Rapid Identification of Intact Staphylococcal Bacteriophages Using Matrix-Assisted Laser Desorption Ionization-Time-of-Flight Mass Spectrometry


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Phages
2.2. Bacteriophage Propagation and Titration
2.3. Concentrating Bacteriophage to Pellets
2.4. Purification of Phage Particles by CsCl Density Gradient Centrifugation
2.5. Fast Protein Liquid Chromatography (FPLC)
2.6. Phage Purification by Ultrafiltration
2.7. Phage Precipitation by Polyethylene Glycol (PEG)
2.8. Sample Preparation for MALDI-MS
2.9. MALDI-MS Profiling Analysis
2.10. MALDI-MS/MS Analysis
3. Results
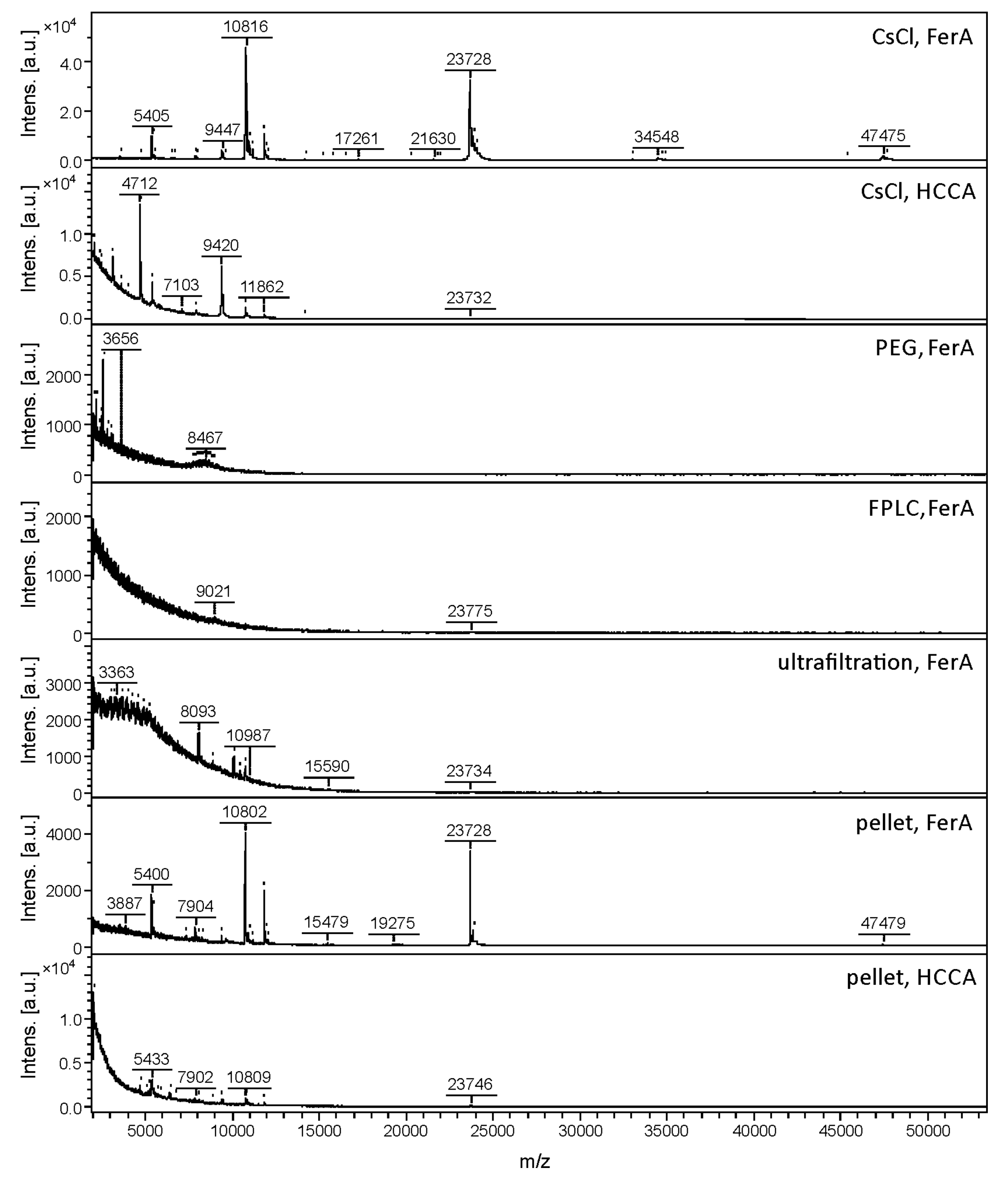
3.1. Comparison of Phage Purification Techniques for Sample Preparation and MALDI-TOF MS Method Development
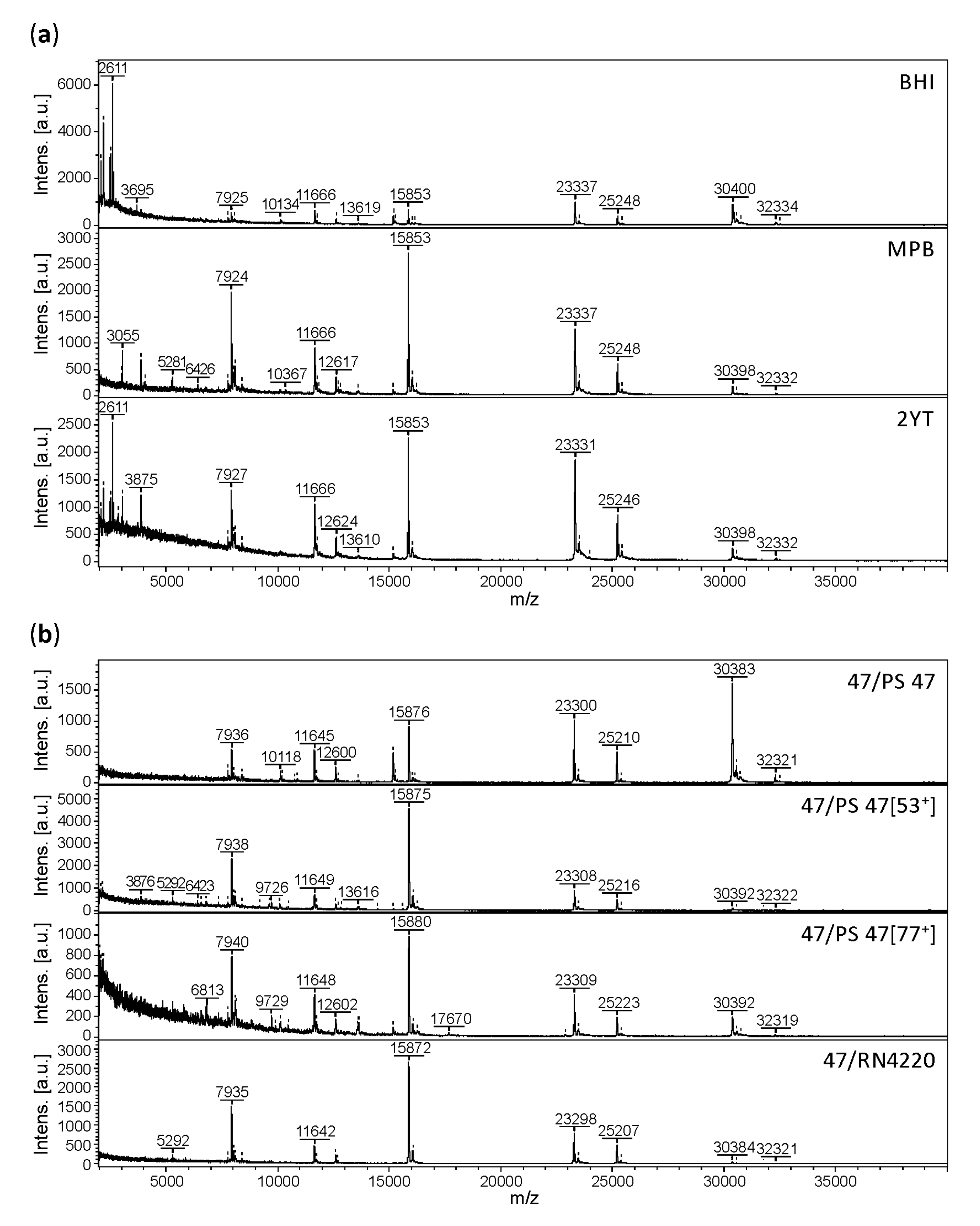
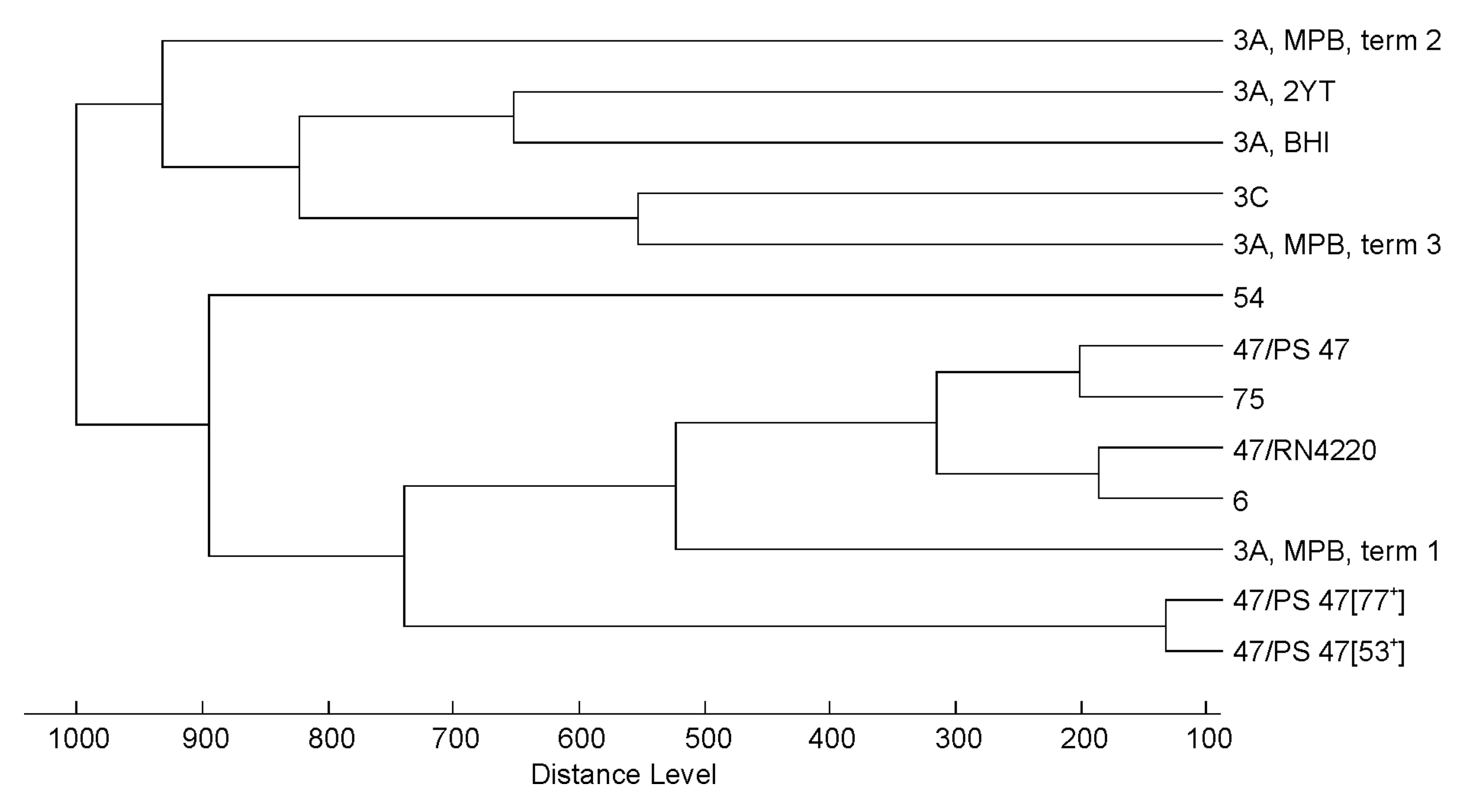
3.2. Effect of Culture Conditions on the Quality of Phage MALDI-TOF Mass Spectra
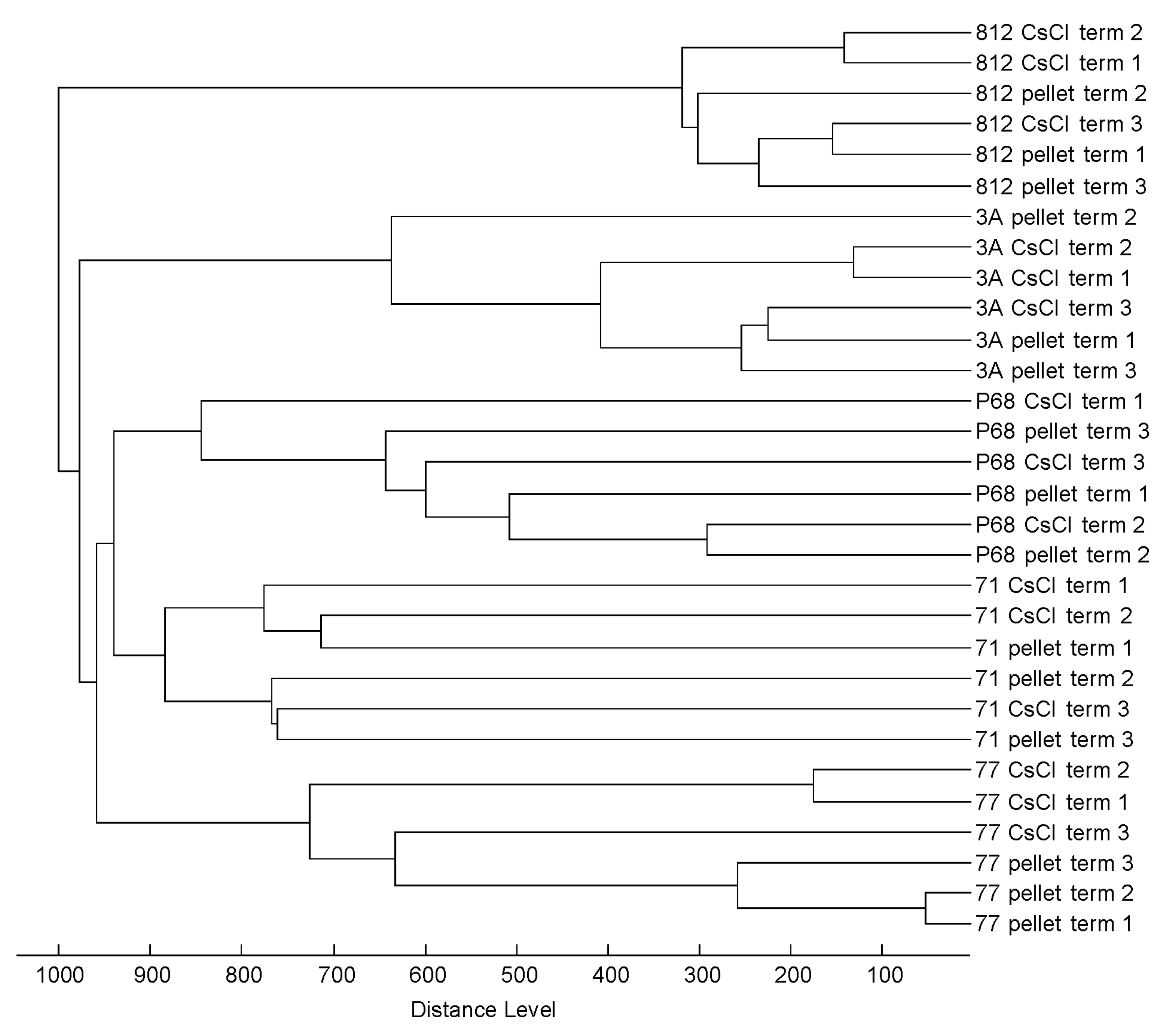
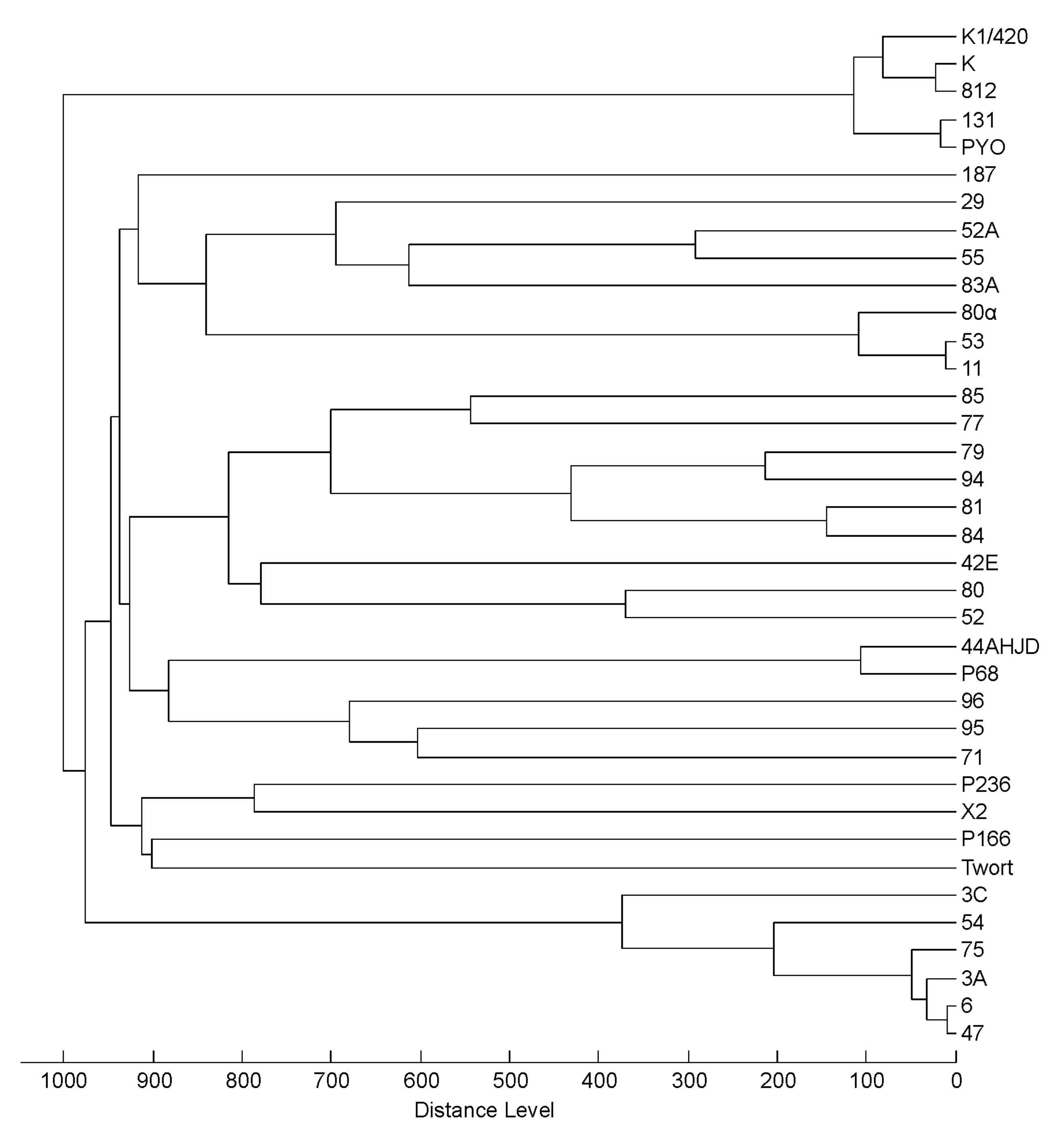
3.3. Profiling of 37 Phages by MALDI-TOF MS
3.4. Practicability of the Method
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Melo, L.D.R.; Oliveira, H.; Santos, S.B.; Sillankorva, S.; Azeredo, J. Phages against infectious diseases. In Bioprospecting; Topics in Biodiversity and Conservation; Springer: Cham, Switzerland, 2017; Volume 16, pp. 269–294. ISBN 978-3-319-47933-0. [Google Scholar]
- Sybesma, P.; Pirnay, J.-P. Expert round table on acceptance and re-implementation of bacteriophage therapy Silk route to the acceptance and re-implementation of bacteriophage therapy. Biotechnol. J. 2016, 11, 595–600. [Google Scholar] [CrossRef]
- Bárdy, P.; Pantůček, R.; Benešík, M.; Doškař, J. Genetically modified bacteriophages in applied microbiology. J. Appl. Microbiol. 2016, 121, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W. Phage classification and characterization. Methods Mol. Biol. 2009, 501, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, B.; Binetti, A.G.; Martín, M.C.; Fernández, M.; Magadán, A.H.; Alvarez, M.A. Multiplex PCR for the detection and identification of dairy bacteriophages in milk. Food Microbiol. 2007, 24, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Kahánková, J.; Pantůček, R.; Goerke, C.; Růžičková, V.; Holochová, P.; Doškař, J. Multilocus PCR typing strategy for differentiation of Staphylococcus aureus siphoviruses reflecting their modular genome structure. Environ. Microbiol. 2010, 12, 2527–2538. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Hendrix, R.W. Phage genomics: Small is beautiful. Cell 2002, 108, 13–16. [Google Scholar] [CrossRef]
- Deghorain, M.; Van Melderen, L. The staphylococci phages family: An overview. Viruses 2012, 4, 3316–3335. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.; Mašlaňová, I.; Indráková, A.; Šiborová, M.; Mikulášek, K.; Bendíčková, K.; Plevka, P.; Vrbovská, V.; Zdráhal, Z.; Doškař, J.; et al. Staphylococcus sciuri bacteriophages double-convert for staphylokinase and phospholipase, mediate interspecies plasmid transduction and package mecA gene. Sci. Rep. 2017, 7, 46319. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Wolz, C. Phages of Staphylococcus aureus and their impact on host evolution. Infect. Genet. Evol. 2014, 21, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Goerke, C.; Pantůček, R.; Holtfreter, S.; Schulte, B.; Zink, M.; Grumann, D.; Bröker, B.M.; Doškař, J.; Wolz, C. Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 2009, 191, 3462–3468. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Haaber, J.; Leisner, J.J.; Cohn, M.T.; Catalan-Moreno, A.; Nielsen, J.B.; Westh, H.; Penadés, J.R.; Ingmer, H. Bacterial viruses enable their host to acquire antibiotic resistance genes from neighbouring cells. Nat. Commun. 2016, 7, 13333. [Google Scholar] [CrossRef] [PubMed]
- Mašlaňová, I.; Doškař, J.; Varga, M.; Kuntová, L.; Mužík, J.; Malúšková, D.; Růžičková, V.; Pantůček, R. Bacteriophages of Staphylococcus aureus efficiently package various bacterial genes and mobile genetic elements including SCCmec with different frequencies. Environ. Microbiol. Rep. 2013, 5, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Mašlaňová, I.; Stříbná, S.; Doškař, J.; Pantůček, R. Efficient plasmid transduction to Staphylococcus aureus strains insensitive to the lytic action of transducing phage. FEMS Microbiol. Lett. 2016, 363, fnw211. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 2016, 161, 2921–2949. [Google Scholar] [CrossRef] [PubMed]
- Doškař, J.; Pallová, P.; Pantůček, R.; Rosypal, S.; Růžičková, V.; Pantůčková, P.; Kailerová, J.; Klepárník, K.; Malá, Z.; Boček, P. Genomic relatedness of phages of the International Typing Set and detection of serogroup A, B and F prophages in lysogenic strains. Can. J. Microbiol. 2000, 46, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Guo, X.; Dong, K.; Zhang, Y.; Li, Q.; Zhu, Y.; Zeng, L.; Tang, R.; Li, L. Safety assessment of Staphylococcus phages of the family Myoviridae based on complete genome sequences. Sci. Rep. 2017, 7, 41259. [Google Scholar] [CrossRef] [PubMed]
- Bizzini, A.; Greub, G. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry, a revolution in clinical microbial identification. Clin. Microbiol. Infect. 2010, 16, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Fenselau, C.; Demirev, P.A. Characterization of intact microorganisms by MALDI mass spectrometry. Mass Spectrom. Rev. 2001, 20, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.J.; Falk, B.; Fenselau, C.; Jackman, J.; Ezzell, J. Viral characterization by direct analysis of capsid proteins. Anal. Chem. 1998, 70, 3863–3867. [Google Scholar] [CrossRef] [PubMed]
- Madonna, A.J.; Van Cuyk, S.; Voorhees, K.J. Detection of Escherichia coli using immunomagnetic separation and bacteriophage amplification coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.C.; Voorhees, K.J. Simultaneous detection of two bacterial pathogens using bacteriophage amplification coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2757–2761. [Google Scholar] [CrossRef] [PubMed]
- McAlpin, C.R.; Cox, C.R.; Matyi, S.A.; Voorhees, K.J. Enhanced matrix-assisted laser desorption/ionization time-of-flight mass spectrometric analysis of bacteriophage major capsid proteins with β-mercaptoethanol pretreatment. Rapid Commun. Mass Spectrom. 2010, 24, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Bourdin, G.; Schmitt, B.; Guy, L.M.; Germond, J.-E.; Zuber, S.; Michot, L.; Reuteler, G.; Brüssow, H. Amplification and purification of T4-like Escherichia coli phages for phage therapy: From laboratory to pilot scale. Appl. Environ. Microbiol. 2014, 80, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Borecká, P.; Rosypal, S.; Pantůček, R.; Doškař, J. Localization of prophages of serological group B and F on restriction fragments defined in the restriction map of Staphylococcus aureus NCTC 8325. FEMS Microbiol. Lett. 1996, 143, 203–210. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, X.; Gerlach, D.; Du, X.; Larsen, J.; Stegger, M.; Kühner, P.; Peschel, A.; Xia, G.; Winstel, V. An accessory wall teichoic acid glycosyltransferase protects Staphylococcus aureus from the lytic activity of Podoviridae. Sci. Rep. 2015, 5, 17219. [Google Scholar] [CrossRef] [PubMed]
- Botka, T.; Růžičková, V.; Konečná, H.; Pantůček, R.; Rychlík, I.; Zdráhal, Z.; Petráš, P.; Doškař, J. Complete genome analysis of two new bacteriophages isolated from impetigo strains of Staphylococcus aureus. Virus Genes 2015, 51, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Pantůček, R.; Rosypalová, A.; Doškař, J.; Kailerová, J.; Růžičková, V.; Borecká, P.; Snopková, Š.; Horváth, R.; Götz, F.; Rosypal, S. The polyvalent staphylococcal phage φ812: Its host-range mutants and related phages. Virology 1998, 246, 241–252. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nováček, J.; Šiborová, M.; Benešík, M.; Pantůček, R.; Doškař, J.; Plevka, P. Structure and genome release of Twort-like Myoviridae phage with a double-layered baseplate. Proc. Natl. Acad. Sci. USA 2016, 113, 9351–9356. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, Y. Isolation and characterization of restriction negative mutants of Staphylococcus aureus. Jikeikai Med. J. 1985, 32, 415–421. [Google Scholar]
- Novick, R. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 1967, 33, 155–166. [Google Scholar] [CrossRef]
- Moša, M.; Boštík, J.; Pantůček, R.; Doškař, J. Medicament in the Form of Anti-Staphylococcus Phage Lysate, Process of Its Preparation and Use. Patent Application CZ201200668-A3, 27 September 2012. [Google Scholar]
- Kramberger, P.; Honour, R.C.; Herman, R.E.; Smrekar, F.; Peterka, M. Purification of the Staphylococcus aureus bacteriophages VDX-10 on methacrylate monoliths. J. Virol. Methods 2010, 166, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Maniatis, T.; Fritsch, E.F. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1987; ISBN 978-0-87969-309-1. [Google Scholar]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Frottin, F.; Martinez, A.; Peynot, P.; Mitra, S.; Holz, R.C.; Giglione, C.; Meinnel, T. The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteom. 2006, 5, 2336–2349. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Traverso, J.A.; Valot, B.; Ferro, M.; Espagne, C.; Ephritikhine, G.; Zivy, M.; Giglione, C.; Meinnel, T. Extent of N-terminal modifications in cytosolic proteins from eukaryotes. Proteomics 2008, 8, 2809–2831. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Humana Press: New York, NY, USA, 2005; pp. 571–607. ISBN 978-1-59259-890-8. [Google Scholar]
- Bonilla, N.; Rojas, M.I.; Netto Flores Cruz, G.; Hung, S.-H.; Rohwer, F.; Barr, J.J. Phage on tap—A quick and efficient protocol for the preparation of bacteriophage laboratory stocks. PeerJ 2016, 4, e2261. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Lehman, S.M.; Vandersteegen, K.; Vandenheuvel, D.; Philippe, D.L.; Cornelissen, A.; Clokie, M.R.J.; Garría, A.J.; De Proft, M.; Maes, M.; et al. CIM® monolithic anion-exchange chromatography as a useful alternative to CsCl gradient purification of bacteriophage particles. Virology 2012, 434, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Valentine, N.; Wunschel, S.; Wunschel, D.; Petersen, C.; Wahl, K. Effect of culture conditions on microorganism identification by matrix-assisted laser desorption ionization mass spectrometry. Appl. Environ. Microbiol. 2005, 71, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [PubMed]
- Salmond, G.P.C.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Pirnay, J.-P.; Merabishvili, M.; Raemdonck, H.V.; Vos, D.D.; Verbeken, G. Bacteriophage production in compliance with regulatory requirements. Methods Mol. Biol. 2018, 1693, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.R.; Rees, J.C.; Voorhees, K.J. Modeling bacteriophage amplification as a predictive tool for optimized MALDI-TOF MS-based bacterial detection. J. Mass Spectrom. 2012, 47, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Pierce, C.L.; Rees, J.C.; Fernández, F.M.; Barr, J.R. Viable Staphylococcus aureus quantitation using 15N metabolically labeled bacteriophage amplification coupled with a multiple reaction monitoring proteomic workflow. Mol. Cell. Proteom. 2012, 11, M111.012849. [Google Scholar] [CrossRef] [PubMed]
- Swatkoski, S.; Russell, S.; Edwards, N.; Fenselau, C. Analysis of a model virus using residue-specific chemical cleavage and MALDI-TOF mass spectrometry. Anal. Chem. 2007, 79, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.C.; Barr, J.R. Detection of methicillin-resistant Staphylococcus aureus using phage amplification combined with matrix-assisted laser desorption/ionization mass spectrometry. Anal. Bioanal. Chem. 2017, 409, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Cargile, B.J.; McLuckey, S.A.; Stephenson, J.L. Identification of bacteriophage MS2 coat protein from E. coli lysates via ion trap collisional activation of intact protein ions. Anal. Chem. 2001, 73, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Wick, C.H.; Elashvili, I.; Stanford, M.F.; McCubbin, P.E.; Deshpande, S.V.; Kuzmanovic, D.; Jabbour, R.E. Mass spectrometry and integrated virus detection system characterization of MS2 bacteriophage. Toxicol. Mech. Methods 2007, 17, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Serafim, V.; Pantoja, L.; Ring, C.J.; Shah, H.; Shah, A.J. Rapid identification of E. coli bacteriophages using mass spectrometry. J. Proteom. Enzymol. 2017, 6, 1000130. [Google Scholar] [CrossRef]
- Goerke, C.; Köller, J.; Wolz, C. Ciprofloxacin and trimethoprim cause phage induction and virulence modulation in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Selva, L.; Viana, D.; Regev-Yochay, G.; Trzcinski, K.; Corpa, J.M.; Lasa, Í.; Novick, R.P.; Penadés, J.R. Killing niche competitors by remote-control bacteriophage induction. Proc. Natl. Acad. Sci. USA 2009, 106, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Nielsen, L.N.; Hvitved, A.; Haaber, J.K.; Wirtz, C.; Andersen, P.S.; Larsen, J.; Wolz, C.; Ingmer, H. Commercial biocides induce transfer of prophage Φ13 from human strains of Staphylococcus aureus to livestock CC398. Front. Microbiol. 2017, 8, 2418. [Google Scholar] [CrossRef] [PubMed]
- Kahánková, J.; Španová, A.; Pantůček, R.; Horák, D.; Doškař, J.; Rittich, B. Extraction of PCR-ready DNA from Staphylococcus aureus bacteriophages using carboxyl functionalized magnetic nonporous microspheres. J. Chromatogr. B 2009, 877, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Czyz, A.; Los, M.; Wrobel, B.; Wegrzyn, G. Inhibition of spontaneous induction of lambdoid prophages in Escherichia coli cultures: Simple procedures with possible biotechnological applications. BMC Biotechnol. 2001, 1, 1. [Google Scholar] [CrossRef]
- O’Flaherty, S.; Ross, R.P.; Meaney, W.; Fitzgerald, G.F.; Elbreki, M.F.; Coffey, A. Potential of the polyvalent anti-Staphylococcus bacteriophage K for control of antibiotic-resistant staphylococci from hospitals. Appl. Environ. Microbiol. 2005, 71, 1836–1842. [Google Scholar] [CrossRef] [PubMed]
- Kvachadze, L.; Balarjishvili, N.; Meskhi, T.; Tevdoradze, E.; Skhirtladze, N.; Pataridze, T.; Adamia, R.; Topuria, T.; Kutter, E.; Rohde, C.; et al. Evaluation of lytic activity of staphylococcal bacteriophage Sb-1 against freshly isolated clinical pathogens. Microb. Biotechnol. 2011, 4, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Vandersteegen, K.; Mattheus, W.; Ceyssens, P.-J.; Bilocq, F.; De Vos, D.; Pirnay, J.-P.; Noben, J.-P.; Merabishvili, M.; Lipinska, U.; Hermans, K.; et al. Microbiological and molecular assessment of bacteriophage ISP for the control of Staphylococcus aureus. PLoS ONE 2011, 6, e24418. [Google Scholar] [CrossRef] [PubMed]
- Łobocka, M.; Hejnowicz, M.S.; Dąbrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dąbrowska, B.; Kwiatek, M.; Parasion, S.; et al. Genomics of staphylococcal Twort-like phages—Potential therapeutics of the post-antibiotic era. Adv. Virus Res. 2012, 83, 143–216. [Google Scholar] [CrossRef] [PubMed]
- Międzybrodzki, R.; Kłak, M.; Jończyk-Matysiak, E.; Bubak, B.; Wójcik, A.; Kaszowska, M.; Weber-Dąbrowska, B.; Łobocka, M.; Górski, A. Means to facilitate the overcoming of gastric juice barrier by a therapeutic staphylococcal bacteriophage A5/80. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Serogroup/Genus | Phage 1 | Propagation Strains (S. aureus) |
|---|---|---|---|
| Siphoviridae | A/Triavirus | 3A * | PS 3A, CCM 4890 |
| 3C * | PS 3C | ||
| 6 * | PS 6 | ||
| 42E * | PS 42E | ||
| 47 * | RN4220, PS 47, PS 47 [53+], PS 47 [77+] | ||
| 54 * | PS 54 | ||
| 75 * | PS 75 | ||
| 79 * | PS 52A | ||
| 81 * | PS 81 | ||
| 94 * | PS 94 | ||
| B/Phietavirus | 11 | CCM 4890 | |
| 29 * | RN4220 | ||
| 52 * | PS 52 | ||
| 52A * | PS 52A, RN4220 | ||
| 53 * | CCM 4890 | ||
| 55 * | RN4220 | ||
| 71 * | PS 71, CCM 4890 | ||
| 80 * | PS 80 | ||
| 80α | RN4220 | ||
| 83A * | PS 83A | ||
| 85 * | PS 85, RN4220 | ||
| 95 * | PS 95 | ||
| 96 * | PS 96, RN4220 | ||
| B166 | CCM 4890 | ||
| B236 | CCM 4890 | ||
| X2 | CCM 4890 | ||
| L/Phietavirus | 187 | PS 187 | |
| F/Biseptimavirus | 77 * | PS 77, CCM 4890 | |
| 84 * | PS 84 | ||
| Myoviridae | D/Kayvirus | 131 | CCM 8428 |
| 812 | CCM 4028 | ||
| K | RN4220 | ||
| K1/420 | CCM 8428 | ||
| PYO | CCM 8428 | ||
| D/Twortvirus | Twort | HER 1048 2 | |
| Podoviridae | G/P68virus | 44AHJD | RN4220 ΔtarM |
| P68 | RN4220 ΔtarM |
| Phage | Isolation Method | MALDI Matrix | One Term | Three Terms | |||
|---|---|---|---|---|---|---|---|
| 100% Peaks | 70–99% Peaks | Maximal Signal-to-Noise | 100% Peaks | 70–99% Peaks | |||
| 3A | CsCl gradient | FerA | 20 | 22 | 504 ± 141 | 12 | 18 |
| CsCl gradient | HCCA | 3 | 2 | 107 ± 41 | - | - | |
| FPLC | FerA | 4 | 5 | 34 ± 11 | - | - | |
| Ultrafiltration | FerA | 1 | 8 | 16 ± 4 | - | - | |
| Pellet | FerA | 23 | 9 | 517 ± 219 | 8 | 8 | |
| Pellet | HCCA | 15 | 4 | 44 ± 4 | - | ||
| 71 | CsCl gradient | FerA | 8 | 13 | 397 ± 307 | 3 | 15 |
| CsCl gradient | HCCA | 2 | 2 | 5 ± 1 | - | - | |
| FPLC | FerA | 5 | 3 | 157 ± 72 | - | - | |
| Ultrafiltration | FerA | 3 | 6 | 17 ± 1 | - | - | |
| Pellet | FerA | 11 | 11 | 557 ± 108 | 9 | 4 | |
| Pellet | HCCA | 14 | 9 | 10 ± 1 | - | - | |
| 77 | CsCl gradient | FerA | 10 | 21 | 591 ± 96 | 6 | 20 |
| CsCl gradient | HCCA | 11 | 3 | 64 ± 11 | - | - | |
| FPLC | FerA | 0 | 0 | N/A | - | - | |
| Ultrafiltration | FerA | 6 | 3 | 19 ± 7 | - | - | |
| Pellet | FerA | 12 | 9 | 247 ± 36 | 8 | 11 | |
| Pellet | HCCA | 9 | 6 | 15 ± 7 | - | - | |
| K1/420 | CsCl gradient | FerA | 41 | 13 | 398 ± 122 | 21 | 12 |
| CsCl gradient | HCCA | 17 | 3 | 429 ± 30 | - | - | |
| FPLC | FerA | 0 | 0 | N/A | - | - | |
| Ultrafiltration | FerA | 0 | 0 | N/A | - | - | |
| Pellet | FerA | 17 | 7 | 125 ± 23 | 8 | 7 | |
| Pellet | HCCA | 5 | 5 | 29 ± 3 | - | - | |
| P68 | CsCl gradient | FerA | 13 | 14 | 452 ± 122 | 7 | 9 |
| CsCl gradient | HCCA | 4 | 5 | 281 ± 18 | - | - | |
| FPLC | FerA | 19 | 11 | 227 ± 23 | - | - | |
| Ultrafiltration | FerA | 5 | 1 | 41 ± 15 | - | - | |
| Pellet | FerA | 9 | 9 | 248 ± 19 | 5 | 4 | |
| Pellet | HCCA | 4 | 4 | 399 ± 166 | - | - | |
| Phage | NCBI Genome Accession No. | Protein Function | NCBI Protein Accession no. | Mw Theoretical | Mw Experimental |
|---|---|---|---|---|---|
| 3A | NC_007053 | Major tail protein | YP_239944 | 23,335 | 23,336 |
| Ig-like domain | YP_239945 | 15,855 | 15,857 | ||
| Unknown | YP_239952 | 10,372 | 10,373 | ||
| 42E | NC_007052 | Major tail protein | YP_239866 | 23,295 | 23,290 |
| Ig-like domain | YP_239868 | 15,871 | 15,873 | ||
| 47 | NC_007054 | Major tail protein | YP_240012 | 23,295 | 23,298 |
| Ig-like domain | YP_240013 | 15,871 | 15,875 | ||
| 11 | NC_004615 | Major tail protein | NP_803292 | 21,382 | 21,376 |
| Head-tail connector protein | NP_803289 | 12,660 | 12,658 | ||
| 80α | NC_009526 | Major tail protein | YP_001285367 | 21,395 | 21,406 |
| Unknown | YP_001285362 | 10,790 | 10,797 | ||
| 53 | NC_007049 | Major tail protein | YP_239653 | 21,395 | 21,405 |
| Head-tail connector protein | YP_239650 | 12,660 | 12,667 | ||
| 55 | NC_007060 | Major capsid protein | YP_240459 | 29,487 | 29,497 |
| 71 | NC_007059 | Head-tail connector protein | YP_240387 | 11,759 | 11,758 |
| Unknown | YP_240388 | 12,857 | 12,857 | ||
| B166 | NC_028859 | Major tail protein | AKC04659 | 20,406 | 20,413 |
| B236 | NC_028915 | Major capsid protein | YP_009209168 | 29,489 | 29,493 |
| 187 | NC_007047 | Major capsid protein | YP_239493 | 32,997 | 32,987 |
| 77 | NC_005356 | Major tail protein | NP_958612 | 23,730 | 23,731 |
| Unknown | NP_958619 | 10,805 | 10,801 | ||
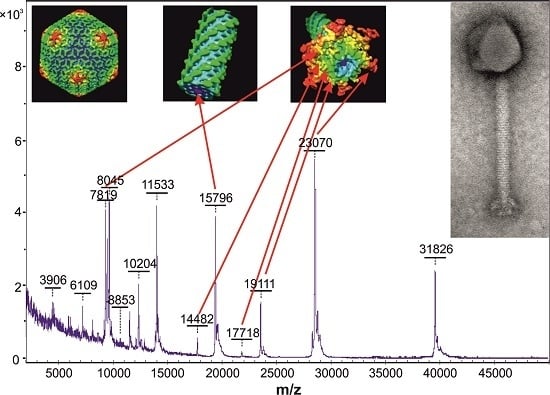
| K1/420 | KJ206563 | Tail tube protein | AHY26502 | 15,794 | 15,796 |
| Baseplate protein | AHY26518 | 19,109 | 19,111 | ||
| Putative baseplate component | AHY26523 | 14,480 | 14,482 | ||
| Ig-like protein | AHY26552 | 23,069 | 23,070 | ||
| Tail morphogenetic protein | AHY26553 | 17,718 | 17,718 | ||
| Major tail protein | AHY26554 | 7818 | 7819 | ||
| 812 | KJ206559 | Tail tube protein | * | 15,794 | 15,800 |
| Baseplate protein | AHY25649 | 19,109 | 19,114 | ||
| Putative baseplate component | AHY25654 | 14,480 | 14,479 | ||
| Ig-like protein | AHY25683 | 23,069 | 23,073 | ||
| Tail morphogenetic protein | * | 17,718 | 17,719 | ||
| Major tail protein | AHY25685 | 7818 | 7817 | ||
| 131 | RAST annotations were used | Tail tube protein | * | 15,794 | 15,792 |
| Baseplate protein | * | 19,109 | 19,109 | ||
| Putative baseplate component | * | 14,480 | 14,484 | ||
| Ig-like protein | * | 23,069 | 23,069 | ||
| Tail morphogenetic protein | * | 17,718 | 17,719 | ||
| K | NC_005880.2 | Tail tube protein | YP_009041323 | 15,794 | 15,802 |
| Baseplate protein | YP_009041339 | 19,109 | 19,104 | ||
| Putative baseplate component | YP_009041343 | 14,480 | 14,488 | ||
| Ig-like protein | YP_009041372 | 23,069 | 23,071 | ||
| Tail morphogenetic protein | YP_009041373 | 17,718 | 17,711 | ||
| Twort | NC_007021 | Structural protein | YP_238577 | 18,759 | 18,763 |
| Unknown | YP_238687 | 3923.5 | 3920 | ||
| Unknown | YP_238618 | 7638 | 7639 | ||
| P68 | NC_004679 | Major capsid protein | NP_817336 | 46,769 | 46,764 |
| Unknown | NP_817338 | 15,112 | 15,104 | ||
| Unknown | NP_817337 | 6916 | 6915 | ||
| 44AHJD | NC_004678 | Major capsid protein | NP_817314 | 46,769 | 46,770 |
| Unknown | NP_817316 | 15,112 | 15,110 | ||
| Unknown | NP_817315 | 6916 | 6917 |
| NCBI Accession No. | Protein Name | Predicted Protein Function | Mw | No. of Matched Peptides | Mascot Score | Sequence Coverage |
|---|---|---|---|---|---|---|
| YP_240967 | ORF189 | Major tail protein | 7818 | 8 | 1276 | 96% |
| YP_240933 | ORF117 | Unknown | 14,480 | 5 | 291 | 51% |
| YP_007112937 | F867_gp192 | Ig-like protein | 23,069 | 3 | 188 | 26% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štveráková, D.; Šedo, O.; Benešík, M.; Zdráhal, Z.; Doškař, J.; Pantůček, R. Rapid Identification of Intact Staphylococcal Bacteriophages Using Matrix-Assisted Laser Desorption Ionization-Time-of-Flight Mass Spectrometry. Viruses 2018, 10, 176. https://doi.org/10.3390/v10040176
Štveráková D, Šedo O, Benešík M, Zdráhal Z, Doškař J, Pantůček R. Rapid Identification of Intact Staphylococcal Bacteriophages Using Matrix-Assisted Laser Desorption Ionization-Time-of-Flight Mass Spectrometry. Viruses. 2018; 10(4):176. https://doi.org/10.3390/v10040176
Chicago/Turabian StyleŠtveráková, Dana, Ondrej Šedo, Martin Benešík, Zbyněk Zdráhal, Jiří Doškař, and Roman Pantůček. 2018. "Rapid Identification of Intact Staphylococcal Bacteriophages Using Matrix-Assisted Laser Desorption Ionization-Time-of-Flight Mass Spectrometry" Viruses 10, no. 4: 176. https://doi.org/10.3390/v10040176
APA StyleŠtveráková, D., Šedo, O., Benešík, M., Zdráhal, Z., Doškař, J., & Pantůček, R. (2018). Rapid Identification of Intact Staphylococcal Bacteriophages Using Matrix-Assisted Laser Desorption Ionization-Time-of-Flight Mass Spectrometry. Viruses, 10(4), 176. https://doi.org/10.3390/v10040176