High-Throughput Sequencing Reveals Further Diversity of Little Cherry Virus 1 with Implications for Diagnostics

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Full Genome Sequencing of LChV-1 Isolates Using HTS Approaches
2.2. Phylogenetic and Sequence Analysis of LChV1 Isolates
2.3. Recombination Analysis
2.4. In Silico Evaluation of Published Primers for the Detection of LChV1
3. Results
3.1. HTS Analyses and Genome Assembly of LChV1 Isolates
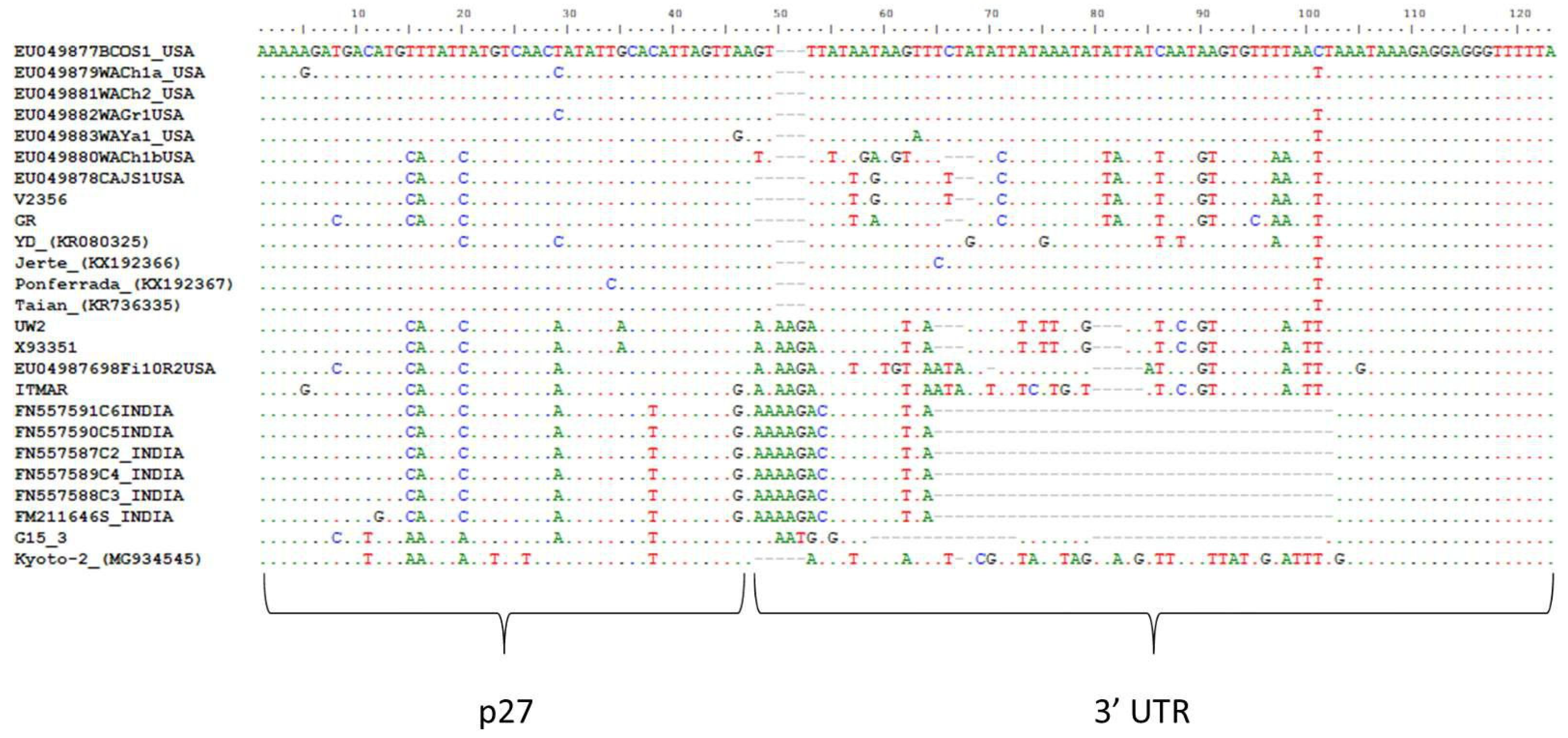
3.2. Genomic Organization and Sequence Similarities of the New LChV1 Isolates
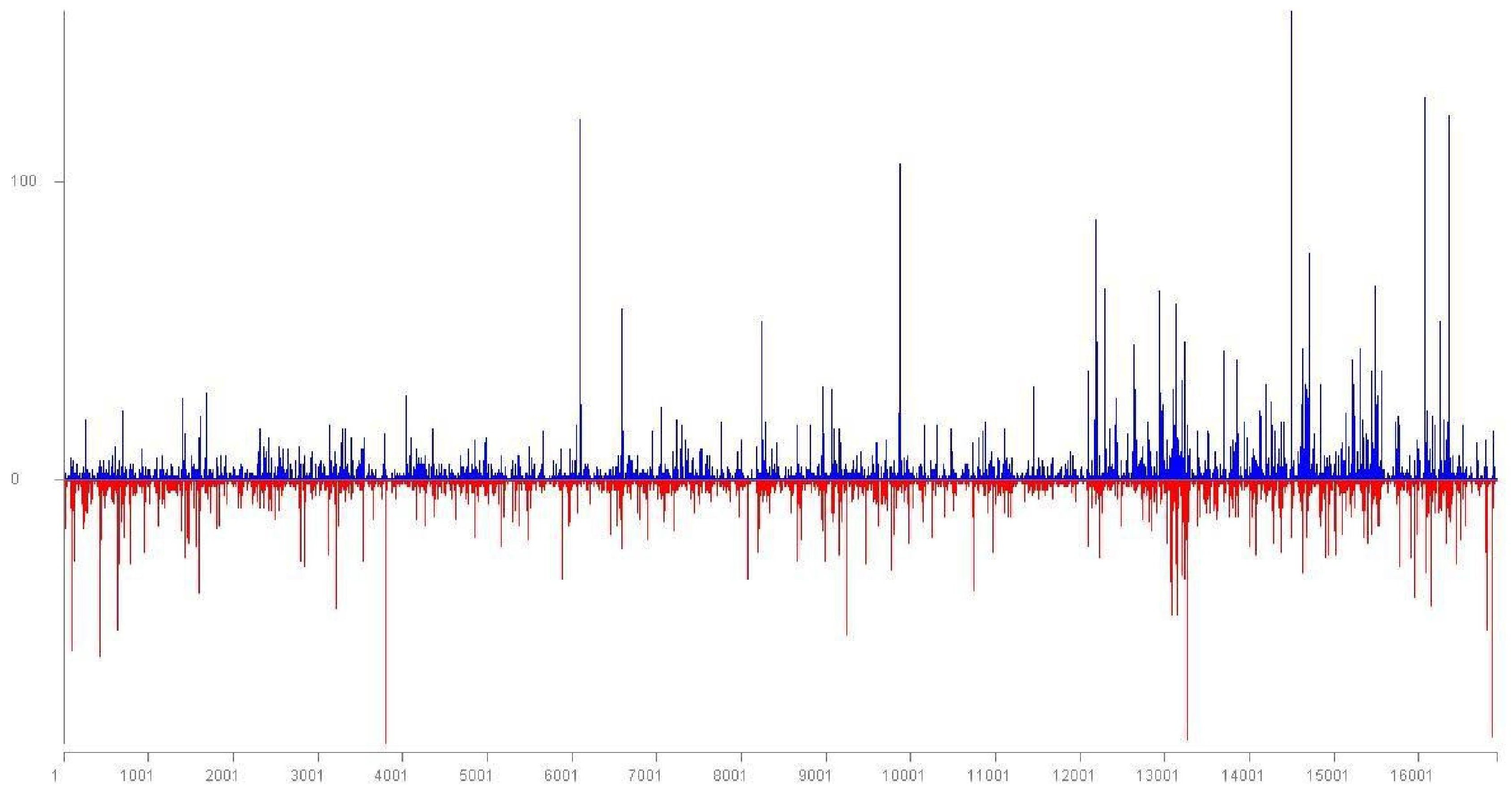
3.3. Intra-Host Genetic Diversity of LChV1
3.4. Recombination Analysis
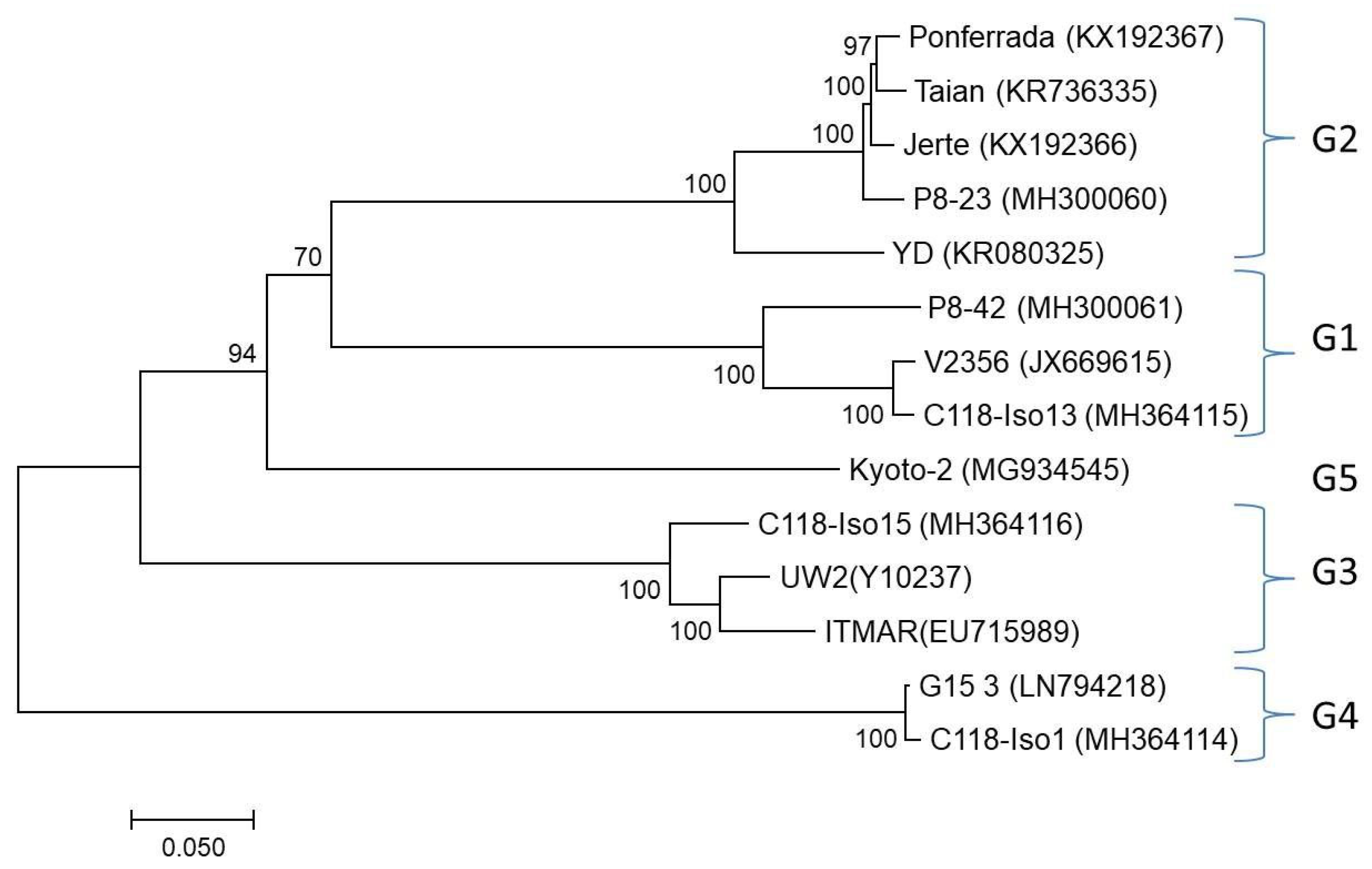
3.5. Phylogenetic Analysis
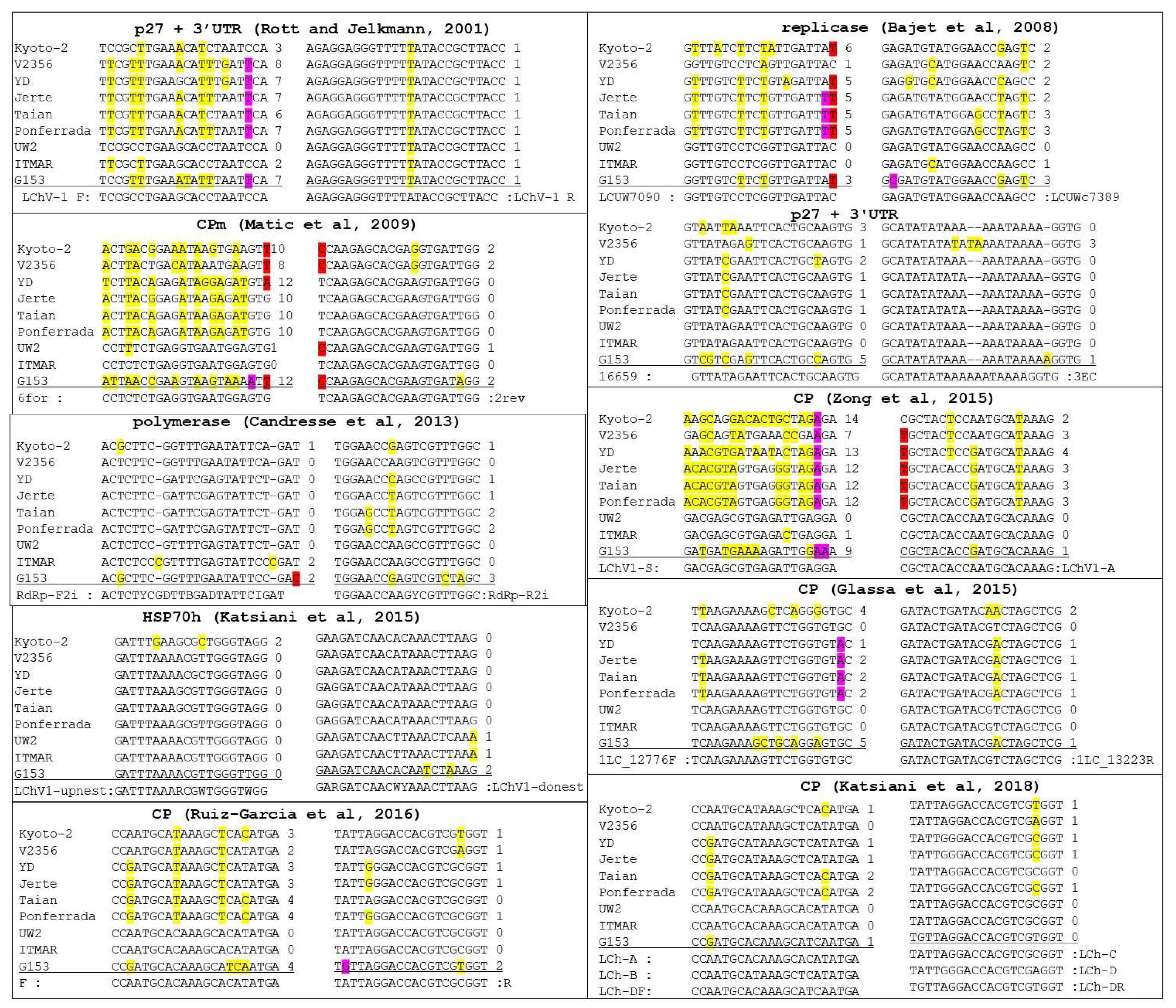
3.6. Detection Range of LChV1 Specific Primers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Martelli, G.P.; AbouGhanem-Sabanadzovic, N.; Agranovsky, A.A.; Al Rwahnih, M.; Dolja, V.V.; Dovas, C.I.; Fuchs, M.; Gugerli, P.; Hu, J.S.; Jelkmann, W.; et al. Taxonomic revision of the family Closteroviridae with special reference to the grapevine leafroll-associated members of the genus Ampelovirus and the putative species unassigned to the family. J. Plant Pathol. 2012, 94, 7–19. [Google Scholar]
- Matic, S.; Minafra, A.; Sanchez-Navarro, J.A.; Pallas, V.; Myrta, A.; Martelli, G.P. ‘Kwanzan Stunting’ syndrome: Detection and molecular characterization of an Italian isolate of Little cherry virus 1. Virus Res. 2009, 143, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Candresse, T.; Marais, A.; Faure, C.; Gentit, P. Association of Little cherry virus 1(LChV1) with the Shirofugen stunt disease and characterization of the genome of a divergent LChV1 isolate. Phytopathology 2013, 103, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W.; Fechtner, B.; Agranovsky, A.A. Complete genome structure and phylogenetic analysis of Little cherry virus, a mealybug transmissible closterovirus. J. Gener. Virol. 1997, 78, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Igori, D.; Yoo, R.H.; Zhao, F.; Cho, I.S.; Choi, G.S.; Lim, H.S.; Lee, S.H.; Moon, J.S. Genomic detection and characterization of a Korean isolate of Little cherry virus 1 sampled from a peach tree. Virus Genes 2015, 51, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, D.; Tan, Y.; Zong, X.; Wei, H.; Hammond, R.W.; Liu, Q. Complete nucleotide sequence of Little cherry virus 1 (LChV1) infecting sweet cherry in China. Arch. Virol. 2016, 161, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-García, A.B.; Martinez, C.; Santiago, R.; Garcia, M.T.; de Prado, N.; Olmos, A. First report of Little cherry virus 1 (LChV1) in sweet cherry in Spain. Plant Dis. 2016, 100, 2340. [Google Scholar] [CrossRef]
- Katsiani, A.T.; Maliogka, V.I.; Amoutzias, G.D.; Efthimiou, K.E.; Katis, N.I. Insights into the genetic diversity and evolution of Little cherry virus 1. Plant Pathol. 2015, 64, 817–824. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hadidi, A.; Flores, R.; Candresse, T.; Barba, M. Next-Generation Sequencing and Genome Editing in Plant Virology. Front. Microbiol. 2016, 7, Article 1325. [Google Scholar] [CrossRef]
- Bag, S.; Al Rwahnih, M.; Li, A.; Gonzalez, A.; Rowhani, A.; Uyemoto, J.K.; Sudarshana, M.R. Detection of a new luteovirus in imported nectarine trees: A case study to propose adoption of metagenomics in post-entry quarantine. Phytopathology 2015, 105, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Villamor, D.E.V.; Mekuria, T.A.; Eastwell, K.C. High-Throughput sequencing identifies novel viruses in nectarine: Insights to the etiology of stem-pitting disease. Phytopathology 2016, 106, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Kinoti, W.M.; Constable, F.E.; Nancarrow, N.; Plummer, K.M.; Rodoni, B. Analysis of intra-host genetic diversity of Prunus necrotic ringspot virus (PNRSV) using amplicon next generation sequencing. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.; Faure, C.; Couture, C.; Bergey, B.; Gentit, P.; Candresse, T. Characterization by deep sequencing of divergent plum bark necrosis stem pitting associated virus isolates and development of a broad spectrum PBNSPaV-specific detection assay. Phytopathology 2014, 104, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, K.B.; Nicholas, H.B., Jr.; Deerfield, D.W., II. GeneDoc: Analysis and Visualization of Genetic Variation. Embnew. News 1997, 4, 14. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rott, M.E.; Jelkmann, W. Detection and partial characterization of a second closterovirus associated with little cherry disease, Little cherry virus-2. Phytopathology 2001, 91, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Bajet, N.B.; Unruh, T.R.; Druffel, K.L.; Eastwell, K.C. Occurrence of two Little cherry viruses in sweet cherry in Washington State. Plant Dis. 2008, 92, 234–238. [Google Scholar] [CrossRef]
- Zong, X.; Wang, W.; Wei, H.; Wang, J.; Yan, X.; Hammond, R.W.; Liu, Q. Incidence of sweet cherry viruses in Shandong province, China and a case study on multiple infection with five viruses. J. Plant Pathol. 2015, 97, 61–68. [Google Scholar]
- Glasa, M.; Benediková, D.; Predajňa, L. First report of Little cherry virus-1 in Slovakia. J. Plant Pathol. 2015, 97, 541–551. [Google Scholar]
- Katsiani, A.T.; Pappi, P.; Olmos, A.; Efthimiou, K.E.; Maliogka, V.I.; Katis, N.I. Development of a Real-Time RT-PCR for the universal detection of LChV1 and study of the seasonal fluctuation of the viral titer in sweet cherry cultivars. Plant Dis. 2018, 102, 899–904. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Roossinck, M.J. Deep sequencing for discovery and evolutionary analysis of plant viruses. Virus Res. 2017, 239, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Martelli, G.P.; Agranovsky, A.A.; Bar-Joseph, M.; Boscia, D.; Candresse, T.; Coutts, R.H.A.; Dolja, V.V.; Hu, J.S.; Jelkmann, W.; Karasev, A.V.; et al. Family ClosteroviridaeIn: Virus Taxonomy-Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Academic Press: San Diego, CA, USA, 2011; pp. 987–1001. [Google Scholar]
- Prosser, S.W.; Goszczynski, D.E.; Meng, B. Molecular analysis of double-stranded RNAs reveals complex infection of grapevines with multiple viruses. Virus Res. 2007, 124, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Mandahar, C.L. Positive Sense Viral RNA. In Multiplication of RNA Plant Viruses; Springer: Dordrecht, The Netherlands, 2006; pp. 29–63. [Google Scholar]
- Tian, T.; Rubio, L.; Yeh, H.H.; Crawford, B.; Falk, B.W. Lettuce infectious yellows virus: In Vitro acquisition analysis using partially purified virions and the whitefly, Bemisiatabaci. J. Gener. Virol. 1999, 80, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayana, T.; Gowda, S.; Mawassi, M.; Albiach-Martí, M.R.; Ayllón, M.A.; Robertson, C.; Garnsey, S.M.; Dawson, W.O. Closterovirus encoded HSP70 homolog and p61 in addition to both coat proteins function in efficient virion assembly. Virology 2000, 278, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Alzhanova, D.V.; Napuli, A.; Creamer, R.; Dolja, V.V. Cell-to-cell movement and assembly of a plant closterovirus: Roles for the capsid proteins and Hsp70 homolog. EMBO J. 2001, 20, 6997–7007. [Google Scholar] [CrossRef] [PubMed]
- Mawassi, M.; Mietkiewska, E.; Gofman, R.; Yang, G.; Bar-Joseph, M. Unusual sequence relationships between two isolates of Citrus tristeza virus. J. Gener. Virol. 1996, 77, 2359–2364. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C.; Kasschau, K.D.; Prokhnevsky, A.I.; Gopinath, K.; Pogue, G.P.; Carrington, J.C.; Dolja, V.V. Suppressor of RNA silencing encoded by Beet yellows virus. Virology 2003, 306, 203–209. [Google Scholar] [CrossRef]
- Lu, R.; Folimonov, A.S.; Shintaku, M.; Li, W.X.; Falk, B.W.; Dawson, W.O.; Ding, S.W. Three distinct suppressors of RNA silencing encoded by a 20-kb viral RNA genome. Proc. Natl. Acad. Sci. USA 2004, 101, 15742–15747. [Google Scholar] [CrossRef] [PubMed]
- Κatsiani, A.; Κatsarou, K.; Kalantidis, K.; Κatis, N.I.; Μaliogka, V.Ι. Identification of an RNA silencing suppressor encoded by the genome of Little Cherry Virus 1. In Proceedings of the 24st International Conference on Virus and other Graft Transmissible Diseases of Fruit Crops, Thessaloniki, Greece, 5–9 June 2017; p. 113. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Plant Species | Country | Symptoms | HTS Template | Viruses Present | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| APCLSV | CVF | PDV | LChV1 | LChV2 | CVA | PBNSPaV | CNRMV | APLPV | |||||
| G15 3 | Prunus avium cv. Tragana-edessis | Greece | no | sRNAs | + | + | + | ||||||
| Kyoto-2 | Prunus serrulata | Japan | yellowish oak-leaf pattern symptoms | dsRNA | + | + | + | + | + | + | |||
| C118 | Prunus avium cv.Larian | Greece | no | Total RNA | + | Multiple genotypes | + | + | |||||
| P8 | Prunus avium cv.Planera | Spain | reddening of leaves | Total RNA | + | Multiple genotypes | + | ||||||
| G15 3 | Identity in nt (aa) with LChV-1 Isolates | |||||||
| Genomic Region | ITMAR | UW2 | V2356 | YD | JERTE | PONFERRADA | TAIAN | KYOTO-2 |
| Full genome | 72 | 72 | 72 | 73 | 73 | 73 | 73 | 73 |
| ORF1a (P-PRO, MET & HEL) | 70 (75) | 70 (76) | 72 (76) | 72 (76) | 72 (77) | 71 (76) | 71 (76) | 71 (76) |
| ORF1b (RdRp) | 80 (90) | 80 (92) | 81 (92) | 80 (92) | 80 (91) | 81 (92) | 80 (90) | 80 (92) |
| ORF2 (p4) | 72 (67) | 70 (67) | 76 (77) | 76 (70) | 74 (70) | 70 (67) | 73 (74) | 81 (83) |
| ORF3 (HSP70h) | 75 (79) | 76 (81) | 74 (81) | 76 (83) | 76 (83) | 76 (83) | 76 (83) | 76 (81) |
| ORF4 (p61) | 72 (75) | 72 (75) | 74 (76) | 72 (75) | 73 (74) | 73 (74) | 73 (74) | 73 (74) |
| ORF5 (CP) | 72 (73) | 72 (72) | 70 (72) | 72 (73) | 72 (74) | 72 (74) | 72 (74) | 71 (72) |
| ORF6 (CPm) | 69 (66) | 70 (68) | 68 (65) | 69 (68) | 69 (67) | 69 (67) | 70 (67) | 70 (68) |
| ORF7 (p21) | 76 (88) | 76 (87) | 76 (85) | 78 (87) | 78 (88) | 78 (88) | 78 (88) | 77 (85) |
| ORF8 (p27) | 71 (69) | 73 (74) | 74 (74) | 73 (71) | 73 (75) | 73 (75) | 72 (75) | 75 (71) |
| KYOTO-2 | Identity in nt (aa) with LChV-1 Isolates | |||||||
| Genomic Region | ITMAR | UW2 | V2356 | YD | JERTE | PONFERRADA | TAIAN | G15 3 |
| Full genome | 75 | 75 | 77 | 77 | 77 | 76 | 77 | 73 |
| ORF1a (P-PRO, MT & HEL) | 74 (79) | 74 (80) | 76 (83) | 75 (83) | 75 (83) | 75 (82) | 75 (82) | 71 (76) |
| ORF1b (RdRp) | 79 (91) | 80 (93) | 83 (94) | 80 (94) | 81 (93) | 81 (93) | 81 (93) | 80 (92) |
| ORF2 (p4) | 79 (70) | 82 (77) | 81 (80) | 81 (70) | 79 (70) | 78 (74) | 78 (74) | 81 (83) |
| ORF3 (HSP70h) | 77 (83) | 78 (84) | 79 (85) | 79 (86) | 80 (87) | 80 (86) | 80 (87) | 76 (81) |
| ORF4 (p61) | 74 (79) | 74 (79) | 76 (80) | 76 (79) | 75 (79) | 76 (79) | 75 (79) | 73 (74) |
| ORF5 (CP) | 74 (76) | 74 (78) | 73 (75) | 77 (78) | 77 (79) | 77 (78) | 78 (78) | 71 (72) |
| ORF6 (CPm) | 72 (70) | 73 (71) | 73 (72) | 73 (72) | 73 (71) | 73 (71) | 73 (70) | 70 (68) |
| ORF7 (p21) | 81 (90) | 80 (90) | 82 (93) | 84 (94) | 84 (92) | 84 (92) | 85 (92) | 77 (85) |
| ORF8 (p27) | 74 (75) | 79 (82) | 81 (85) | 80 (85) | 79 (82) | 79 (81) | 79 (82) | 75 (71) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsiani, A.; Maliogka, V.I.; Katis, N.; Svanella-Dumas, L.; Olmos, A.; Ruiz-García, A.B.; Marais, A.; Faure, C.; Theil, S.; Lotos, L.; et al. High-Throughput Sequencing Reveals Further Diversity of Little Cherry Virus 1 with Implications for Diagnostics. Viruses 2018, 10, 385. https://doi.org/10.3390/v10070385
Katsiani A, Maliogka VI, Katis N, Svanella-Dumas L, Olmos A, Ruiz-García AB, Marais A, Faure C, Theil S, Lotos L, et al. High-Throughput Sequencing Reveals Further Diversity of Little Cherry Virus 1 with Implications for Diagnostics. Viruses. 2018; 10(7):385. https://doi.org/10.3390/v10070385
Chicago/Turabian StyleKatsiani, Asimina, Varvara I. Maliogka, Nikolaos Katis, Laurence Svanella-Dumas, Antonio Olmos, Ana B. Ruiz-García, Armelle Marais, Chantal Faure, Sébastien Theil, Leonidas Lotos, and et al. 2018. "High-Throughput Sequencing Reveals Further Diversity of Little Cherry Virus 1 with Implications for Diagnostics" Viruses 10, no. 7: 385. https://doi.org/10.3390/v10070385
APA StyleKatsiani, A., Maliogka, V. I., Katis, N., Svanella-Dumas, L., Olmos, A., Ruiz-García, A. B., Marais, A., Faure, C., Theil, S., Lotos, L., & Candresse, T. (2018). High-Throughput Sequencing Reveals Further Diversity of Little Cherry Virus 1 with Implications for Diagnostics. Viruses, 10(7), 385. https://doi.org/10.3390/v10070385