Rhabdoviruses, Antiviral Defense, and SUMO Pathway

Abstract
1. Introduction
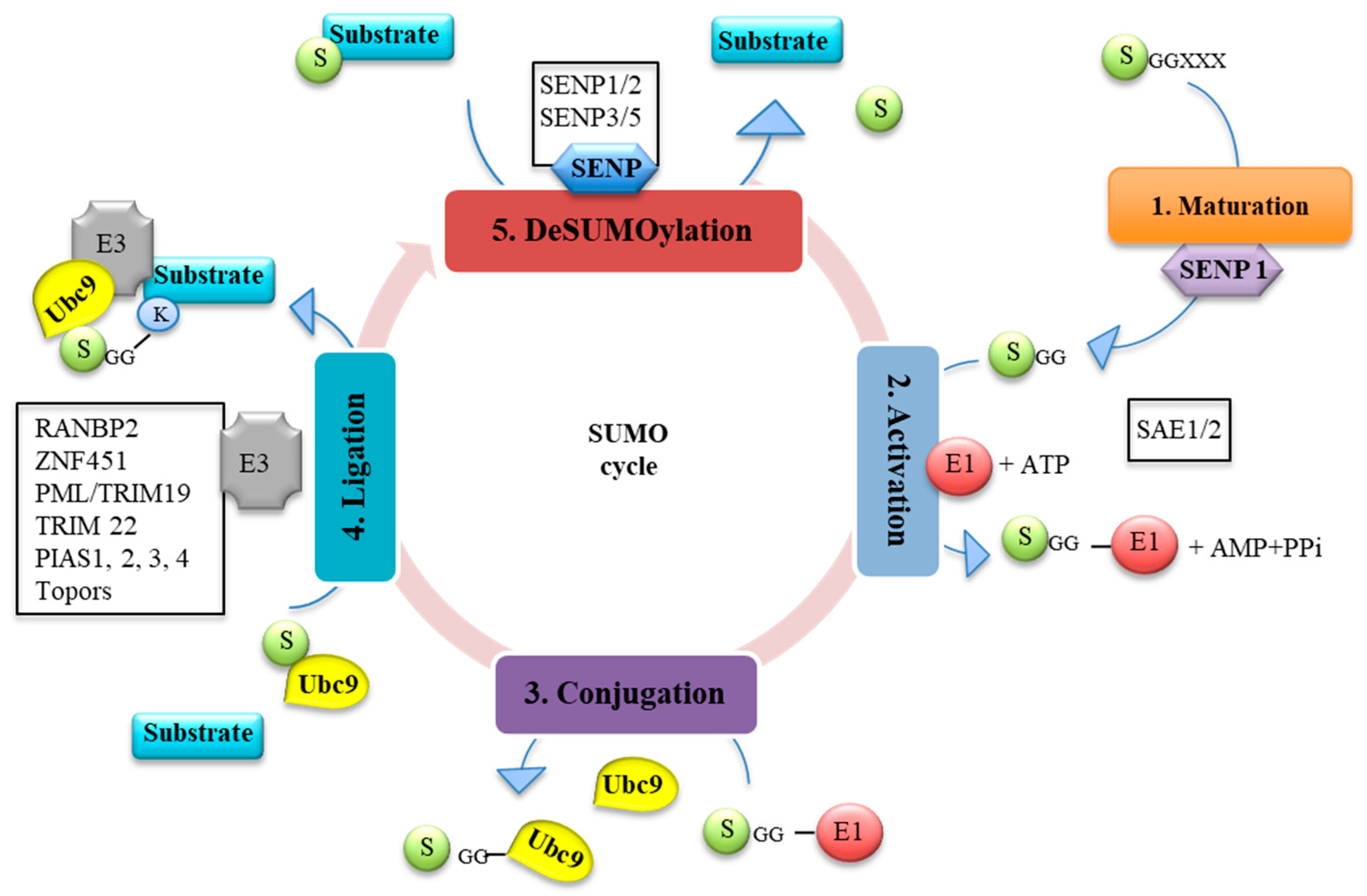
1.1. SUMO Pathway
1.2. Regulation of SUMO and SUMOylation by Viruses and IFN
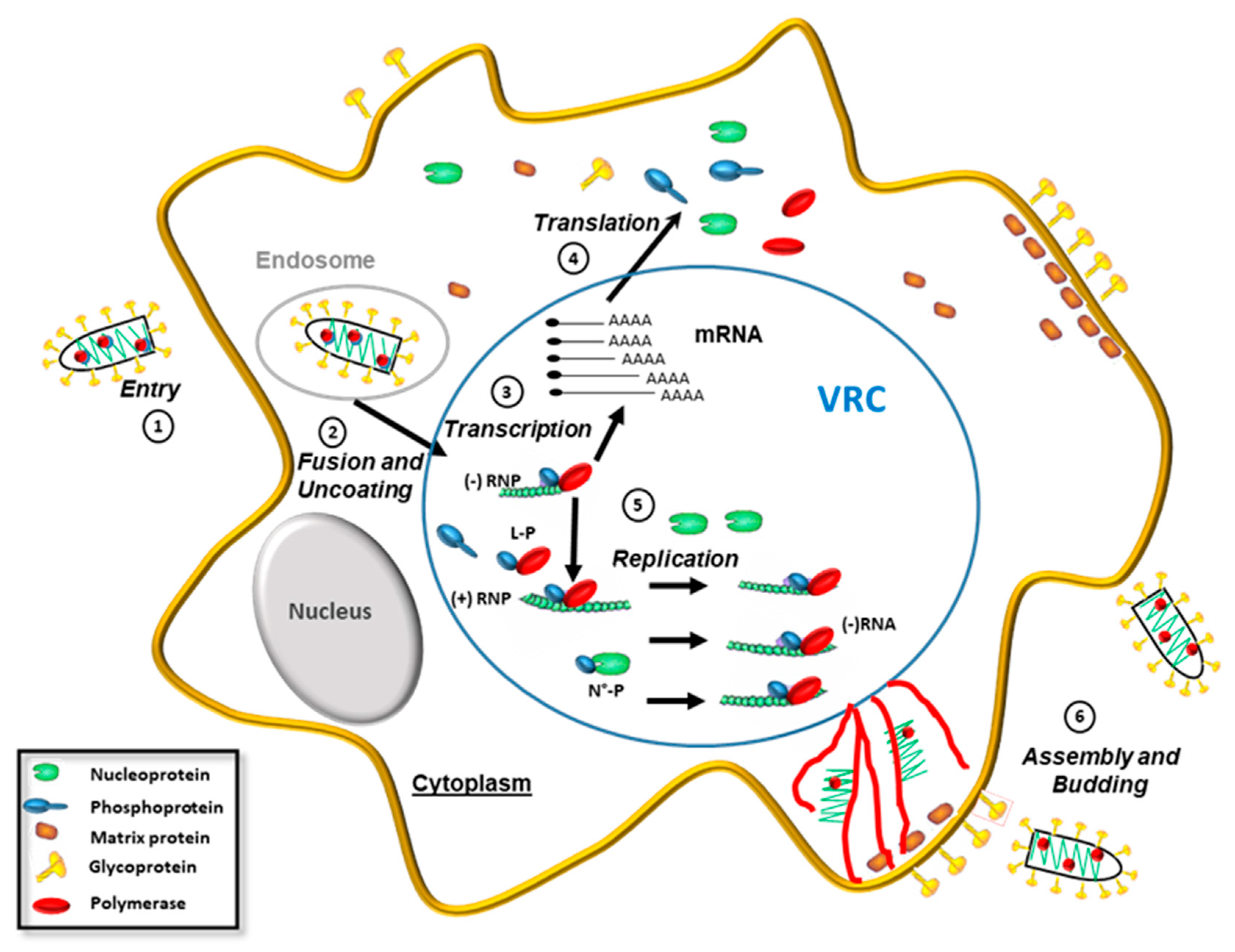
2. Rhabdoviruses
3. Effects of SUMO on IFN Signaling and Production
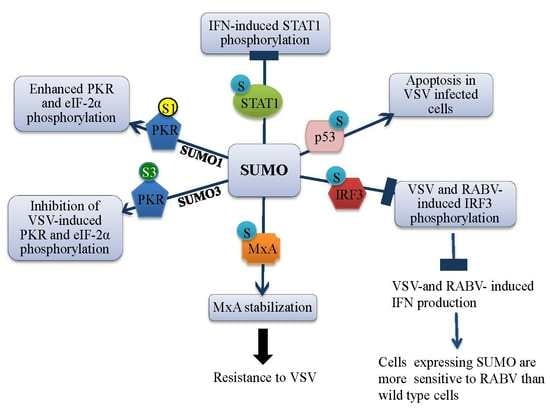
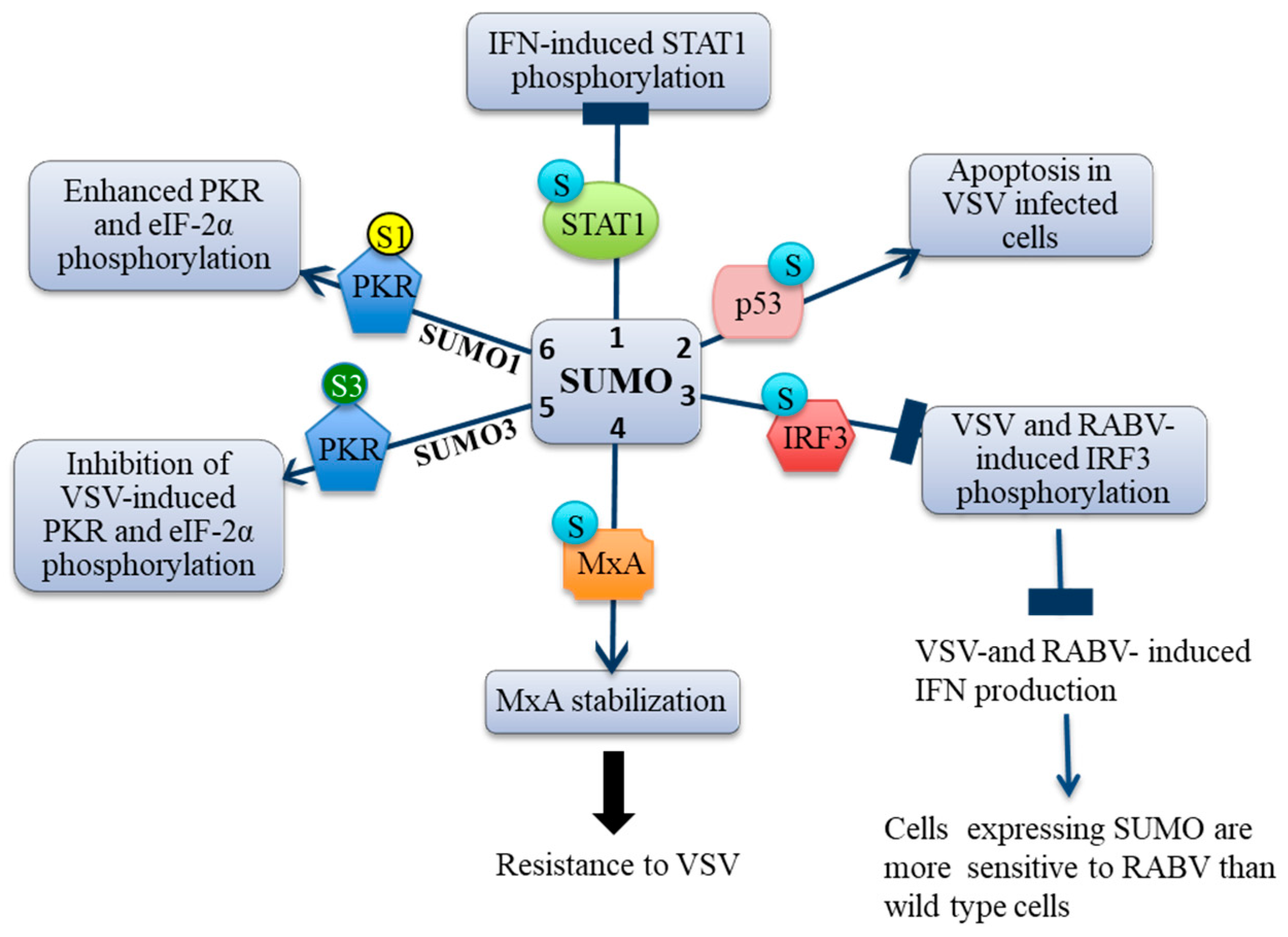
3.1. SUMO Inhibits IFN-Induced STAT1 Activation
3.2. SUMO Inhibits VSV-Induced and RABV-Induced IFN
4. Effect of SUMO on Restriction Factors Conferring Resistance to Rhabdoviruses
4.1. Restriction Factors Conferring Resistance to Rhabdoviruses
4.2. VSV Restriction Factors and SUMO
4.3. SUMO Confers an Intrinsic Resistance to VSV and Not to RABV
4.4. MxA Is Conjugated to SUMO
4.5. MxA Mediates SUMO-Induced VSV Resistance
5. SUMO Paralogs Differentially Alter PKR Activation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hannoun, Z.; Maarifi, G.; Chelbi-Alix, M.K. The implication of SUMO in intrinsic and innate immunity. Cytokine Growth Factor Rev. 2016, 29, 3–16. [Google Scholar] [CrossRef]
- Maarifi, G.; Hannoun, Z.; Geoffroy, M.C.; El Asmi, F.; Zarrouk, K.; Nisole, S.; Blondel, D.; Chelbi-Alix, M.K. MxA Mediates SUMO-Induced Resistance to Vesicular Stomatitis Virus. J. Virol. 2016, 90, 6598–6610. [Google Scholar] [CrossRef] [PubMed]
- Maarifi, G.; Maroui, M.A.; Dutrieux, J.; Dianoux, L.; Nisole, S.; Chelbi-Alix, M.K. Small Ubiquitin-like Modifier Alters IFN Response. J. Immunol. 2015, 195, 2312–2324. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.C.; Hay, R.T. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat. Rev. Mol. Cell. Biol. 2009, 10, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.T. Decoding the SUMO signal. BioChem. Soc. Trans. 2013, 41, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Adorisio, S.; Fierabracci, A.; Muscari, I.; Liberati, A.M.; Ayroldi, E.; Migliorati, G.; Thuy, T.T.; Riccardi, C.; Delfino, D.V. SUMO proteins: Guardians of immune system. J. Autoimmun. 2017, 84, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell. Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef]
- Tatham, M.H.; Geoffroy, M.C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell. Biol. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- Maarifi, G.; El Asmi, F.; Maroui, M.A.; Dianoux, L.; Chelbi-Alix, M.K. Differential effects of SUMO1 and SUMO3 on PKR activation and stability. Sci. Rep. 2018, 8, 1277. [Google Scholar] [CrossRef] [PubMed]
- Matic, I.; van Hagen, M.; Schimmel, J.; Macek, B.; Ogg, S.C.; Tatham, M.H.; Hay, R.T.; Lamond, A.I.; Mann, M.; Vertegaal, A.C.O. In vivo identification of human small ubiquitin-like modifier polymerization sites by high accuracy mass spectrometry and an in vitro to in vivo strategy. Mol. Cell Proteomics 2008, 7, 132–144. [Google Scholar] [CrossRef]
- Ulrich, H.D. The fast-growing business of SUMO chains. Mol. Cell. 2008, 32, 301–305. [Google Scholar] [CrossRef]
- Vertegaal, A.C. SUMO chains: Polymeric signals. BioChem. Soc. Trans. 2010, 38, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.S. Protein modification by SUMO. Annu Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.; Gast, A.; Seeler, J.S.; Dejean, A.; Melchior, F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 2002, 108, 109–120. [Google Scholar] [CrossRef]
- Cappadocia, L.; Pichler, A.; Lima, C.D. Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nat. Struct. Mol. Biol. 2015, 22, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Weger, S.; Hammer, E.; Heilbronn, R. Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Lett. 2005, 579, 5007–5012. [Google Scholar] [CrossRef]
- Chu, Y.; Yang, X. SUMO E3 ligase activity of TRIM proteins. Oncogene 2011, 30, 1108–1116. [Google Scholar] [CrossRef]
- Duan, Z.; Gao, B.; Xu, W.; Xiong, S. Identification of TRIM22 as a RING finger E3 ubiquitin ligase. Biochem. Biophys. Res. Commun. 2008, 374, 502–506. [Google Scholar] [CrossRef]
- Song, J.; Durrin, L.K.; Wilkinson, T.A.; Krontiris, T.G.; Chen, Y. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 14373–14378. [Google Scholar] [CrossRef]
- Lowrey, A.J.; Cramblet, W.; Bentz, G.L. Viral manipulation of the cellular sumoylation machinery. Cell. Commun. Signal. 2017, 15, 27. [Google Scholar] [CrossRef] [PubMed]
- Wilson, V.G. Viral Interplay with the Host Sumoylation System. In SUMO Regulation of Cellular Processes; Wilson, V.G., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; pp. 359–388. ISBN 978-3-319-50044-7. [Google Scholar]
- Domingues, P.; Golebiowski, F.; Tatham, M.H.; Lopes, A.M.; Taggart, A.; Hay, R.T.; Hale, B.G. Global Reprogramming of Host SUMOylation during Influenza Virus Infection. Cell Rep. 2015, 13, 1467–1480. [Google Scholar] [CrossRef]
- Kubota, T.; Matsuoka, M.; Chang, T.H.; Tailor, P.; Sasaki, T.; Tashiro, M.; Kato, A.; Ozato, K. Virus infection triggers SUMOylation of IRF3 and IRF7, leading to the negative regulation of type I interferon gene expression. J. Biol. Chem. 2008, 283, 25660–25670. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Villar, L.; Perez-Giron, J.V.; Vilas, J.M.; Soto, A.; de la Cruz-Hererra, C.F.; Lang, V.; Collado, M.; Vidal, A.; Rodriguez, M.S.; Munoz-Fontela, C.; Rivas, C. SUMOylation of p53 mediates interferon activities. Cell Cycle 2013, 12, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Santamaria, J.; Campagna, M.; Ortega-Molina, A.; Marcos-Villar, L.; de la Cruz-Herrera, C.F.; Gonzalez, D.; Gallego, P.; Lopitz-Otsoa, F.; Esteban, M.; Rodriguez, M.S.; Serrano, M.; Rivas, C. Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 2012, 3, e393. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Herrera, C.F.; Campagna, M.; Garcia, M.A.; Marcos-Villar, L.; Lang, V.; Baz-Martinez, M.; Gutierrez, S.; Vidal, A.; Rodriguez, M.S.; Esteban, M.; Rivas, C. Activation of the double-stranded RNA-dependent protein kinase PKR by small ubiquitin-like modifier (SUMO). J. Biol. Chem. 2014, 289, 26357–26367. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; Maarifi, G.; McManus, F.P.; Lamoliatte, F.; Thibault, P.; Chelbi-Alix, M.K. Promyelocytic Leukemia Protein (PML) Requirement for Interferon-induced Global Cellular SUMOylation. Mol. Cell. Proteomics 2018, 17, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- McManus, F.P.; Bourdeau, V.; Acevedo, M.; Lopes-Paciencia, S.; Mignacca, L.; Lamoliatte, F.; Pino, J.W.R.; Ferbeyre, G.; Thibault, P. Quantitative SUMO proteomics reveals the modulation of several PML nuclear body associated proteins and an anti-senescence function of UBC9. Sci. Rep. 2018, 8, 7754. [Google Scholar] [CrossRef]
- Nanos-Webb, A.; Deyrieux, A.; Bian, X.; Rosas-Acosta, G.; Wilson, V.G. Cloning the human SUMO1 promoter. Mol. Biol. Rep. 2010, 37, 1155–1163. [Google Scholar] [CrossRef]
- Sahin, U.; Ferhi, O.; Carnec, X.; Zamborlini, A.; Peres, L.; Jollivet, F.; Vitaliano-Prunier, A.; de Thé, H.; Lallemand-Breitenbach, V. Interferon controls SUMO availability via the Lin28 and let-7 axis to impede virus replication. Nat. Commun. 2014, 5, 4187. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Kondo, H.; Goodin, M.M.; Kurath, G.; Vasilakis, N. The family Rhabdoviridae: Mono- and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 2017, 227, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Cureton, D.K.; Massol, R.H.; Saffarian, S.; Kirchhausen, T.L.; Whelan, S.P.J. Vesicular Stomatitis Virus Enters Cells through Vesicles Incompletely Coated with Clathrin That Depend upon Actin for Internalization. PLoS Pathog. 2009, 5, e1000394. [Google Scholar] [CrossRef] [PubMed]
- Johannsdottir, H.K.; Mancini, R.; Kartenbeck, J.; Amato, L.; Helenius, A. Host Cell Factors and Functions Involved in Vesicular Stomatitis Virus Entry. J. Virol. 2009, 83, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Piccinotti, S.; Whelan, S.P.J. Rabies Internalizes into Primary Peripheral Neurons via Clathrin Coated Pits and Requires Fusion at the Cell Body. PLoS Pathog. 2016, 12, e1005753. [Google Scholar] [CrossRef] [PubMed]
- Albertini, A.A.V.; Baquero, E.; Ferlin, A.; Gaudin, Y.; Albertini, A.A.V.; Baquero, E.; Ferlin, A.; Gaudin, Y. Molecular and Cellular Aspects of Rhabdovirus Entry. Viruses 2012, 4, 117–139. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional Characterization of Negri Bodies (NBs) in Rabies Virus-Infected Cells: Evidence that NBs Are Sites of Viral Transcription and Replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.S.; Cureton, D.K.; Rahmeh, A.A.; Whelan, S.P. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 2010, 6, e1000958. [Google Scholar] [CrossRef]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudriere-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.A.; Whelan, S.P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. MBio 2018, 9, e02290-17. [Google Scholar] [CrossRef]
- Albertini, A.A.V.; Ruigrok, R.W.H.; Blondel, D. Chapter 1—Rabies Virus Transcription and Replication. In Advances in Virus Research; Jackson, A.C., Ed.; Research Advances in Rabies; Academic Press: Amsterdam, The Netherlands, 2011; Volume 79, pp. 1–22. [Google Scholar]
- Chelbi-Alix, M.K.; Wietzerbin, J. Interferon, a growing cytokine family: 50 years of interferon research. Biochimie 2007, 89, 713–718. [Google Scholar] [CrossRef]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.C.; Gamero, A.M. The role of signal transducer and activator of transcription-2 in the interferon response. J. Interferon Cytokine Res. 2012, 32, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, K.; Nowicka, H.; Kostyrko, K.; Antonczyk, A.; Wesoly, J.; Bluyssen, H.A. The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses. Cytokine Growth Factor Rev. 2016, 29, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Fink, K.; Grandvaux, N. STAT2 and IRF9: Beyond ISGF3. Jakstat 2013, 2, e27521. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.C.; Gamero, A.M. STAT2 phosphorylation and signaling. Jakstat 2013, 2, e25790. [Google Scholar] [CrossRef] [PubMed]
- Mi, Z.; Fu, J.; Xiong, Y.; Tang, H. SUMOylation of RIG-I positively regulates the type I interferon signaling. Protein Cell 2010, 1, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Xiong, Y.; Xu, Y.; Cheng, G.; Tang, H. MDA5 is SUMOylated by PIAS2beta in the upregulation of type I interferon signaling. Mol. Immunol. 2011, 48, 415–422. [Google Scholar] [CrossRef]
- Blondel, D.; Maarifi, G.; Nisole, S.; Chelbi-Alix, M.K. Resistance to Rhabdoviridae Infection and Subversion of Antiviral Responses. Viruses 2015, 7, 3675–3702. [Google Scholar] [CrossRef]
- Weidner, J.M.; Jiang, D.; Pan, X.B.; Chang, J.; Block, T.M.; Guo, J.T. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010, 84, 12646–12657. [Google Scholar] [CrossRef]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; Freiberg, A.N.; Su, L.; Lee, B.; Cheng, G. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, J.; Zurcher, T.; Haller, O.; Staeheli, P. Resistance to influenza virus and vesicular stomatitis virus conferred by expression of human MxA protein. J. Virol. 1990, 64, 3370–3375. [Google Scholar] [PubMed]
- Staeheli, P.; Pavlovic, J. Inhibition of vesicular stomatitis virus mRNA synthesis by human MxA protein. J. Virol. 1991, 65, 4498–4501. [Google Scholar] [PubMed]
- Espert, L.; Degols, G.; Gongora, C.; Blondel, D.; Williams, B.R.; Silverman, R.H.; Mechti, N. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J. Biol. Chem. 2003, 278, 16151–16158. [Google Scholar] [CrossRef] [PubMed]
- Blondel, D.; Kheddache, S.; Lahaye, X.; Dianoux, L.; Chelbi-Alix, M.K. Resistance to rabies virus infection conferred by the PMLIV isoform. J. Virol. 2010, 84, 10719–10726. [Google Scholar] [CrossRef]
- El Asmi, F.; Maroui, M.A.; Dutrieux, J.; Blondel, D.; Nisole, S.; Chelbi-Alix, M.K. Implication of PMLIV in both intrinsic and innate immunity. PLoS Pathog. 2014, 10, e1003975. [Google Scholar] [CrossRef] [PubMed]
- Chelbi-Alix, M.K.; Quignon, F.; Pelicano, L.; Koken, M.H.; de The, H. Resistance to virus infection conferred by the interferon-induced promyelocytic leukemia protein. J. Virol. 1998, 72, 1043–1051. [Google Scholar] [PubMed]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Fensterl, V.; Wetzel, J.L.; Sen, G.C. Interferon-induced protein Ifit2 protects mice from infection of the peripheral nervous system by vesicular stomatitis virus. J. Virol. 2014, 88, 10303–10311. [Google Scholar] [CrossRef]
- Fensterl, V.; Wetzel, J.L.; Ramachandran, S.; Ogino, T.; Stohlman, S.A.; Bergmann, C.C.; Diamond, M.S.; Virgin, H.W.; Sen, G.C. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog. 2012, 8, e1002712. [Google Scholar] [CrossRef]
- Davis, B.M.; Fensterl, V.; Lawrence, T.M.; Hudacek, A.W.; Sen, G.C.; Schnell, M.J. Ifit2 Is a Restriction Factor in Rabies Virus Pathogenicity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, H.; Mejido, J.; Balinsky, C.A.; Morrow, A.N.; Clark, C.R.; Zhao, T.; Zoon, K.C. Identification of alpha interferon-induced genes associated with antiviral activity in Daudi cells and characterization of IFIT3 as a novel antiviral gene. J. Virol. 2010, 84, 10671–10680. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.L.; Carton, J.M.; Lou, J.; Xing, L.; Rubin, B.Y. Interferon-induced guanylate binding protein-1 (GBP-1) mediates an antiviral effect against vesicular stomatitis virus and encephalomyocarditis virus. Virology 1999, 256, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; Taniguchi, T. Integration of interferon-α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Fontela, C.; Macip, S.; Martinez-Sobrido, L.; Brown, L.; Ashour, J.; Garcia-Sastre, A.; Lee, S.W.; Aaronson, S.A. Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 2008, 205, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, C.; Hu, X.; Shang, Y.; Wu, L. MicroRNA-21: A Positive Regulator for Optimal Production of Type I and Type III Interferon by Plasmacytoid Dendritic Cells. Front. Immunol. 2017, 8, 947. [Google Scholar] [CrossRef]
- Moussavi, M.; Fazli, L.; Tearle, H.; Guo, Y.; Cox, M.; Bell, J.; Ong, C.; Jia, W.; Rennie, P.S. Oncolysis of prostate cancers induced by vesicular stomatitis virus in PTEN knockout mice. Cancer Res. 2010, 70, 1367–1376. [Google Scholar] [CrossRef]
- Kamitani, T.; Nguyen, H.P.; Kito, K.; Fukuda-Kamitani, T.; Yeh, E.T. Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 1998, 273, 3117–3120. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef]
- Haller, O.; Kochs, G. Interferon-Induced Mx Proteins: Dynamin-Like GTPases with Antiviral Activity. Traffic 2002, 3, 710–717. [Google Scholar] [CrossRef]
- Janzen, C.; Kochs, G.; Haller, O. A Monomeric GTPase-Negative MxA Mutant with Antiviral Activity. J. Virol. 2000, 74, 8202–8206. [Google Scholar] [CrossRef] [PubMed]
- Nigg, P.E.; Pavlovic, J. Oligomerization and GTP-binding Requirements of MxA for Viral Target Recognition and Antiviral Activity against Influenza A Virus. J. Biol. Chem. 2015, 290, 29893–29906. [Google Scholar] [CrossRef] [PubMed]
- Brantis-de-Carvalho, C.E.; Maarifi, G.; Goncalves Boldrin, P.E.; Zanelli, C.F.; Nisole, S.; Chelbi-Alix, M.K.; Valentini, S.R. MxA interacts with and is modified by the SUMOylation machinery. Exp. Cell. Res. 2015, 330, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. MicroBiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Structure and function of the protein kinase R. Curr Top. MicroBiol. Immunol 2007, 316, 253–292. [Google Scholar]
- Proud, C.G. PKR: A new name and new roles. Trends BioChem. Sci. 1995, 20, 241–246. [Google Scholar] [CrossRef]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Galabru, J. The double-stranded RNA-dependent protein kinase is also activated by heparin. Eur. J. Biochem. 1987, 167, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Okumura, A.J.; Uematsu, K.; Hatakeyama, S.; Zhang, D.E.; Kamura, T. Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 2013, 288, 2839–2847. [Google Scholar] [CrossRef]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Restriction Factors and SUMO Inhibiting VSV | Inhibition | References |
|---|---|---|
| IFITM3 | Entry | [52] |
| Ch25h | Fusion and uncoating | [53] |
| Primary transcription | [54,55] | |
| [2] | ||
| Secondary transcription | [56] | |
| [57,58,59] | ||
| PKR | Translation | [60] |
| IFIT2 4 | Replication | [61,62,63] |
| IFIT3 | Production | [64] |
| Tetherin | Assembly and budding | [52] |
| GBP1 | Production | [65] |
| p53 | Production | [25,66] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Asmi, F.; Brantis-de-Carvalho, C.E.; Blondel, D.; Chelbi-Alix, M.K. Rhabdoviruses, Antiviral Defense, and SUMO Pathway. Viruses 2018, 10, 686. https://doi.org/10.3390/v10120686
El Asmi F, Brantis-de-Carvalho CE, Blondel D, Chelbi-Alix MK. Rhabdoviruses, Antiviral Defense, and SUMO Pathway. Viruses. 2018; 10(12):686. https://doi.org/10.3390/v10120686
Chicago/Turabian StyleEl Asmi, Faten, Carlos Eduardo Brantis-de-Carvalho, Danielle Blondel, and Mounira K. Chelbi-Alix. 2018. "Rhabdoviruses, Antiviral Defense, and SUMO Pathway" Viruses 10, no. 12: 686. https://doi.org/10.3390/v10120686
APA StyleEl Asmi, F., Brantis-de-Carvalho, C. E., Blondel, D., & Chelbi-Alix, M. K. (2018). Rhabdoviruses, Antiviral Defense, and SUMO Pathway. Viruses, 10(12), 686. https://doi.org/10.3390/v10120686