |
| Ixodes scapularis associated virus 2 | 2.0% | 0% | 0.9% | 96.4% | 2 |
| Bacteria | |||||
| Rickettsia spp. | 5.9% | 80.3% | 46.4% | 98.8% | 21 |
| Borreliella burgdorferi | 33.3% | 47.5% | 41.1% | 99.7% | 154 |
| Anaplasma phagocytophilum | 11.8% | 9.8% | 10.7% | 98.9% | 28 |
| Wolbachia spp. | 5.9% | 9.8% | 8.0% | 97.5% | 10 |
| Ehrlichia spp. | 5.9% | 1.6% | 3.6% | 96.6% | 45 |
| Borrelia miyamotoi | 0% | 3.3% | 1.8% | 100.0% | 64 |
| Borrelia mayonii | 0% | 1.6% | 0.9% | 99.4% | 1 |
| Eukaryotes | |||||
| Novel filarial worm | 21.6% | 14.8% | 17.9% | 88.7% | 113 |
| Babesia odocoilei | 5.9% | 13.1% | 9.8% | 100.0% | 22 |
| Babesia microti | 3.9% | 13.1% | 8.9% | 99.7% | 52 |
| Closest Related Sequence | Contig Length (nt) | Accession | BLAST Percent Identity 1 | |
|---|---|---|---|---|
| Norway mononegavirus 1-like sequence | Norway mononegavirus 1 (MF141072.1) | 5020 | MH560586 | 71% |
| Blacklegged tick phlebovirus-like sequence | Blacklegged tick phlebovirus 1 (KX184201.1) | 874 | MH560584 | 77% |
| Norway phlebovirus 1-like sequence | Norway phlebovirus 1 (MF141061.1) | 791 | MH560585 | 78% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cross, S.T.; Kapuscinski, M.L.; Perino, J.; Maertens, B.L.; Weger-Lucarelli, J.; Ebel, G.D.; Stenglein, M.D. Co-Infection Patterns in Individual Ixodes scapularis Ticks Reveal Associations between Viral, Eukaryotic and Bacterial Microorganisms. Viruses 2018, 10, 388. https://doi.org/10.3390/v10070388
Cross ST, Kapuscinski ML, Perino J, Maertens BL, Weger-Lucarelli J, Ebel GD, Stenglein MD. Co-Infection Patterns in Individual Ixodes scapularis Ticks Reveal Associations between Viral, Eukaryotic and Bacterial Microorganisms. Viruses. 2018; 10(7):388. https://doi.org/10.3390/v10070388
Chicago/Turabian StyleCross, Shaun T., Marylee L. Kapuscinski, Jacquelyn Perino, Bernadette L. Maertens, James Weger-Lucarelli, Gregory D. Ebel, and Mark D. Stenglein. 2018. "Co-Infection Patterns in Individual Ixodes scapularis Ticks Reveal Associations between Viral, Eukaryotic and Bacterial Microorganisms" Viruses 10, no. 7: 388. https://doi.org/10.3390/v10070388
APA StyleCross, S. T., Kapuscinski, M. L., Perino, J., Maertens, B. L., Weger-Lucarelli, J., Ebel, G. D., & Stenglein, M. D. (2018). Co-Infection Patterns in Individual Ixodes scapularis Ticks Reveal Associations between Viral, Eukaryotic and Bacterial Microorganisms. Viruses, 10(7), 388. https://doi.org/10.3390/v10070388