Pseudomonas PB1-Like Phages: Whole Genomes from Metagenomes Offer Insight into an Abundant Group of Bacteriophages


Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Viral DNA Extraction
2.3. PCR Amplification and Amplicon Analysis
2.4. Determination of the Presence of PB1 and Pbunaviruses in Publicly Available Metagenomic Datasets
2.5. Assembly and Annotation of Uncultivated PB1-Like Genomes
2.6. Comparative Genomics
3. Results
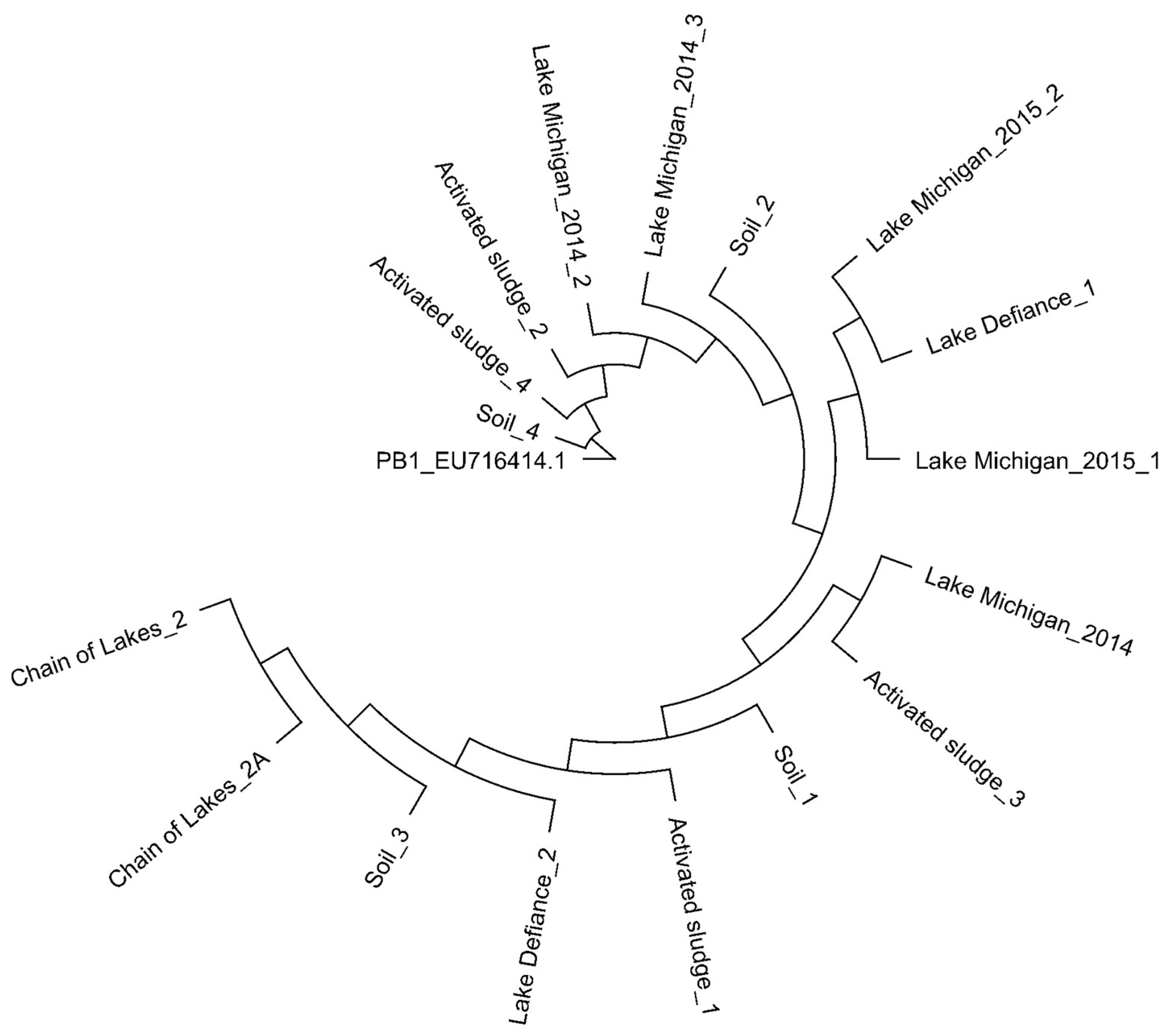
3.1. Hunting for Pbunaviruses in Soil, Freshwater and Activated Sludge
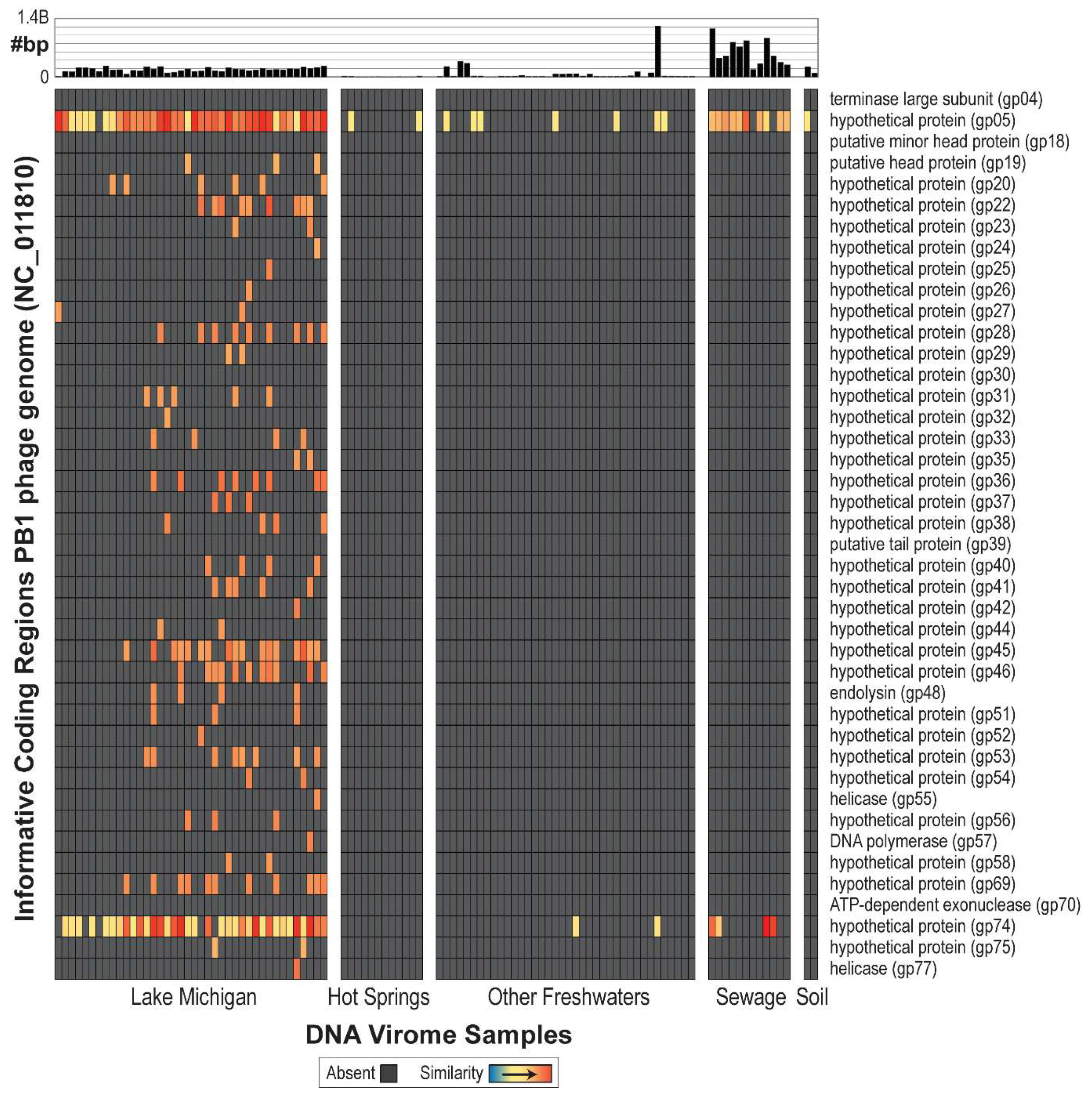
3.2. Searching for PB1 in Soil, Freshwater, and Activated Sludge Viral Metagenomes
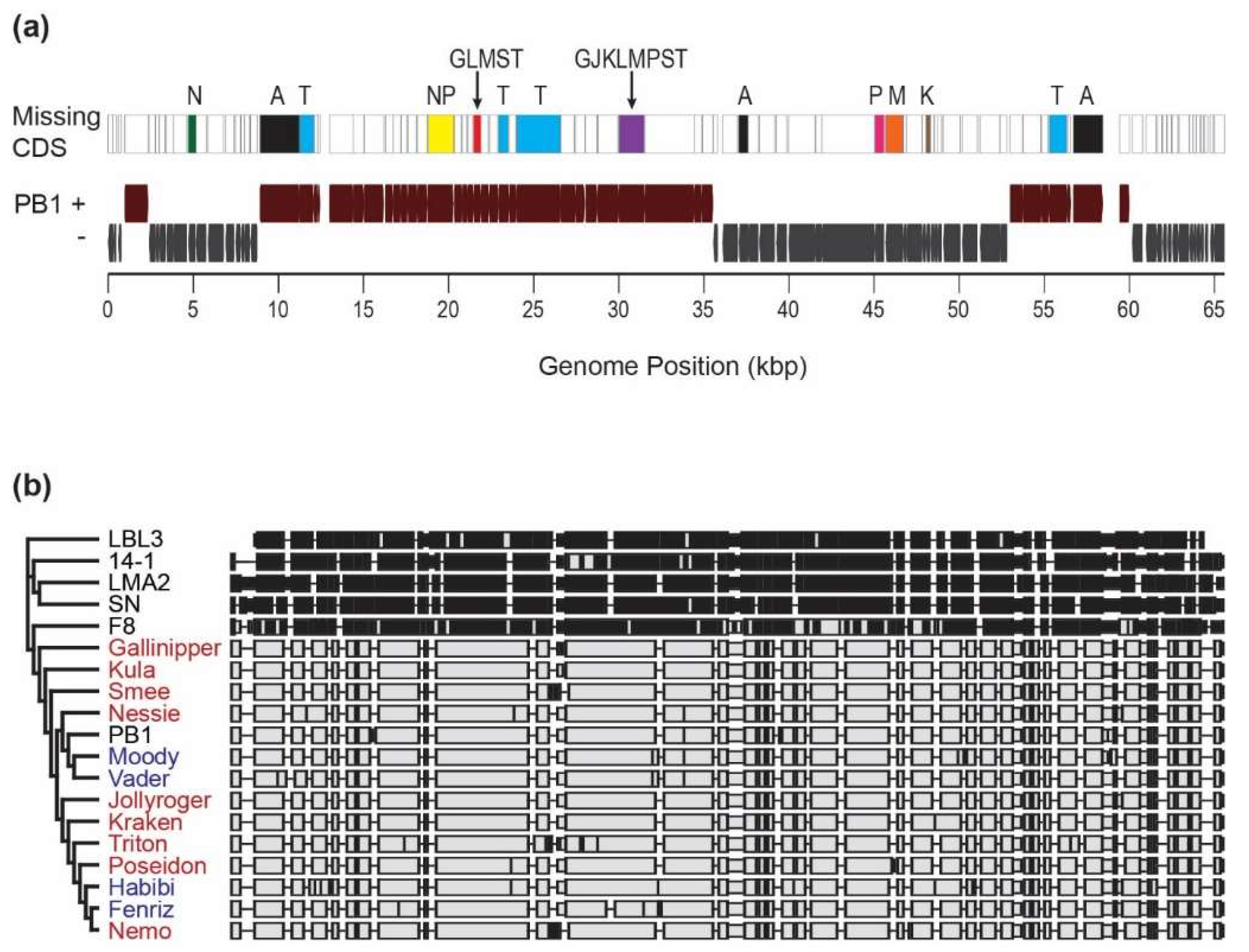
3.3. Unique PB1-Like Phage Genomes Can Be Assembled and Closed from Viral Metagenome Data Generated from Lake Michigan
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Salmond, G.P.C.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Thurber, R.V. Viruses manipulate the marine environment. Nature 2009, 459, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Balcazar, J.L. Bacteriophages as vehicles for antibiotic resistance genes in the environment. PLoS Pathog. 2014, 10, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.R.; Kropinski, A.M.; Clokie, M.R. Bacteriophage behavioral ecology: How phages alter their bacterial host’s habits. Bacteriophage 2014, 4, e29866. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M. Marine viruses: Truth or dare. Ann. Rev. Mar. Sci. 2012, 4, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef] [PubMed]
- Bruder, K.; Malki, K.; Cooper, A.; Sible, E.; Shapiro, J.W.; Watkins, S.C.; Putonti, C. Freshwater Metaviromics and Bacteriophages: A Current Assessment of the State of the Art in Relation to Bioinformatic Challenges. Evol. Bioinform. 2016, 12, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Perez Sepulveda, B.; Redgwell, T.; Rihtman, B.; Pitt, F.; Scanlan, D.J.; Millard, A. Marine phage genomics: The tip of the iceberg. FEMS Microbiol. Lett. 2016, 363, fnw158. [Google Scholar] [CrossRef] [PubMed]
- Pratama, A.A.; van Elsas, J.D. The “Neglected” Soil Virome–Potential Role and Impact. Trends Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 2015, 4, e08490. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, F.H.; Silveira, C.B.; Gregoracci, G.B.; Thompson, C.C.; Edwards, R.A.; Brussaard, C.P.D.; Dutilh, B.E.; Thompson, F.L. Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 2017, 8, 15955. [Google Scholar] [CrossRef] [PubMed]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Labonté, J.M.; Suttle, C.A. Metagenomic and whole-genome analysis reveals new lineages of gokushoviruses and biogeographic separation in the sea. Front. Microbiol. 2013, 4, 404. [Google Scholar] [CrossRef] [PubMed]
- Hanson, C.A.; Marston, M.F.; Martiny, J.B.H. Biogeographic Variation in Host Range Phenotypes and Taxonomic Composition of Marine Cyanophage Isolates. Front. Microbiol. 2016, 7, 983. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.-E.T.; Suttle, C.A. Biogeography of Viruses in the Sea. Annu. Rev. Virol. 2015, 2, 41–66. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.-E.T.; Winget, D.M.; White, R.A.; Hallam, S.J.; Suttle, C.A. Combining genomic sequencing methods to explore viral diversity and reveal potential virus-host interactions. Front. Microbiol. 2015, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hernandez, F.; Fornas, O.; Gomez, M.L.; Bolduc, B.; de La Cruz Peña, M.J.; Martínez, J.M.; Anton, J.; Gasol, J.M.; Rosselli, R.; Rodriguez-Valera, F.; et al. Single-virus genomics reveals hidden cosmopolitan and abundant viruses. Nat. Commun. 2017, 8, 15892. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz Peña, M.J.; Martinez-Hernandez, F.; Garcia-Heredia, I.; Lluesma Gomez, M.; Fornas, Ò.; Martinez-Garcia, M. Deciphering the human virome with single-virus genomics and metagenomics. Viruses 2018, 10, 113. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Westveld, A.H.; Brum, J.R.; Sullivan, M.B. Modeling ecological drivers in marine viral communities using comparative metagenomics and network analyses. Proc. Natl. Acad. Sci. USA 2014, 111, 10714–10719. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel K-MER based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Garza, D.R.; Dutilh, B.E. From cultured to uncultured genome sequences: Metagenomics and modeling microbial ecosystems. Cell Mol. Life Sci. 2015, 72, 4287–4308. [Google Scholar] [CrossRef] [PubMed]
- Iverson, V.; Morris, R.M.; Frazar, C.D.; Berthiaume, C.T.; Morales, R.L.; Armbrust, E.V. Untangling genomes from metagenomes: Revealing an uncultured class of marine Euryarchaeota. Science 2012, 335, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Miller, C.S.; Castelle, C.J.; VerBerkmoes, N.C.; Wilkins, M.J.; Hettich, R.L.; Lipton, M.S.; Williams, K.H.; et al. Fermentation, Hydrogen, and Sulfur Metabolism in Multiple Uncultivated Bacterial Phyla. Science 2012, 337, 1661–1665. [Google Scholar] [CrossRef] [PubMed]
- Inskeep, W.P.; Jay, Z.J.; Herrgard, M.J.; Kozubal, M.A.; Rusch, D.B.; Tringe, S.G.; Macur, R.E.; Boyd, E.S.; Spear, J.R.; Roberto, F.F. Phylogenetic and Functional Analysis of Metagenome Sequence from High-Temperature Archaeal Habitats Demonstrate Linkages between Metabolic Potential and Geochemistry. Front. Microbiol. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Sharon, I.; Banfield, J.F. Genomes from Metagenomics. Science 2013, 342, 1057–1058. [Google Scholar] [CrossRef] [PubMed]
- Handley, K.M.; Bartels, D.; O’Loughlin, E.J.; Williams, K.H.; Trimble, W.L.; Skinner, K.; Gilbert, J.A.; Desai, N.; Glass, E.M.; Paczian, T.; et al. The complete genome sequence for putative H2- and S-oxidizer Candidatus Sulfuricurvum sp., assembled de novo from an aquifer-derived metagenome. Environ. Microbiol. 2014, 16, 3443–3462. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T. Strain recovery from metagenomes. Nat. Biotechnol. 2015, 33, 1041–1043. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Saw, J.H.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, K.; Breier, J.A.; Dick, G.J. Metagenomic resolution of microbial functions in deep-sea hydrothermal plumes across the Eastern Lau Spreading Center. ISME J. 2016, 10, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Kumar, S.; Prasoodanan, V.P.K.; Harish, K.; Sharma, A.K.; Sharma, V.K. Reconstruction of Bacterial and Viral Genomes from Multiple Metagenomes. Front. Microbiol. 2016, 7, 469. [Google Scholar] [CrossRef] [PubMed]
- Haroon, M.F.; Thompson, L.R.; Parks, D.H.; Hugenholtz, P.; Stingl, U. A catalogue of 136 microbial draft genomes from Red Sea metagenomes. Sci. Data 2016, 3, 160050. [Google Scholar] [CrossRef] [PubMed]
- Laczny, C.C.; Muller, E.E.L.; Heintz-Buschart, A.; Herold, M.; Lebrun, L.A.; Hogan, A.; May, P.; de Beaufort, C.; Wilmes, P. Identification, Recovery, and Refinement of Hitherto Undescribed Population-Level Genomes from the Human Gastrointestinal Tract. Front. Microbiol. 2016, 7, 884. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Svartström, O.; Alneberg, J.; Terrapon, N.; Lombard, V.; de Bruijn, I.; Malmsten, J.; Dalin, A.-M.; EL Muller, E.; Shah, P.; Wilmes, P.; et al. Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J. 2017, 11, 2538–2551. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and Diversity of the Microviridae Viral Family through a Collection of 81 New Complete Genomes Assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; Bodewes, R.; Ruiz-González, A.; Baumgärtner, W.; Koopmans, M.P.; Osterhaus, A.D.M.E.; Schürch, A.C. Recovering full-length viral genomes from metagenomes. Front. Microbiol. 2015, 6, 1069. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.F.; Banfield, J.F. Virus population dynamics and acquired virus resistance in natural microbial communities. Science 2008, 320, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Labonté, J.M.; Suttle, C.A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013, 7, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Garcia-Heredia, I.; Martin-Cuadrado, A.-B.; Ghai, R. Reconstruction of novel cyanobacterial siphovirus genomes from Mediterranean metagenomic fosmids. Appl. Environ. Microbiol. 2013, 79, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Rodriguez-Valera, F.; Kimes, N.E.; Ghai, R. Expanding the marine virosphere using metagenomics. PLoS Genet. 2013, 9, e1003987. [Google Scholar] [CrossRef] [PubMed]
- Bellas, C.M.; Anesio, A.M.; Barker, G. Analysis of virus genomes from glacial environments reveals novel virus groups with unusual host interactions. Front. Microbiol. 2015, 6, 656. [Google Scholar] [CrossRef] [PubMed]
- Skvortsov, T.; de Leeuwe, C.; Quinn, J.P.; McGrath, J.W.; Allen, C.C.R.; McElarney, Y.; Watson, C.; Arkhipova, K.; Lavigne, R.; Kulakov, L.A. Metagenomic characterisation of the viral community of Lough Neagh, the largest freshwater lake in Ireland. PLoS ONE 2016, 11, e0150361. [Google Scholar] [CrossRef] [PubMed]
- Voorhies, A.A.; Eisenlord, S.D.; Marcus, D.N.; Duhaime, M.B.; Biddanda, B.A.; Cavalcoli, J.D.; Dick, G.J. Ecological and genetic interactions between cyanobacteria and viruses in a low-oxygen mat community inferred through metagenomics and metatranscriptomics: Cyanobacteria-virus interactions in a low-O2 mat community. Environ. Microbiol. 2016, 18, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Ghai, R.; Mehrshad, M.; Mizuno, C.M.; Rodriguez-Valera, F. Metagenomic recovery of phage genomes of uncultured freshwater actinobacteria. ISME J. 2017, 11, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics: Consensus statement. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Malki, K.; Kula, A.; Bruder, K.; Sible, E.; Hatzopoulos, T.; Steidel, S.; Watkins, S.C.; Putonti, C. Bacteriophages isolated from Lake Michigan demonstrate broad host-range across several bacterial phyla. Virol. J. 2015, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.-J.; Miroshnikov, K.; Mattheus, W.; Krylov, V.; Robben, J.; Noben, J.-P.; Vanderschraeghe, S.; Sykilinda, N.; Kropinski, A.M.; Volckaert, G.; et al. Comparative analysis of the widespread and conserved PB1-like viruses infecting Pseudomonas aeruginosa. Environ. Microbiol. 2009, 11, 2874–2883. [Google Scholar] [CrossRef] [PubMed]
- Krylov, V.N.; Tolmachova, T.O.; Akhverdian, V.Z. DNA homology in species of bacteriophages active on Pseudomonas aeruginosa. Arch. Virol. 1993, 131, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Garbe, J.; Wesche, A.; Bunk, B.; Kazmierczak, M.; Selezska, K.; Rohde, C.; Sikorski, J.; Rohde, M.; Jahn, D.; Schobert, M. Characterization of JG024, a pseudomonas aeruginosa PB1-like broad host range phage under simulated infection conditions. BMC Microbiol. 2010, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Danis-Wlodarczyk, K.; Olszak, T.; Arabski, M.; Wasik, S.; Majkowska-Skrobek, G.; Augustyniak, D.; Gula, G.; Briers, Y.; Jang, H.B.; Vandenheuvel, D.; et al. Characterization of the Newly Isolated Lytic Bacteriophages KTN6 and KT28 and Their Efficacy against Pseudomonas aeruginosa Biofilm. PLoS ONE 2015, 10, e0127603. [Google Scholar] [CrossRef]
- Pires, D.P.; Sillankorva, S.; Kropinski, A.M.; Lu, T.K.; Azeredo, J. Complete Genome Sequence of Pseudomonas aeruginosa Phage vB_PaeM_CEB_DP1. Genome Announc. 2015, 3, e00918-15. [Google Scholar] [CrossRef] [PubMed]
- Pourcel, C.; Midoux, C.; Latino, L.; Petit, M.-A.; Vergnaud, G. Complete Genome Sequences of Pseudomonas aeruginosa Phages vB_PaeP_PcyII-10_P3P1 and vB_PaeM_PcyII-10_PII10A. Genome Announc. 2016, 4, e00916-16. [Google Scholar] [CrossRef] [PubMed]
- Neves, P.R.; Cerdeira, L.T.; Mitne-Neto, M.; Oliveira, T.G.M.; McCulloch, J.A.; Sampaio, J.L.M.; Mamizuka, E.M.; Levy, C.E.; Sato, M.I.Z.; Lincopan, N. Complete Genome Sequence of an F8-Like Lytic Myovirus (SPM-1) That Infects Metallo-Lactamase-Producing Pseudomonas aeruginosa. Genome Announc. 2014, 2, e00061-14. [Google Scholar] [CrossRef] [PubMed]
- Zwart, G.; Crump, B.; Kamst-van Agterveld, M.; Hagen, F.; Han, S. Typical freshwater bacteria: An analysis of available 16S rRNA gene sequences from plankton of lakes and rivers. Aquat. Microb. Ecol. 2002, 28, 141–155. [Google Scholar] [CrossRef]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A Guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; de Vargas, C.; Gasol, J.M.; et al. Ocean plankton. Patterns and ecological drivers of ocean viral communities. Science 2015, 348, 1261498. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; Sullivan, M.B. The Pacific Ocean Virome (POV): A Marine Viral Metagenomic Dataset and Associated Protein Clusters for Quantitative Viral Ecology. PLoS ONE 2013, 8, e57355. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Suzuki, M.T.; Teeling, H.; Weber, M.; Venter, J.C.; Rusch, D.B.; Béjà, O. Assessing diversity and biogeography of aerobic anoxygenic phototrophic bacteria in surface waters of the Atlantic and Pacific Oceans using the Global Ocean Sampling expedition metagenomes. Environ. Microbiol. 2007, 9, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.; Rodriguez-Brito, B.; Bangor, D.; McNairnie, P.; Breitbart, M.; Salamon, P.; Felts, B.; Nulton, J.; Mahaffy, J.; Rohwer, F. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinform. 2005, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.C.; Putonti, C. The use of informativity in the development of robust viromics-based examinations. PeerJ 2017, 5, e3281. [Google Scholar] [CrossRef] [PubMed]
- Sible, E.; Cooper, A.; Malki, K.; Bruder, K.; Watkins, S.C.; Fofanov, Y.; Putonti, C. Survey of viral populations within Lake Michigan nearshore waters at four Chicago area beaches. Data Brief 2015, 5, 9–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.C.; Kuehnle, N.; Ruggeri, C.A.; Malki, K.; Bruder, K.; Elayyan, J.; Damisch, K.; Vahora, N.; O’Malley, P.; Ruggles-Sage, B.; et al. Assessment of a metaviromic dataset generated from nearshore Lake Michigan. Mar. Freshw. Res. 2016, 67, 1700–1708. [Google Scholar] [CrossRef]
- Magill, D.J.; Krylov, V.N.; Shaburova, O.V.; McGrath, J.W.; Allen, C.C.R.; Quinn, J.P.; Kulakov, L.A. Pf16 and phiPMW: Expanding the realm of Pseudomonas putida bacteriophages. PLoS ONE 2017, 12, e0184307. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Tsementzi, D.; Kyrpides, N.C.; Konstantinidis, K.T. Individual genome assembly from complex community short-read metagenomic datasets. ISME J. 2012, 6, 898–901. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Primer Pair | Forward Primer (5′–3′) | Reverse Primer (3′–5′) | Annotated Coding Sequence (CDS) in Amplicon | Strains/Species Expected to Amplify |
|---|---|---|---|---|
| 1 | CTACGGCCGTGCAGAC | CTCCATGTGTGGCATCC | PB1 gp10 (HP), PB1 gp11 (HP) | SPM-1, PB1, F8 |
| 2 | ACCTTCTTCGGCATCCTC | TGTGGTCACCGTATTCCA | PB1 gp23 (HP), PB1 gp24 (HP) | DL60, DL52, vB_PaeM_C1-14_Ab28, SPM-1, PB1, F8 |
| 3 | CGCCATAATAGGCTCCAA | CGAGACATTCGCTGATGA | PB1 gp55 (helicase) | SPM-1, PB1, F8 |
| 4 | ACCGACTCACGACGATGG | CGGCAAGGTGTTCGCTTA | PB1 gp68 (HP), PB1 gp69 (HP) | PB1 |
| 5 | CGTCGAGGATGCTGATGG | GGCAGGTCCGAAGGCTAC | PB1 gp84 (HP), PB1 gp85 (HP), PB1 gp86 (HP) | vB_PaeM_LS1, vB_PaeM_E215, KPP22M1, vB_PaeM_E217, NP3, phiKTN6, KPP22M3, KPP22M2, KP22, vB_Pae_PS44, LMA2, vB_PaeM_CEB_DP1, vB_PaeM_PAO1_Ab29, vB_PaeM_PAO1_Ab27, KPP12, NH-4, PB1 |
| Phage | Isolation (Beach/Date) | Coverage (Average) | Length (bp) | # Annotated Genes | Accession Number | SRA Accession (Raw Reads) |
|---|---|---|---|---|---|---|
| Gallinipper | 95th Street/10 June 2014 | 30 | 65,917 | 89 | KT372690 | SRX956318 |
| Jollyroger | 57th Street/8 July 2014 | 200 | 65,795 | 90 | KT372691 | SRX995828 |
| Kraken | 95th Street/5 August 2014 (Biological Replicate 1) | 15 | 65,762 | 88 | KT372692 | SRX995836 |
| Kula | 95th Street/5 August 2014 (Biological Replicate 2) | 20 | 65,762 | 89 | KT372693 | SRX995836 |
| Nemo | 95th Street/5 August 2014 (Biological Replicate 3) | 20 | 66,283 | 88 | KT372694 | SRX995836 |
| Nessie | Wilmette/13 May 2014 | 220 | 65,762 | 89 | KT372695 | SRX995816 |
| Poseidon | Wilmette/5 August 2014 | 130 | 65,762 | 88 | KT372696 | SRX995833 |
| Smee | Montrose/8 July 2014 | 115 | 66,278 | 89 | KT372697 | SRX995827 |
| Triton | 57th/5 August 2014 | 125 | 65,762 | 85 | KT372698 | SRX995835 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watkins, S.C.; Sible, E.; Putonti, C. Pseudomonas PB1-Like Phages: Whole Genomes from Metagenomes Offer Insight into an Abundant Group of Bacteriophages. Viruses 2018, 10, 331. https://doi.org/10.3390/v10060331
Watkins SC, Sible E, Putonti C. Pseudomonas PB1-Like Phages: Whole Genomes from Metagenomes Offer Insight into an Abundant Group of Bacteriophages. Viruses. 2018; 10(6):331. https://doi.org/10.3390/v10060331
Chicago/Turabian StyleWatkins, Siobhan C., Emily Sible, and Catherine Putonti. 2018. "Pseudomonas PB1-Like Phages: Whole Genomes from Metagenomes Offer Insight into an Abundant Group of Bacteriophages" Viruses 10, no. 6: 331. https://doi.org/10.3390/v10060331
APA StyleWatkins, S. C., Sible, E., & Putonti, C. (2018). Pseudomonas PB1-Like Phages: Whole Genomes from Metagenomes Offer Insight into an Abundant Group of Bacteriophages. Viruses, 10(6), 331. https://doi.org/10.3390/v10060331