Parechovirus A Detection by a Comprehensive Approach in a Clinical Laboratory


 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Virus and Cell Lines, Chemicals
2.3. Antibodies
2.4. Immunofluorescence Assay
2.5. Immunoblotting Analysis
2.6. HPeV3 VP0 Expression Vector
2.7. Laboratory Diagnosis
3. Results
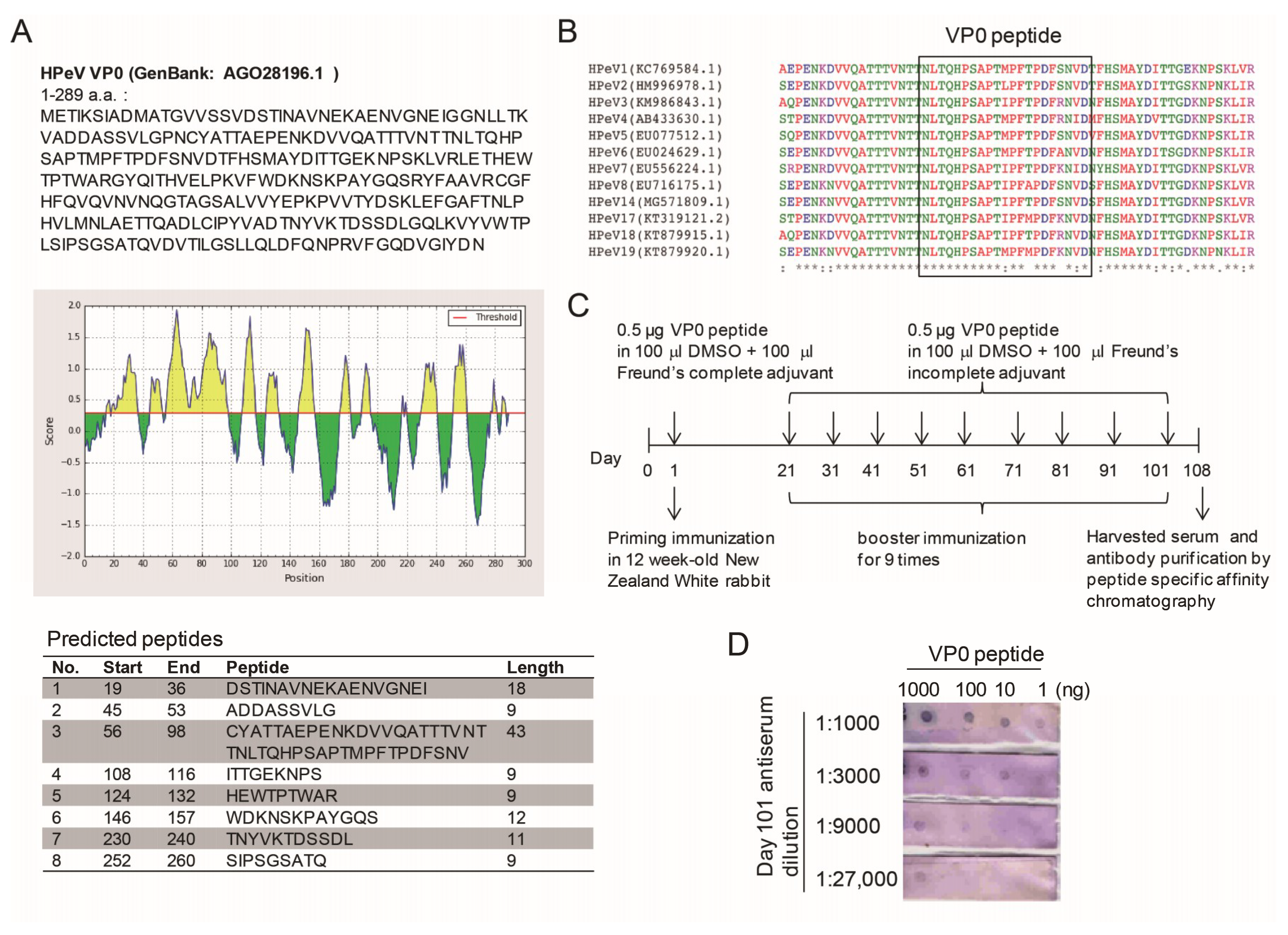
3.1. VP0 Antigeneticity/Epitope Analysis and Antibody Production
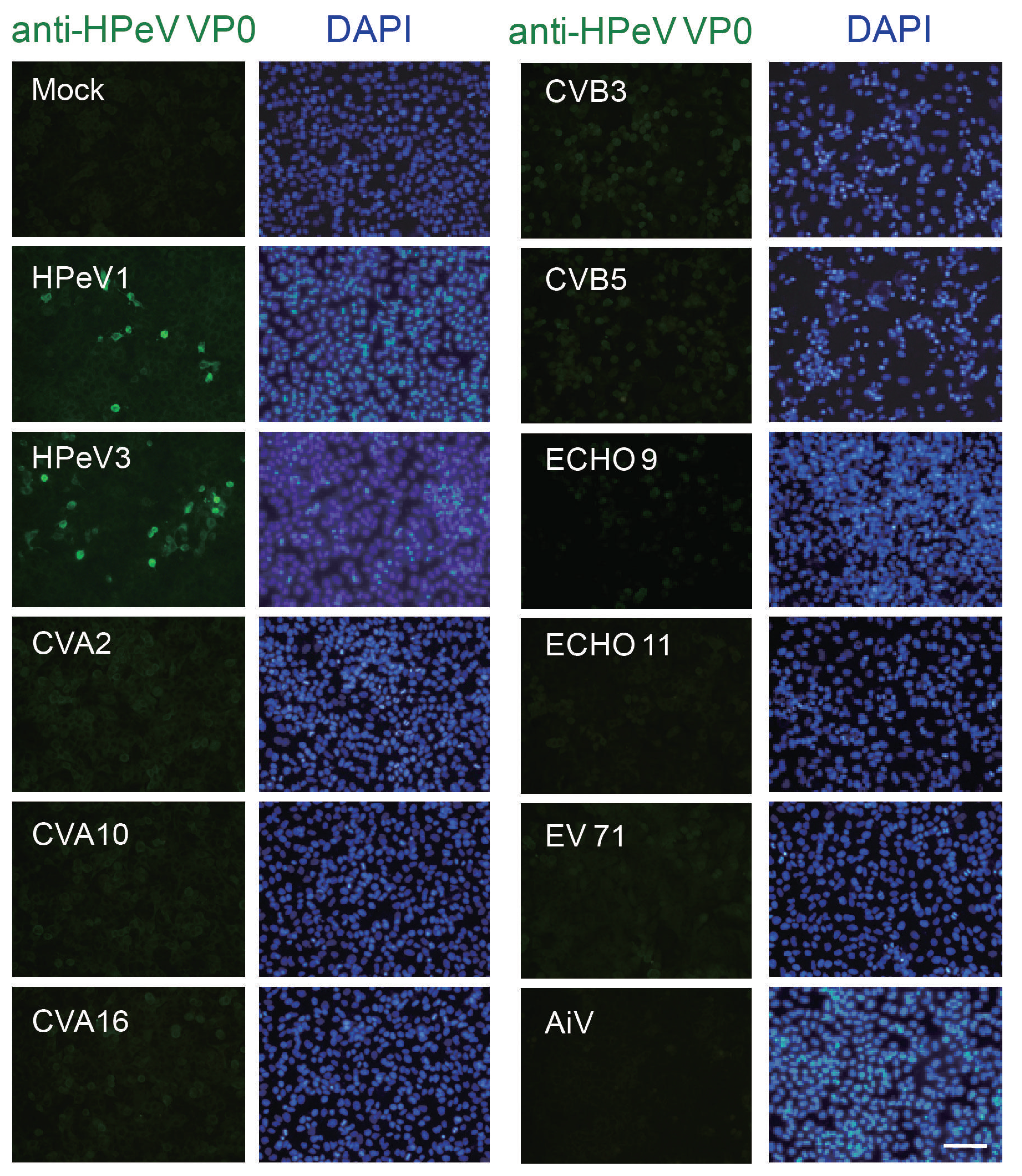
3.2. Antibody Specificity Analysis by IFA and Immunoblotting
3.3. HPeV Detection by RT-PCR and IFA
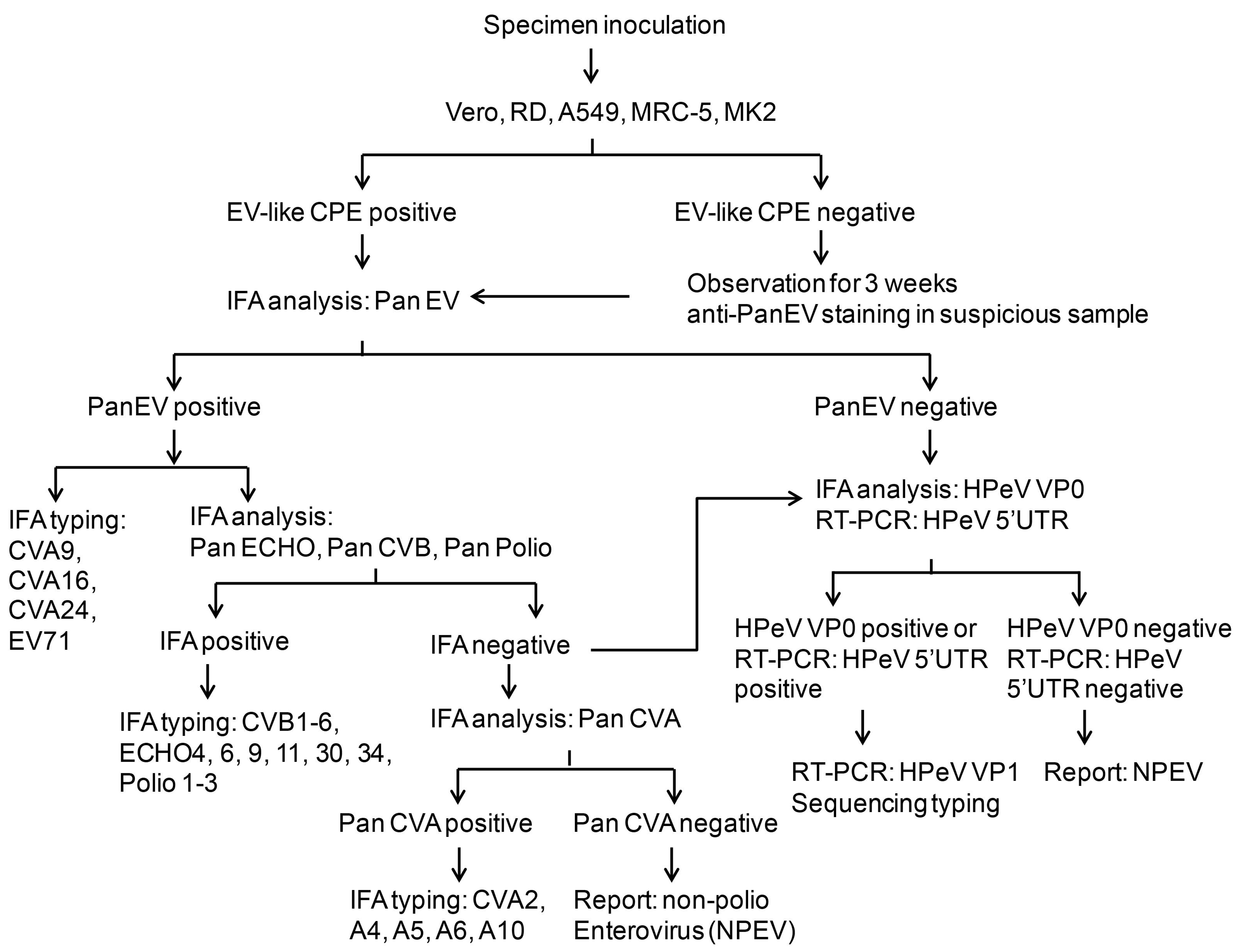
3.4. HPeV Diagnosis Protocol
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nateri, A.S.; Hughes, P.J.; Stanway, G. In vivo and in vitro identification of structural and sequence elements of the human parechovirus 5′ untranslated region required for internal initiation. J. Virol. 2000, 74, 6269–6277. [Google Scholar] [CrossRef] [PubMed]
- Stanway, G.; Hyypia, T. Parechoviruses. J. Virol. 1999, 73, 5249–5254. [Google Scholar]
- Benschop, K.; Molenkamp, R.; van der Ham, A.; Wolthers, K.; Beld, M. Rapid detection of human parechoviruses in clinical samples by real-time PCR. J. Clin. Virol. 2008, 41, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Tapia, G.; Cinek, O.; Witso, E.; Kulich, M.; Rasmussen, T.; Grinde, B.; Ronningen, K.S. Longitudinal observation of parechovirus in stool samples from Norwegian infants. J. Med. Virol. 2008, 80, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Khetsuriani, N.; Lamonte-Fowlkes, A.; Oberst, S.; Pallansch, M.A. Enterovirus surveillance--United States, 1970-2005. MMWR Surveill. Summ. 2006, 55, 1–20. [Google Scholar] [PubMed]
- Van der Sanden, S.; de Bruin, E.; Vennema, H.; Swanink, C.; Koopmans, M.; van der Avoort, H. Prevalence of human parechovirus in the Netherlands in 2000 to 2007. J. Clin. Microbiol. 2008, 46, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Abed, Y.; Boivin, G. Human parechovirus types 1, 2 and 3 infections in Canada. Emerg. Infect. Dis. 2006, 12, 969–975. [Google Scholar] [CrossRef]
- Benschop, K.S.; Schinkel, J.; Minnaar, R.P.; Pajkrt, D.; Spanjerberg, L.; Kraakman, H.C.; Berkhout, B.; Zaaijer, H.L.; Beld, M.G.; Wolthers, K.C. Human parechovirus infections in Dutch children and the association between serotype and disease severity. Clin. Infect. Dis. 2006, 42, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Harvala, H.; Simmonds, P. Human parechoviruses: Biology, epidemiology and clinical significance. J. Clin. Virol. 2009, 45, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Britton, P.N.; Khandaker, G.; Khatami, A.; Teutsch, S.; Francis, S.; McMullan, B.J.; Jones, C.A. High prevalence of developmental concern amongst infants at 12 months following hospitalised parechovirus infection. J. Paediatr. Child Health 2018, 54, 289–295. [Google Scholar] [CrossRef]
- Berkovich, S.; Pangan, J. Recoveries of virus from premature infants during outbreaks of respiratory disease: The relation of ECHO virus type 22 to disease of the upper and lower respiratory tract in the premature infant. Bull N. Y. Acad. Med. 1968, 44, 377–387. [Google Scholar] [PubMed]
- Birenbaum, E.; Handsher, R.; Kuint, J.; Dagan, R.; Raichman, B.; Mendelson, E.; Linder, N. Echovirus type 22 outbreak associated with gastro-intestinal disease in a neonatal intensive care unit. Am. J. Perinatol. 1997, 14, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Strenger, V.; Diedrich, S.; Boettcher, S.; Richter, S.; Maritschnegg, P.; Gangl, D.; Fuchs, S.; Grangl, G.; Resch, B.; Urlesberger, B. Nosocomial Outbreak of Parechovirus 3 Infection among Newborns, Austria, 2014. Emerg. Infect. Dis. 2016, 22, 1631–1634. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Chen, Y.S.; Chen, B.C.; Huang, T.S.; Chang, T.H. Human Parechovirus Infection in Children in Taiwan: A Retrospective, Single-Hospital Study. Jpn. J. Infect. Dis. 2018, 71, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.C.; Cheng, M.F.; Huang, T.S.; Liu, Y.C.; Tang, C.W.; Chen, C.S.; Chen, Y.S. Detection and identification of human parechoviruses from clinical specimens. Diagn. Microbiol. Infect. Dis. 2009, 65, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yamashita, T.; Tsuzuki, H.; Takeda, N.; Sakae, K. Isolation and identification of a novel human parechovirus. J. Gen. Virol. 2004, 85, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Wildenbeest, J.G.; Benschop, K.S.; Minnaar, R.P.; Bouma-de Jongh, S.; Wolthers, K.C.; Pajkrt, D. Clinical relevance of positive human parechovirus type 1 and 3 PCR in stool samples. Clin. Microbiol. Infect. 2014, 20, O640–O647. [Google Scholar] [CrossRef]
- Nix, W.A.; Maher, K.; Pallansch, M.A.; Oberste, M.S. Parechovirus typing in clinical specimens by nested or semi-nested PCR coupled with sequencing. J. Clin. Virol. 2010, 48, 202–207. [Google Scholar] [CrossRef]
- Hematian, A.; Sadeghifard, N.; Mohebi, R.; Taherikalani, M.; Nasrolahi, A.; Amraei, M.; Ghafourian, S. Traditional and Modern Cell Culture in Virus Diagnosis. Osong. Public Health Res.Perspect. 2016, 7, 77–82. [Google Scholar] [CrossRef]
- Hodinka, R.L. Point: Is the era of viral culture over in the clinical microbiology laboratory? J. Clin. Microbiol. 2013, 51, 2–4. [Google Scholar] [CrossRef]
- Wolf, J.M.; Gregianini, T.S.; Seadi, C.M.; Tumioto, G.L.; Dambros, B.P.; Lehmann, F.K.; Carli, S.D.; Ikuta, N.; Lunge, V.R. Performance of direct immunofluorescence assay for the detection of human metapneumovirus under clinical laboratory settings. Rev. Soc. Bras. Med. Trop. 2015, 48, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Abbott, W.M.; Damschroder, M.M.; Lowe, D.C. Current approaches to fine mapping of antigen-antibody interactions. Immunology 2014, 142, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Joki-Korpela, P.; Roivainen, M.; Lankinen, H.; Poyry, T.; Hyypia, T. Antigenic properties of human parechovirus 1. J. Gen. Virol. 2000, 81, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Abed, Y.; Wolf, D.; Dagan, R.; Boivin, G. Development of a serological assay based on a synthetic peptide selected from the VP0 capsid protein for detection of human parechoviruses. J. Clin. Microbiol. 2007, 45, 2037–2039. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Yang, C.S.; Chen, Y.S.; Chen, B.C.; Chiang, A.J.; Chang, Y.H.; Tsai, W.L.; Lin, Y.S.; Chao, D.; Chang, T.H. Genome and infection characteristics of human parechovirus type 1: The interplay between viral infection and type I interferon antiviral system. PLoS ONE 2015, 10, e0116158. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Kawakami, A.; Osaka, M.; Uematsu, S.; Akira, S.; Shimokado, K.; Sacks, F.M.; Yoshida, M. Apolipoprotein CIII induces monocyte chemoattractant protein-1 and interleukin 6 expression via Toll-like receptor 2 pathway in mouse adipocytes. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2242–2248. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Dilnessa, T.; Zeleke, H. Cell Culture, Cytopathic Effect and Immunofluorescence Diagnosis of Viral Infection. J. Microbiol. Modern Tech. 2017, 2, 102. [Google Scholar]
- Cobo, F. Application of molecular diagnostic techniques for viral testing. Open Virol. J. 2012, 6, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Westerhuis, B.M.; Koen, G.; Wildenbeest, J.G.; Pajkrt, D.; de Jong, M.D.; Benschop, K.S.; Wolthers, K.C. Specific cell tropism and neutralization of human parechovirus types 1 and 3: Implications for pathogenesis and therapy development. J. Gen. Virol. 2012, 93, 2363–2370. [Google Scholar] [CrossRef] [PubMed]
- Soria-Guerra, R.E.; Nieto-Gomez, R.; Govea-Alonso, D.O.; Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J.Biomed. Inform. 2015, 53, 405–414. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolates | Total | RT-PCR: 5′UTR | IFA: Anti-VP0 | RT-PCR: VP1 | ||||
|---|---|---|---|---|---|---|---|---|
| Pos. (n, %) | Neg. (n, %) | Pos. (n, %) | Neg. (n, %) | Pos. (n, %) | Neg. (n,%) | ND (n) | ||
| HPeV | 74 | 74 (100) | 0 | 74 (100) | 0 | 49 (75.4) # | 16 (24.6) # | 9 |
| HPeV negative control | 12 | 0 | 12 (100) | 0 | 12 (100) | 0 | 12 (100) | 0 |
| NPEV | 7 | 0 | 7 (100) | 0 | 0 | 0 | 7 (100) | 0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, B.-C.; Chang, J.-T.; Huang, T.-S.; Chen, J.-J.; Chen, Y.-S.; Jan, M.-W.; Chang, T.-H. Parechovirus A Detection by a Comprehensive Approach in a Clinical Laboratory. Viruses 2018, 10, 711. https://doi.org/10.3390/v10120711
Chen B-C, Chang J-T, Huang T-S, Chen J-J, Chen Y-S, Jan M-W, Chang T-H. Parechovirus A Detection by a Comprehensive Approach in a Clinical Laboratory. Viruses. 2018; 10(12):711. https://doi.org/10.3390/v10120711
Chicago/Turabian StyleChen, Bao-Chen, Jenn-Tzong Chang, Tsi-Shu Huang, Jih-Jung Chen, Yao-Shen Chen, Ming-Wei Jan, and Tsung-Hsien Chang. 2018. "Parechovirus A Detection by a Comprehensive Approach in a Clinical Laboratory" Viruses 10, no. 12: 711. https://doi.org/10.3390/v10120711
APA StyleChen, B.-C., Chang, J.-T., Huang, T.-S., Chen, J.-J., Chen, Y.-S., Jan, M.-W., & Chang, T.-H. (2018). Parechovirus A Detection by a Comprehensive Approach in a Clinical Laboratory. Viruses, 10(12), 711. https://doi.org/10.3390/v10120711