An Evolutionary Insight into Zika Virus Strains Isolated in the Latin American Region


Abstract
1. Introduction
2. Materials and Methods
2.1. Sequences
2.2. Recombination Analysis
2.3. Bayesian Coalescent Markov Chain Monte Carlo (MCMC) Analysis
2.4. Compositional Analyses
2.5. Prediction of Exposed Residues and Structural Regions of E and prM Proteins
3. Results
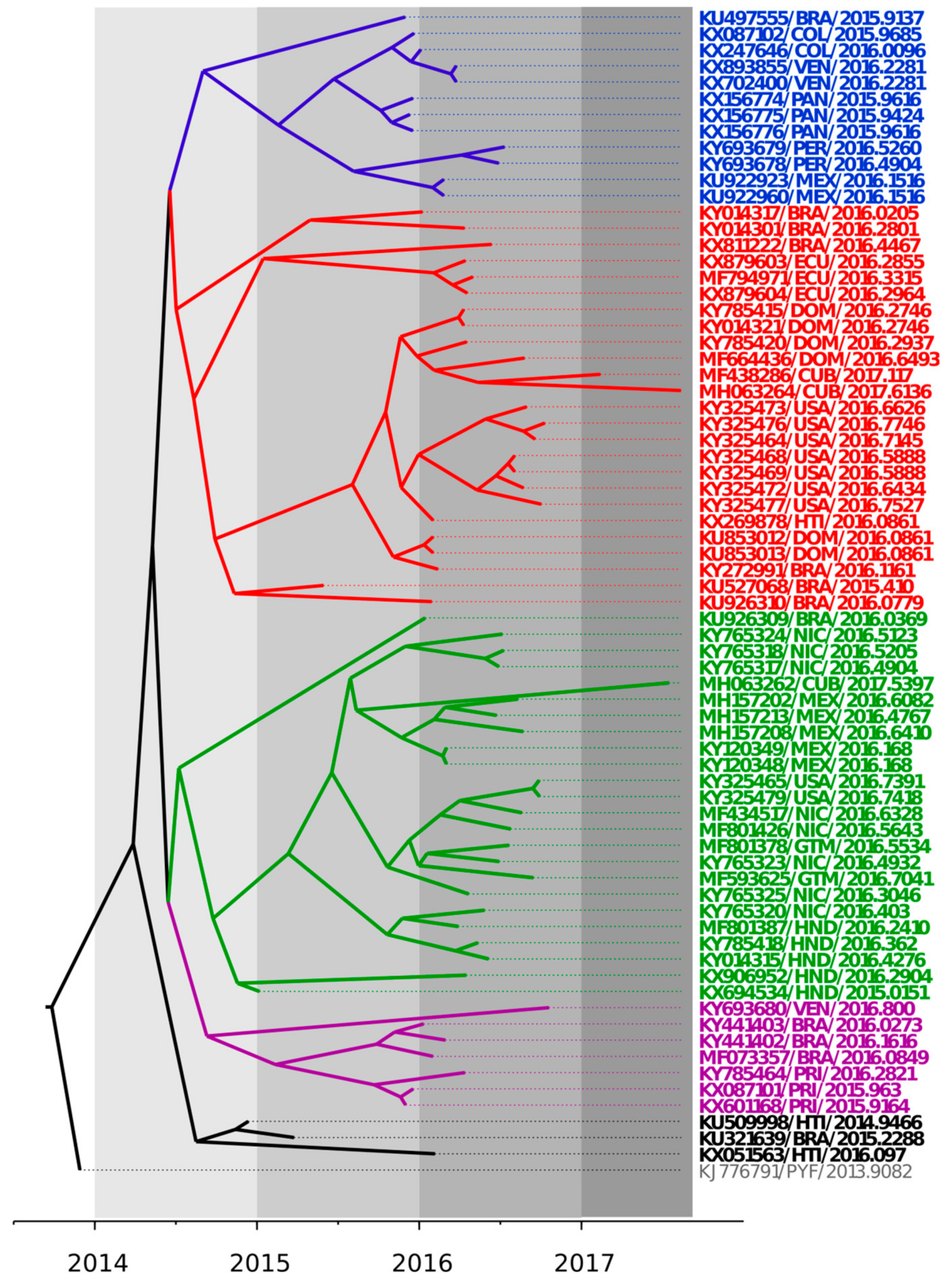
3.1. Bayesian Coalescent Analysis of ZIKV Strains Recently Isolated in the Latin American Region
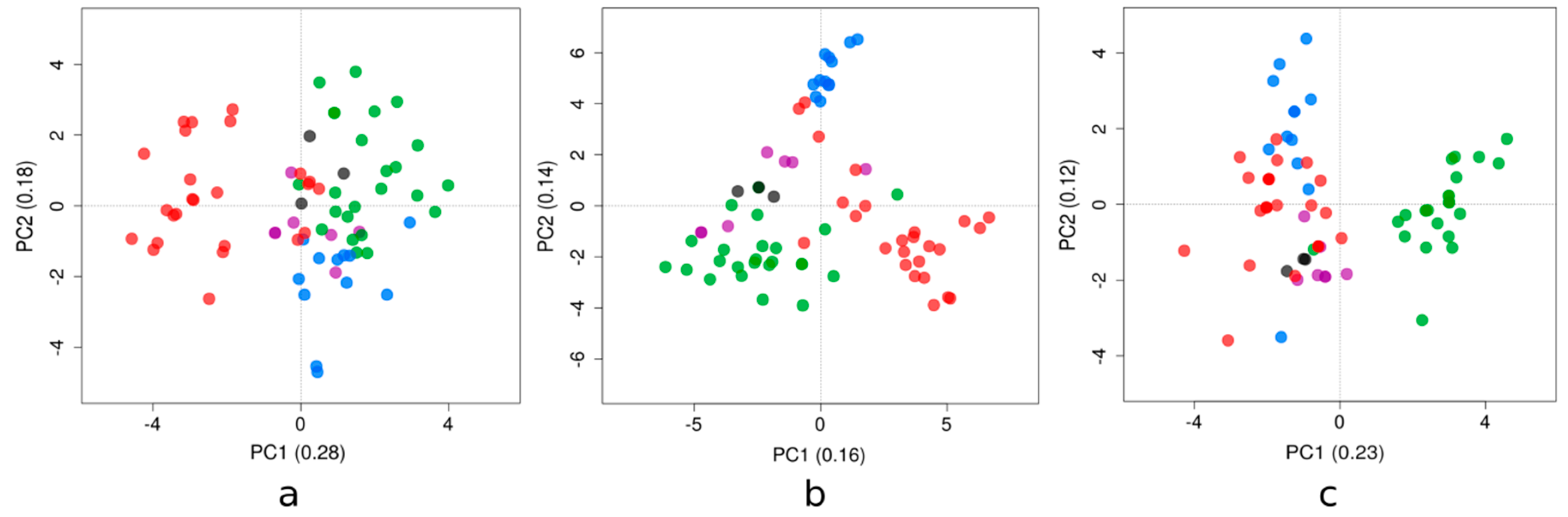
3.2. Trends in Compositional Properties Across ZIKV Strains Isolated in the Latin American Region
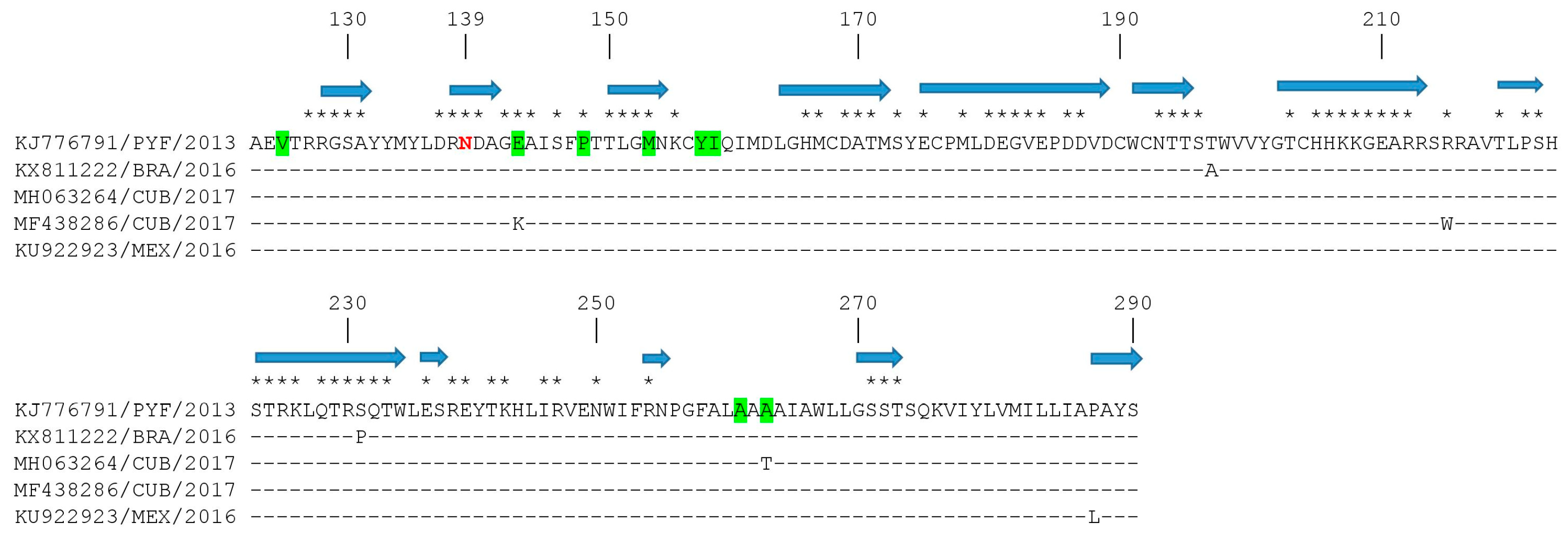
3.3. Mapping of Amino Acid Substitutions in the ZIKV E Protein
3.4. Mapping Amino Acid Substitutions in the ZIKV prM Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value a | HPD b | ESS c |
|---|---|---|---|
| Log likelihood | −21,802.85 | −21,852.01 to −21,744.02 | 3179.90 |
| Clock rate d | 1.21 × 10−3 | 7.55 × 10−4 to 1.66 × 10−3 | 4823.21 |
| tMRCA e All | 4.20 | 3.70 to 5.03 | 291.75 |
| 29/6/2013 | 3/10/2012 to 2/13/2014 | ||
| tMRCA e Latin American clade | 3.37 | 2.87 to 6.33 | |
| 3/27/2014 | 10/24/2013 to 11/13/2014 |
Appendix B
| Strain a | Amino Acid Position b | |||||||
|---|---|---|---|---|---|---|---|---|
| 23 | 51 | 68 | 260 | 330 | 335 | 369 | 443 | |
| KU321639/BRA/2015 | I | |||||||
| MH157202/MEX/2016 | T | |||||||
| MF801378/GMT/2016 | T | |||||||
| KU497555/BRA/2015 | T | |||||||
| KX087101/PRI/2015 | L | |||||||
| KU926310/BRA/2016 | A | |||||||
| KY014317/BRA/2016 | S | |||||||
| KX694534/HND/2015 | R | |||||||
References
- Fajardo, A.; Cristina, J.; Moreno, P. Emergence and Spreading Potential of Zika Virus. Front. Microbiol. 2016, 7, 1667. [Google Scholar] [CrossRef] [PubMed]
- Kuno, G.; Chang, G. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch. Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika Virus Outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Musso, D. Zika virus transmission from French Polynesia to Brazil. Emerg. Infect. Dis. 2015, 21, 1887. [Google Scholar] [CrossRef] [PubMed]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika virus outbreak, Bahia, Brazil. Emerg. Infect. Dis. 2015, 21, 1885–1886. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, M.; Fischer, M.; Staples, E. Zika Virus Spreads to New Areas—Region of the Americas, May 2015–January 2016. Morb. Mortal. Wkly. Rep. 2016, 65, 55–58. [Google Scholar] [CrossRef]
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martínez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N.; WHO Zika Causality Working Group. Zika Virus Infection as a Cause of Congenital Brain Abnormalities and Guillain-Barré Syndrome: Systematic Review. PLoS Med. 2017, 14, e1002203. [Google Scholar] [CrossRef]
- Faye, O.; Freire, C.C.; Iamarino, A.; Faye, O.; de Oliveira, J.V.; Diallo, M. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl. Trop. Dis. 2014, 8, 2636. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef]
- Zhu, Z.; Chan, J.F.; Tee, K.M.; Choi, G.K.; Lau, S.K.; Woo, P.C.; Tse, H.; Yuen, K.Y. Comparative genomic analysis of pre-epidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg. Microbes Infect. 2016, 5, e22. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, A.; Soñora, M.; Moreno, P.; Moratorio, G.; Cristina, J. Bayesian coalescent inference reveals high evolutionary rates and diversification of Zika virus populations. J. Med. Virol. 2016, 88, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Nambala, P.; Su, W.C. Role of Zika Virus prM Protein in Viral Pathogenicity and Use in Vaccine Development. Front. Microbiol. 2018, 9, 1797. [Google Scholar] [CrossRef]
- Goo, L.; DeMaso, C.R.; Pelc, R.S.; Ledgerwood, J.E.; Graham, B.S.; Kuhn, R.J.; Pierson, T.C. The Zika virus envelope protein glycan loop regulates virion antigenicity. Virology 2018, 515, 191–202. [Google Scholar] [CrossRef]
- Holmes, E.C. The Evolution and Emergence of RNA Viruses, 1st ed.; Oxford University Press: Oxford, UK, 2009; pp. 113–155. ISBN 978-0-19-921112-8. [Google Scholar]
- Domingo, E.; Perales, C. Quasispecies and viruses. Eur. Biophys. J. 2018, 47, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Kosakovski-Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Simón, D.; Fajardo, A.; Sóñora, M.; Delfraro, A.; Musto, H. Host influence in the genomic composition of flaviviruses: A multivariate approach. Biochem. Biophys. Res. Commun. 2017, 492, 572–578. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A Contributed Package to the R Project for Statistical Computing Devoted to Biological Sequences Retrieval and Analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations, 1st ed.; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Springer: New York, NY, USA, 2007; pp. 207–232. ISBN 978-3-540-35306-5. [Google Scholar]
- Larsen, J.E.P.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, e2. [Google Scholar] [CrossRef][Green Version]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Massad, E.; Burattini, M.N.; Khan, K.; Struchiner, C.J.; Coutinho, F.A.B.; Wilder-Smith, A. On the origin and timing of Zika virus introduction in Brazil. Epidemiol. Infect. 2017, 15, 1–10. [Google Scholar] [CrossRef]
- Aldunate, F.; Gámbaro, F.; Fajardo, A.; Soñora, M.; Cristina, J. Evidence of increasing diversification of Zika virus strains isolated in the American continent. J. Med. Virol. 2017, 89, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Stoletzki, N.; Eyre-Walker, A. Synonymous codon usage in Escherichia coli: Selection for translational accuracy. Mol. Biol. Evol. 2007, 24, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Costafreda, M.I.; Pérez-Rodriguez, F.J.; D’Andrea, L.; Guix, S.; Ribes, E.; Bosch, A.; Pintó, R.M. Hepatitis A virus adaptation to cellular shutoff is driven bydynamic adjustments of codon usage and results in the selection of populations with altered capsids. J. Virol. 2014, 88, 5029–5041. [Google Scholar] [CrossRef] [PubMed]
- Heinz, F.X.; Stiasny, K. The Antigenic Structure of Zika Virus and Its Relation to Other Flaviviruses: Implications for Infection and Immunoprophylaxis. Microbiol. Mol. Biol. Rev. 2017, 81, e00055-16. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Song, J.; Lu, X.; Deng, Y.Q.; Musyoki, A.M.; Cheng, H.; Zhang, Y.; Yuan, Y.; Song, H.; Haywood, J.; et al. Structures of the Zika Virus Envelope Protein and Its Complex with a Flavivirus Broadly Protective Antibody. Cell Host Microbe 2016, 19, 696–704. [Google Scholar] [CrossRef]
- Zhao, H.; Fernandez, E.; Dowd, K.A.; Speer, S.D.; Platt, D.J.; Gorman, M.J.; Govero, J.; Nelson, C.A.; Pierson, T.C.; Diamond, M.S.; et al. Structural Basis of Zika Virus-Specific Antibody Protection. Cell 2016, 166, 1016–1027. [Google Scholar] [CrossRef]
- Lin, H.H.; Yip, B.S.; Huang, L.M.; Wu, S.C. Zika virus structural biology and progress in vaccine development. Biotechnol. Adv. 2018, 36, 47–53. [Google Scholar] [CrossRef]
- Yuan, L.; Huang, X.Y.; Liu, Z.Y.; Zhang, F.; Zhu, X.L.; Yu, J.Y.; Ji, X.; Xu, Y.P.; Li, G.; Li, C.; et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef]
- Bos, S.; Viranaicken, W.; Turpin, J.; El-Kalamouni, C.; Roche, M.; Krejbich-Trotot, P.; Desprès, P.; Gadea, G. The structural proteins of epidemic and historical strains of Zika virus differ in their ability to initiate viral infection in human host cells. Virology 2018, 516, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Lim, E.X.; Zhang, S.; Fibriansah, G.; Ng, T.S.; Ooi, J.S.; Shi, J.; Lok, S.M. Structure of the thermally stable Zika virus. Nature 2016, 533, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Lambert, A.J.; Holodniy, M.; Saavedra, S.; Signor, L. Phylogeny of Zika virus in Western Hemisphere, 2015. Emerg. Infect. Dis. 2016, 22, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, A.; Dai, L.; Contreras, D.; Sinha, S.; Sun, R.; Arumugaswami, V. Comparative analysis of protein evolution in the genome of pre-epidemic and epidemic Zika virus. Infect. Genet. Evol. 2017, 51, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, S.; Zhang, B.; Wei, W. Analysis of Synonymous Codon Usage Bias of Zika Virus and Its Adaption to the Hosts. PLoS ONE 2016, 11, e0166260. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, e107. [Google Scholar] [CrossRef] [PubMed]
- Khrustalev, V.V.; Khrustaleva, T.A.; Sharma, N.; Giri, R. Mutational pressure in Zika virus: Local ADAR-editing areas associated with pauses in translation and replication. Front. Cell Infect. Microbiol. 2017, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Piontkivska, H.; Frederick, M.; Miyamoto, M.M.; Wayne, M.L. RNA editing by the host ADAR system affects the molecular evolution of the Zika virus. Ecol. Evol. 2017, 7, 4475–4485. [Google Scholar] [CrossRef]
- Martina, B.E.; Koraka, P.; van den Doel, P.; van Amerongen, G.; Rimmelzwaan, G.F.; Osterhaus, A.D. Immunization with West Nile virus envelope domain III protects mice against lethal infection with homologous and heterologous virus. Vaccine 2008, 26, 153–157. [Google Scholar] [CrossRef]
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef]
- Yoshii, K.; Igarashi, M.; Ichii, O.; Yokozawa, K.; Ito, K.; Kariwa, H.; Takashima, I. A conserved region in the prM protein is a critical determinant in the assembly of flavivirus particles. J. Gen. Virol. 2012, 93, 27–38. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Vaccine Pipeline Tracker. 2018. Available online: http://www.who.int/immunization/research/vaccine_pipeline_tracker_spreadsheet/en/ (accessed on 18 November 2018).
- Oliveira, E.R.A.; de Alencastro, R.B.; Horta, B.A.C. New insights into flavivirus biology: The influence of pH over interactions between prM and E. proteins. J. Comput. Aided Mol. Des. 2017, 31, 1009–1019. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simón, D.; Fajardo, A.; Moreno, P.; Moratorio, G.; Cristina, J. An Evolutionary Insight into Zika Virus Strains Isolated in the Latin American Region. Viruses 2018, 10, 698. https://doi.org/10.3390/v10120698
Simón D, Fajardo A, Moreno P, Moratorio G, Cristina J. An Evolutionary Insight into Zika Virus Strains Isolated in the Latin American Region. Viruses. 2018; 10(12):698. https://doi.org/10.3390/v10120698
Chicago/Turabian StyleSimón, Diego, Alvaro Fajardo, Pilar Moreno, Gonzalo Moratorio, and Juan Cristina. 2018. "An Evolutionary Insight into Zika Virus Strains Isolated in the Latin American Region" Viruses 10, no. 12: 698. https://doi.org/10.3390/v10120698
APA StyleSimón, D., Fajardo, A., Moreno, P., Moratorio, G., & Cristina, J. (2018). An Evolutionary Insight into Zika Virus Strains Isolated in the Latin American Region. Viruses, 10(12), 698. https://doi.org/10.3390/v10120698