There is a growing interest in understanding the diversity, evolution, and disease associations of viruses in natural populations [
1]. Although sampling of many terrestrial species is relatively straightforward, there may be serious logistical challenges for animals that live in inaccessible habitats. Marine environments are one such habitat [
2,
3,
4]. It has recently been shown that wild populations can be sampled using Unmanned Aerial Vehicles (UAVs) [
5,
6]. UAVs are rapidly transforming wildlife science, allowing sampling from dangerous and inaccessible environments to address questions previously only approached by theory. Here, we show how UAVs can be used to sample viruses. This approach may ultimately enable a better understanding of the patterns and drivers of disease emergence in wild populations.
There is evidence that marine mammal health is deteriorating as anthropogenic stressors on the world’s oceans increase [
7]. However, contemporary assessments of marine mammal health are strongly biased towards animals whose health is already compromised, such as stranded animals, which in part reflects the difficulties in sampling aquatic environments. Sampling from free-ranging marine mammals is therefore critical to assess whether healthy animal populations are potential reservoirs of viruses and other transmittable agents.
Following the use of UAV technology for sampling, we employed a meta-transcriptomic approach [
8,
9] to help characterize the virome of an important marine mammal, the Eastern Australian humpback whale (
Megaptera novaeangliae), which serves as a model for work in this area. Recent analyses of whale breath, or “blow”, have revealed an extraordinary diversity and abundance of microbiota. Importantly, the microbial communities observed were divergent from those present in the surrounding seawater such that they could be considered as distinctly whale blow associated [
5,
6]. To date, however, these studies have not included virus sampling, and little is known about the diversity of the whale virome and whether this differs fundamentally from that seen in terrestrial mammals.
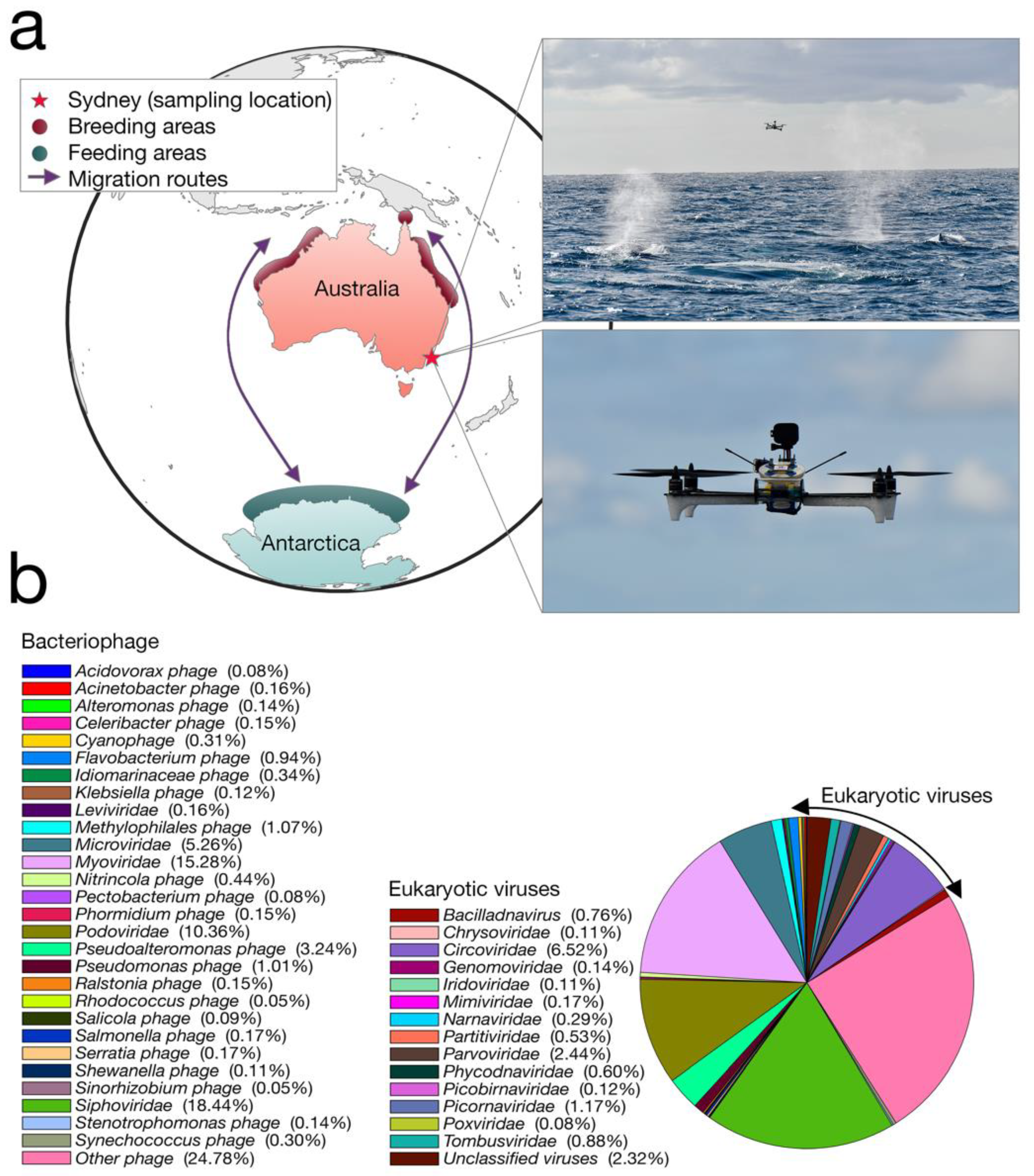
We collected whale blow samples from 19 humpbacks during the 2017 annual northward migration from Antarctica to northern Australia (
Figure 1a). To adhere to all Australian legislative requirements, our UAVs were registered with the Civil Aviation Safety Authority (CASA) and operated by a CASA-certified remote pilot. All flights were conducted in good weather (no rain, Beaufort < 3), from a small research vessel, where the UAV was launched and landed on a launch pad at the stern of the boat. A closed, sterile petri dish was placed on eight suction cups on the UAV before each flight.
Members of the team visually scanned the area for humpback whales. Once an individual or pod was chosen, the vessel was driven at a constant speed and distance from the whale. Once the respiratory rhythm was determined (i.e., downtime length), the UAV was launched to coincide with surfacing. The UAV pilot was directed by spotters on the vessel and positioned the UAV with the aid of the live feed from a forward-facing camera. To minimize sample contamination, the petri dish remained closed until immediately before the whale surfaced. The dish was remotely opened as the UAV accelerated towards and through the densest part of the whale blow, collecting the maximum amount of sample in the dish and lid (see
Video S1). The petri dish was immediately closed and the UAV was returned to the vessel. The petri dish containing the sample was removed from the UAV and secured with Parafilm
®. All samples were stored immediately in a portable −80 °C freezer. A different whale was sampled each flight. Different individuals within a pod were chosen based upon unique distinctive markings (e.g., white flanks and barnacle arrangements).
RNA was extracted using an RNeasy Plus Universal mini kit (Qiagen, Australia). Due to low RNA concentration, all 19 samples were pooled and concentrated using a NucleoSpin RNA Clean-up XS kit (Macherey-Nagel, Australia). A single library was produced for RNA sequencing using the Low-Input SMARTer Stranded Total RNA Sample Prep Kit with Mammalian rRNA depletion (Clontech, Australia), with 1 ng of the pooled whale blow RNA as input. Paired-end (100 bp) sequencing of the RNA library was performed on the HiSeq 2500 platform (Illumina, Australia) at the Australian Genome Research Facility.
RNA sequencing of the rRNA-depleted library resulted in 19,389,378 paired reads (100 nt in length) that were assembled de novo into 107,681 contigs. Sequencing reads were first quality trimmed then assembled using Trinity [
10]. The assembled transcriptome was annotated based on similarity searches against the NCBI nucleotide (nt) and non-redundant protein (nr) databases using BLASTn [
11] and Diamond (BLASTX) [
12], respectively, and an e-value threshold of 1 × 10
−5. Transcript abundance was estimated using RSEM [
13] implemented within Trinity.
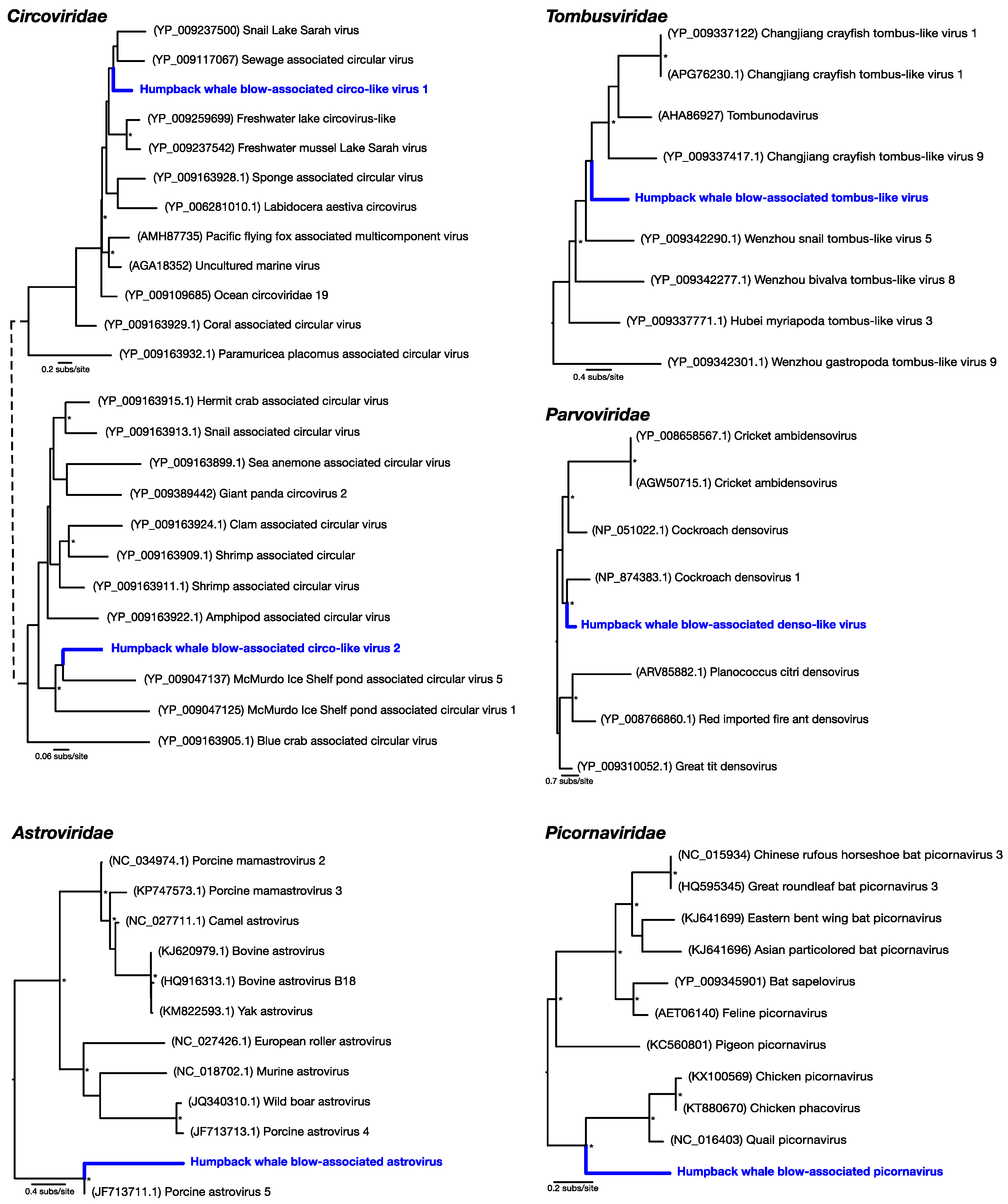
Our transcriptome data revealed that the humpback whale blow contains a wide diversity of DNA and RNA viruses (that we refer to “whale-blow-associated” viruses). BLAST analysis revealed the relative abundance of taxonomic classes present in the non-rRNA transcriptome data, of which bacteria occupied ~45%, while ciliates were the second-most abundant source at ~29%. Importantly, Baleen whale species contributed 0.9% of the transcriptome data and were the most abundant source of mammalian RNA, indicating our sample is indeed whale associated. Viruses occupied ~0.01% of the non-rRNA transcriptome, which falls within the range of other meta-transcriptome studies of vertebrates [
9]. Despite this relatively low abundance, the viral contigs observed fell into 42 classified viral families, including 29 families of bacteriophage (
Figure 1b). The most relatively abundant bacteriophages included the
Siphoviridae (18.4% of all viruses) and the
Myoviridae (15.2% of all viruses). Among the most abundant viral families that are known to infect eukaryotes were small single-stranded (ss) DNA viruses, specifically the
Circoviridae (and
Circoviridae-like viruses) (6.5% of all viruses), as well as members of the
Parvoviridae (2.4%) and an RNA virus family, the
Tombusviridae (0.9%).
We next inferred the evolutionary relationships of the viruses contained in whale blow with their closest phylogenetic relatives. Translated open reading frame segments were combined with protein sequences obtained from GenBank, using the top search results from BLAST (see
Table 1 for more details of the sequences analyzed). Sequences were aligned using MAFFT v.3.4 [
14], employing the E-INS-I algorithm with poorly aligned regions removed using trimAl v.1.2 [
15]. To estimate phylogenetic trees for the virus data sets, we selected the optimal amino acid substitution model identified using the Bayesian Information Criterion as implemented in Modelgenerator v.0.85 [
16] and analyzed the data using the maximum likelihood approach available in PhyML v3.1 [
17] with 1000 bootstrap replicates. Phylogenetic trees were annotated with FigTree v.1.4.2.
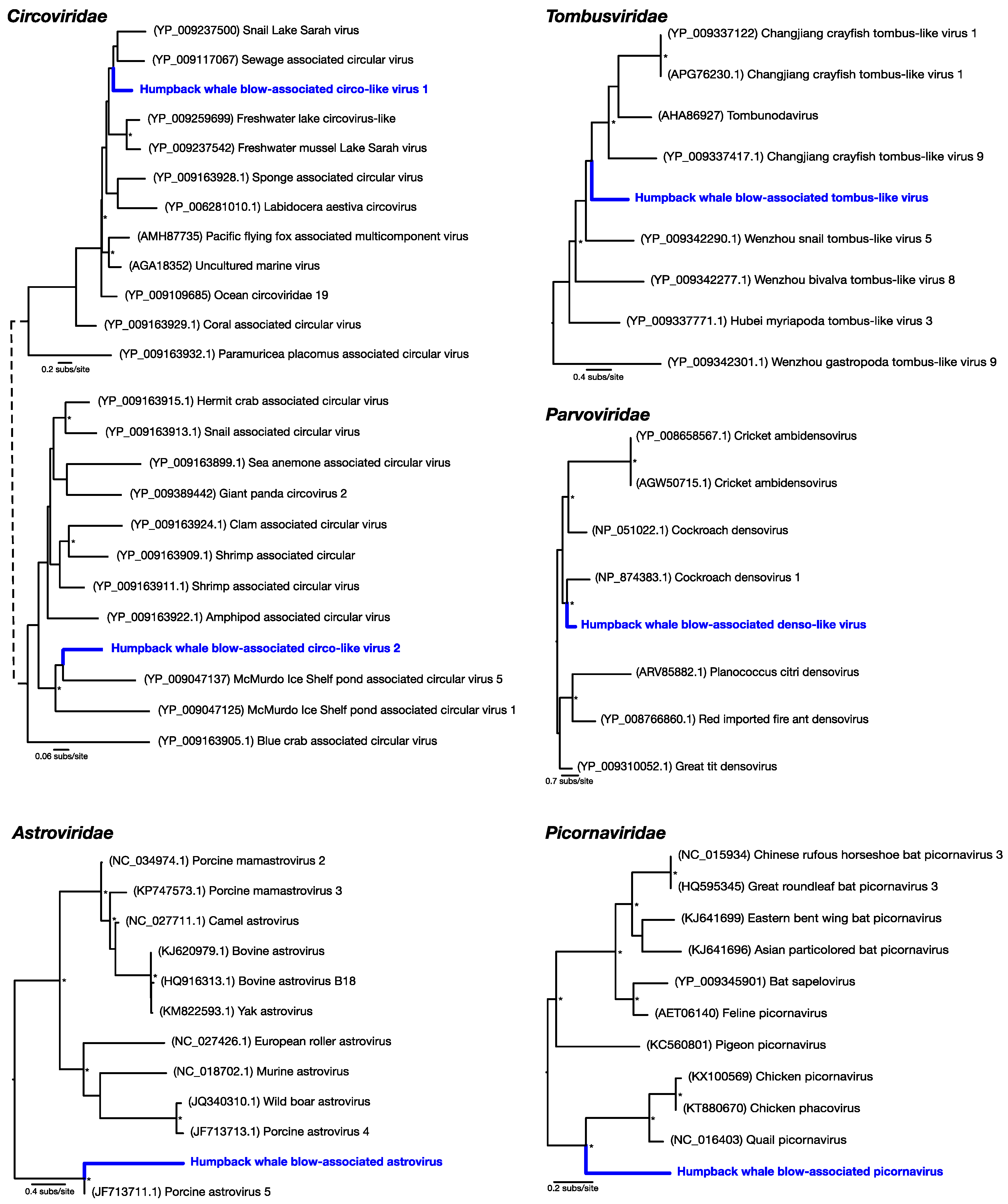
Of the most abundant eukaryotic viruses, two novel (as determined by phylogenetic analysis) circular Rep-encoding ssDNA viruses (CRESS-DNA viruses)
Circoviridae-like viruses were identified, denoted here as humpback whale blow-associated circo-like virus 1 and 2 (
Table 1;
Figure 2). Related viruses have previously been identified in many aquatic systems, for which marine invertebrates, particularly crustaceans, are thought to be a primary host [
18]. Humpback whale blow-associated circo-like virus-1 exhibited 51% amino acid identity to the replication-associated protein (Rep) of its closest genetic relative, sewage-associated circular DNA virus-29, and 46% amino acid identity to the Rep of Lake Sarah associated circular virus-32. Humpback whale blow-associated circo-like virus-2 shared 46% amino acid identity to the Rep of McMurdo Ice Shelf virus-5, isolated from a freshwater pond in Antarctica [
19]. As these ssDNA viruses appear to be major virome components in many aquatic environments [
18], they are likely associated with aquatic ecosystems in general.
Another relatively abundant viral contig was a partial genome of a novel densovirus (family
Parvoviridae). The most similar amino acid sequence to this new virus, denoted here as humpback whale blow-associated denso-like virus, was a densovirus isolated from a
Periplaneta fuliginosa (i.e., a cockroach), sharing only 47% sequence similarity to the nonstructural protein (
Table 1;
Figure 2). Similarly, a novel tombus-like viral partial genome, falling into the
Tombusviridae, was identified and was closely related to Changjiang tombus-like virus-9 isolated from crayfish, with 41% sequence similarity to the RNA-dependent RNA polymerase (RdRp). We denote this virus humpback whale blow-associated tombus-like virus (
Table 1;
Figure 2).
To reveal viruses at very low relative abundance, a Diamond BLAST [
12] analysis was performed against the raw 100 bp sequencing reads. This process identified several sequencing reads that matched viruses, later assembled into short contigs, that comprised two potentially new RNA viruses from the
Picornaviridae and the
Astroviridae. Humpback whale blow-associated picornavirus shared 61% amino acid similarity to the RdRp of the most closely related
Coturnix coturnix (quail) picornavirus (
Table 1;
Figure 2). Similarly, humpback whale blow-associated astrovirus shared 76% amino acid identity with the nonstructural protein 1a of porcine astrovirus-5 (
Figure 2). Both picornaviruses and astroviruses are single-stranded, positive-sense RNA viruses with small icosahedral capsids and no external envelope which may aid their preservation in harsh marine environments, and viruses from these families are commonly found in aquatic vertebrates [
9]. As only short fragments of these viruses’ genomes were identified in our data set, their phylogenetic position requires confirmation. This is likely due to the low quantity of RNA isolated from the whale blow samples and the pooling of individual samples. However, that both these viruses were most closely related to other vertebrate viruses suggested that they are likely whale associated rather than sampled from the surrounding seawater. Further sampling of the sea water virome is required to understand the potential enormous diversity that comprises the aquatic virosphere.
Little is known about the transmission of whale viruses. Analyses of whale influenza viruses suggest that they likely originated from gulls and that feeding activities of gulls and whales often place them in close contact, such that oral–fecal transmission through seawater is a likely route [
20] and which might explain our observation of viruses associated with aquatic ecosystems. In addition, given the vast aerosol produced by whales, and their close contact within migrating pods as well as at feeding and breeding grounds, respiratory transmission may also play an important role in the movement of viruses in whales.
In sum, we show that drone-based virological surveys of previously inaccessible wildlife populations has the potential to help reveal the diversity of the virosphere, facilitating the detection of viruses infecting wildlife and aiding evaluation of their pathogenic and zoonotic potential.
Supplementary Materials
The following are available online at
https://www.mdpi.com/1999-4915/10/6/300/s1. Video S1: GoPro footage from UAV demonstrates whale blow sampling. All sequence reads generated in this project are available under the NCBI Short Read Archive (SRA) under accession number SRP149185 and virus sequences have been deposited in GenBank.
Author Contributions
Conceptualization, J.L.G., V.P., R.H., E.C.H.; Methodology, J.L.G., V.P., E.H., A.S., R.H., E.C.H.; Validation, J.L.G., J.P.B., J.-S.E., E.H., E.C.H.; Formal Analysis, J.L.G., E.H., J.S.E., J.P.B.; Investigation, J.L.G., V.P., E.H., A.S., M.O., R.H., E.C.H.; Resources, V.P., A.S., R.H., E.C.H.; Writing—Original Draft Preparation, J.L.G., V.P., R.H., E.C.H.; Writing—Review & Editing, J.L.G., V.P., E.H., M.O., R.H., E.C.H.; Visualization, J.L.G.; Supervision, J.L.G., R.H., E.C.H.; Project Administration, J.L.G.; Funding Acquisition, R.H., E.C.H, A.S, J.L.G..
Funding
V.P. is supported by a Macquarie Research Excellence scholarship. E.C.H. is funded by an ARC Australian Laureate Fellowship (FL170100022).
Acknowledgments
This research was conducted under the Macquarie University Animal Ethics Committee Animal Research Authority 2016/010 and the NSW Office of Environment and Heritage Scientific Research permit SL101743. We thank Heliguy and Guy Alexander for providing drone manufacturing and testing support, CASA certification, and operational insurances, and NSW Maritime and Oliver Masens for their support and use of vessels for sampling. We also thank the Cape Solander Whale Migration Study—particularly Wayne Reynolds, Sue Rennie Wright, and Mark Mcgeachie—and Sally Browning and Air Pig Productions for their donation of drone batteries.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Geoghegan, J.L.; Holmes, E.C. Predicting virus emergence amid evolutionary noise. Open Biol. 2017, 7, 170189. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. Metagenomic analysis of coastal RNA virus communities. Science 2006, 312, 1795–1798. [Google Scholar] [CrossRef] [PubMed]
- Bogomolni, A.L.; Gast, R.J.; Ellis, J.C.; Dennett, M.; Pugliares, K.R.; Lentell, B.J.; Moore, M.J. Victims or vectors: A survey of marine vertebrate zoonoses from coastal waters of the Northwest Atlantic. Dis. Aquat. Organ. 2008, 81, 13–38. [Google Scholar] [CrossRef] [PubMed]
- Pirotta, V.; Smith, A.; Ostrowski, M.; Russell, D.; Jonsen, I.D.; Grech, A.; Harcourt, R. An economical custom-built drone for assessing whale health. Front. Marine Sci. 2017, 4, 425. [Google Scholar] [CrossRef]
- Apprill, A.; Miller, C.A.; Moore, M.J.; Durban, J.W.; Fearnbach, H.; Barrett-Lennard, L.G. Extensive core microbiome in drone-captured whale blow supports a framework for health monitoring. mSystems 2017, 2, e00119-17. [Google Scholar] [CrossRef] [PubMed]
- Gulland, F.M.D.; Hall, A.J. Is marine mammal health deteriorating? Trends in the global reporting of marine mammal disease. EcoHealth 2007, 4, 135–150. [Google Scholar] [CrossRef]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-Seq: Reference generation and analysis with Trinity. Nature Protocol. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 15, 403–410. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nature Meth. 2014, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Creevey, C.J.; Pentony, M.M.; Naughton, T.J.; Mclnerney, J.O. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 2006, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [PubMed]
- Rosario, K.; Schenck, R.O.; Harbeitner, R.C.; Lawler, S.N.; Breitbart, M. Novel circular single-stranded DNA viruses identified in marine invertebrates reveal high sequence diversity and consistent predicted intrinsic disorder patterns within putative structural proteins. Front. Microbiol. 2015, 6, 696. [Google Scholar] [CrossRef] [PubMed]
- Zawar-Reza, P.; Arguello-Astorga, G.R.; Kraberger, S.; Julian, L.; Stainton, D.; Broady, P.A.; Varsani, A. Diverse small circular single-stranded DNA viruses identified in a freshwater pond on the McMurdo Ice Shelf (Antarctica). Infect. Genet. Evol. 2014, 26, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, V.S.; Bean, W.J.; Geraci, J.; Fiorelli, P.; Early, G.; Webster, R.G. Characterization of two influenza A viruses from a pilot whale. J. Virol. 1986, 58, 655–656. [Google Scholar] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).

 ,
, 

{kind=link}
{kind=link}