Analysis of 19 Highly Conserved Vibrio cholerae Bacteriophages Isolated from Environmental and Patient Sources Over a Twelve-Year Period


Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Genome Characteristics and Phylogeny
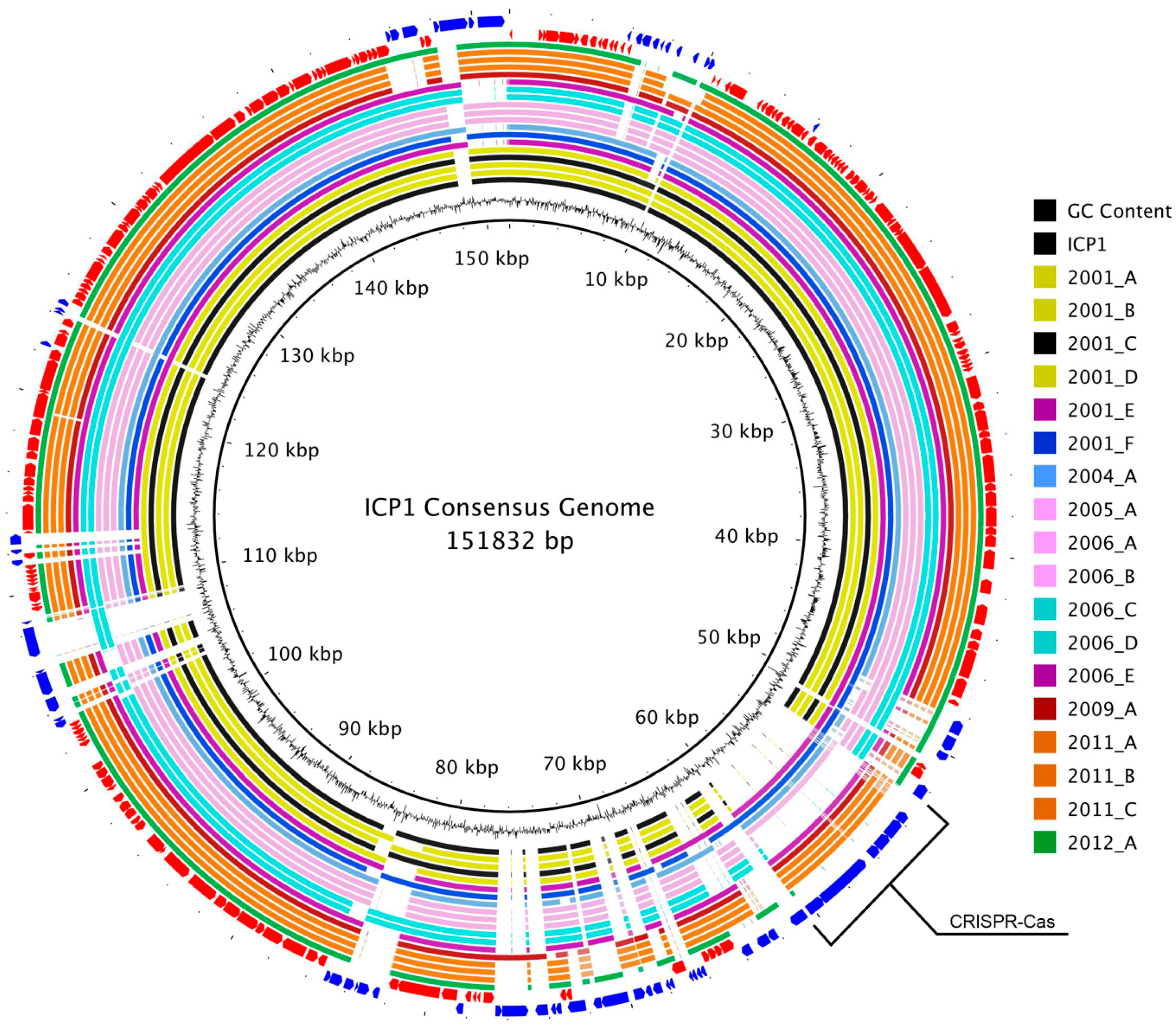
3.2. Genome Alignment Visualization
3.3. ORF Annotation
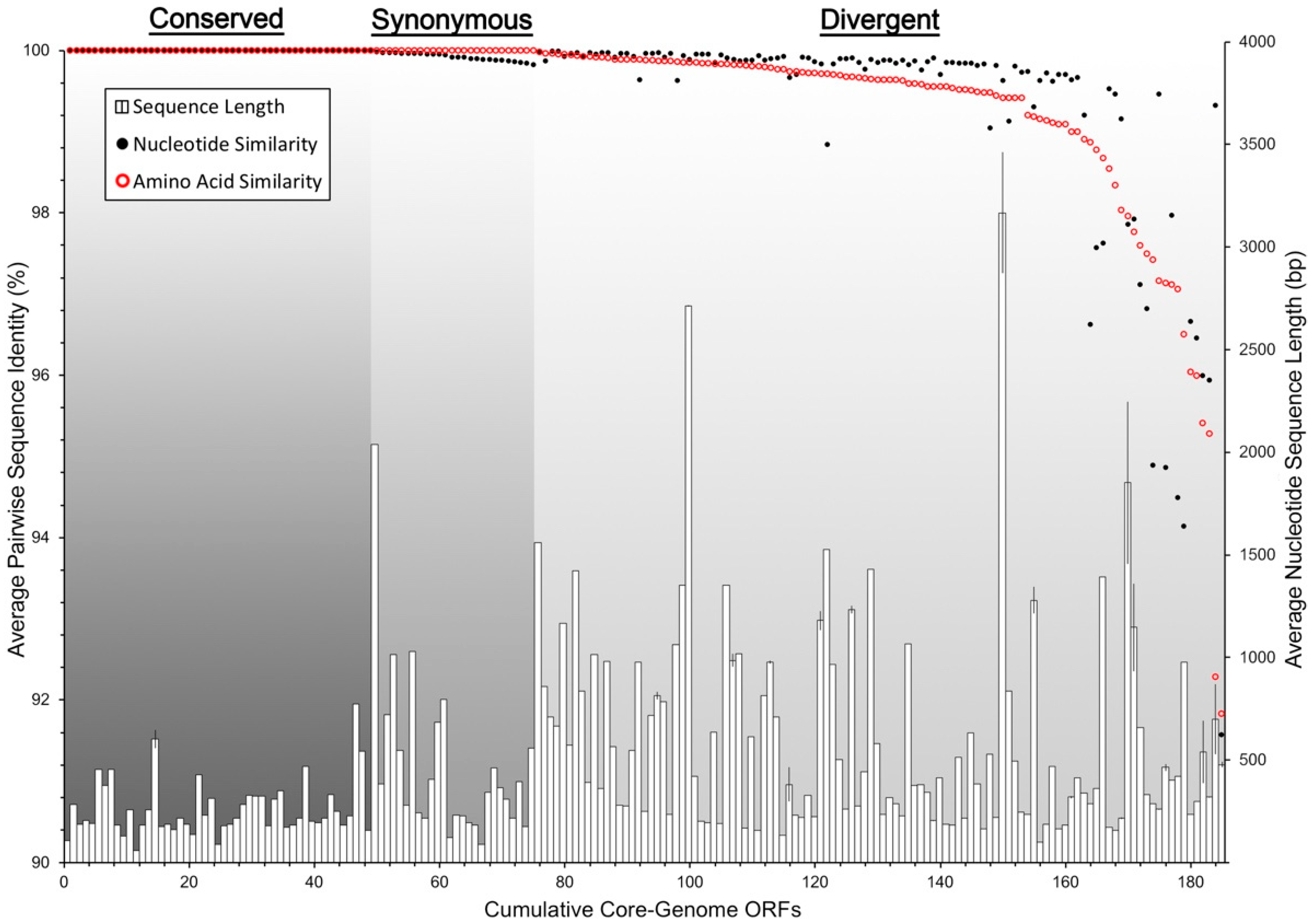
3.4. Core and Accessory Genome Analysis
3.5. Conserved Functional Domains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ali, M.; Nelson, A.R.; Lopez, A.L.; Sack, D.A. Updated global burden of cholera in endemic countries. PLoS Negl. Trop. Dis. 2015, 9, e0003832. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.; Nair, G.B.; Kong, R.Y.C. Evolution of new variants of Vibrio cholerae O1. Trends Microbiol. 2010, 18, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Mutreja, A.; Kim, D.W.; Thomson, N.R.; Connor, T.R.; Lee, J.H.; Kariuki, S.; Croucher, N.J.; Choi, S.Y.; Harris, S.R.; Lebens, M.; et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 2011, 477, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Thomson, N.; Mutreja, A.; Piarroux, R. Widespread epidemic cholera caused by a restricted subset of Vibrio cholerae clones. Clin. Microbiol. Infect. 2014, 20, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Weill, F.-X.; Domman, D.; Njamkepo, E.; Tarr, C.; Rauzier, J.; Fawal, N.; Keddy, K.H.; Salje, H.; Moore, S.; Mukhopadhyay, A.K.; et al. Genomic history of the seventh pandemic of cholera in Africa. Science 2017, 358, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-J.; Yi, H.; Lee, J.H.; Kim, D.W.; Chun, J. Genomic evolution of Vibrio cholerae. Curr. Opin. Microbiol. 2010, 13, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Domman, D.; Quilici, M.-L.; Dorman, M.J.; Njamkepo, E.; Mutreja, A.; Mather, A.E.; Delgado, G.; Morales-Espinosa, R.; Grimont, P.A.D.; Lizárraga-Partida, M.L.; et al. Integrated view of Vibrio cholerae in the Americas. Science 2017, 358, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Parikka, K.J.; Le Romancer, M.; Wauters, N.; Jacquet, S. Deciphering the virus-to-prokaryote ratio (VPR): Insights into virus-host relationships in a variety of ecosystems. Biol. Rev. Camb. Philos. Soc. 2017, 92, 1081–1100. [Google Scholar] [CrossRef] [PubMed]
- Ofir, G.; Sorek, R. Contemporary Phage Biology: From Classic Models to New Insights. Cell 2018, 172, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Seed, K.D.; Faruque, S.M.; Mekalanos, J.J.; Calderwood, S.B.; Qadri, F.; Camilli, A. Phase variable O antigen biosynthetic genes control expression of the major protective antigen and bacteriophage receptor in Vibrio cholerae O1. PLoS Pathog. 2012, 8, e1002917-13. [Google Scholar] [CrossRef] [PubMed]
- Seed, K.D.; Bodi, K.L.; Kropinski, A.M.; Ackermann, H.-W.; Calderwood, S.B.; Qadri, F.; Camilli, A. Evidence of a dominant lineage of Vibrio cholerae-specific lytic bacteriophages shed by cholera patients over a 10-year period in Dhaka, Bangladesh. mBio 2011, 2, e00334-10. [Google Scholar] [CrossRef] [PubMed]
- Faruque, S.M.; Naser, I.B.; Islam, M.J.; Faruque, A.S.G.; Ghosh, A.N.; Nair, G.B.; Sack, D.A.; Mekalanos, J.J. Seasonal epidemics of cholera inversely correlate with the prevalence of environmental cholera phages. Proc. Natl. Acad. Sci. USA 2005, 102, 1702–1707. [Google Scholar] [CrossRef] [PubMed]
- Naser, I.B.; Hoque, M.M.; Nahid, M.A.; Tareq, T.M.; Rocky, M.K.; Faruque, S.M. Analysis of the CRISPR-Cas system in bacteriophages active on epidemic strains of Vibrio cholerae in Bangladesh. Sci. Rep. 2017, 7, 14880. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, B.J.; Barth, Z.K.; McKitterick, A.C.; Seed, K.D. A highly specific phage defense system is a conserved feature of the Vibrio cholerae mobilome. PLoS Genet. 2017, 13, e1006838. [Google Scholar] [CrossRef] [PubMed]
- Seed, K.D.; Lazinski, D.W.; Calderwood, S.B.; Camilli, A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature 2013, 494, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Weil, A.; Ryan, E.T.; Calderwood, S.B.; Harris, J.B.; Chowdhury, F.; Begum, Y.; Qadri, F.; LaRocque, R.C.; Turnbaugh, P.J. Gut microbial succession follows acute secretory diarrhea in humans. mBio 2015, 6, e00381-15. [Google Scholar] [CrossRef] [PubMed]
- Das, M.M.; Bhotra, T.; Zala, D.; Singh, D.V. Phenotypic and genetic characteristics of Vibrio cholerae O1 carrying Haitian ctxB and attributes of classical and El Tor biotypes isolated from Silvassa, India. J. Med. Microbiol. 2016, 65, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.-F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Liu, B.; Feng, L.; Ding, P.; Guo, X.; Wang, M.; Cao, B.; Reeves, P.R.; Wang, L. Origins of the current seventh cholera pandemic. Proc. Natl. Acad. Sci. USA 2016, 113, E7730–E7739. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Ravet, V.; Pereira, O.; Sullivan, M.B. Genomic characteristics and environmental distributions of the uncultivated Far-T4 phages. Front. Microbiol. 2015, 6, 199. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Skurnik, M. Genomic characterization of sixteen Yersinia enterocolitica-infecting Podoviruses of pig origin. Viruses 2018, 10, 174. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Standardized Name | Previous Name | Isolation Year | Isolation Source | Genome Size (bp) | GenBank Accession | Genome Citation |
|---|---|---|---|---|---|---|
| ICP1 | - | 2001 | Stool | 125,956 | HQ641347 | Seed et al., 2011 |
| ICP1_2001_A | - | 2001 | Stool | 124,826 | HQ641353 | Seed et al., 2011 |
| ICP1_2001_B | JSF1 | 2001 | Water | 126,082 | KY883636 | Naser et al., 2017 |
| ICP1_2001_C | JSF2 | 2001 | Water | 126,082 | KY883637 | Naser et al., 2017 |
| ICP1_2001_D | JSF4 | 2001 * | Water * | 124,261 | KY065147 | Naser et al., 2017 |
| ICP1_2001_E | JSF5 | 2001 * | Water | 132,142 | KY883634 | Naser et al., 2017 |
| ICP1_2001_F | JSF6 | 2001 * | Water | 133,685 | KY883635 | Naser et al., 2017 |
| ICP1_2004_A | - | 2004 | Stool | 128,083 | HQ641354 | Seed et al., 2011 |
| ICP1_2005_A | - | 2005 | Stool | 129,373 | HQ641352 | Seed et al., 2011 |
| ICP1_2006_A | - | 2006 | Stool | 123,104 | HQ641351 | Seed et al., 2011 |
| ICP1_2006_B | - | 2006 | Stool | 123,097 | HQ641350 | Seed et al., 2011 |
| ICP1_2006_C | - | 2006 | Stool | 124,497 | HQ641349 | Seed et al., 2011 |
| ICP1_2006_D | - | 2006 | Stool | 124,497 | HQ641348 | Seed et al., 2011 |
| ICP1_2006_E | - | 2006 | Stool | 128,298 | MH310934 | This study |
| ICP1_2009_A | JSF13 | 2009 | Water | 128,814 | KY883638 | Naser et al., 2017 |
| ICP1_2011_A | - | 2011 | Stool | 126,861 | MH310933 | This study |
| ICP1_2011_B | - | 2011 | Stool | 125,128 | MH310935 | This study |
| ICP1_2011_C | JSF14 | 2011 | Water | 125,096 | KY883639 | Naser et al., 2017 |
| ICP1_2012_A | - | 2012 | Stool | 121,418 | MH310936 | This study |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angermeyer, A.; Das, M.M.; Singh, D.V.; Seed, K.D. Analysis of 19 Highly Conserved Vibrio cholerae Bacteriophages Isolated from Environmental and Patient Sources Over a Twelve-Year Period. Viruses 2018, 10, 299. https://doi.org/10.3390/v10060299
Angermeyer A, Das MM, Singh DV, Seed KD. Analysis of 19 Highly Conserved Vibrio cholerae Bacteriophages Isolated from Environmental and Patient Sources Over a Twelve-Year Period. Viruses. 2018; 10(6):299. https://doi.org/10.3390/v10060299
Chicago/Turabian StyleAngermeyer, Angus, Moon Moon Das, Durg Vijai Singh, and Kimberley D. Seed. 2018. "Analysis of 19 Highly Conserved Vibrio cholerae Bacteriophages Isolated from Environmental and Patient Sources Over a Twelve-Year Period" Viruses 10, no. 6: 299. https://doi.org/10.3390/v10060299
APA StyleAngermeyer, A., Das, M. M., Singh, D. V., & Seed, K. D. (2018). Analysis of 19 Highly Conserved Vibrio cholerae Bacteriophages Isolated from Environmental and Patient Sources Over a Twelve-Year Period. Viruses, 10(6), 299. https://doi.org/10.3390/v10060299