Detection of a Conspecific Mycovirus in Two Closely Related Native and Introduced Fungal Hosts and Evidence for Interspecific Virus Transmission


Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolates Used in the Study
2.2. Fungal Cultivation and RNA Extraction
2.3. Screening for HfMV1 and Viral Sequencing
2.4. Phylogenetic Analyses
2.5. Genetic Diversity, Differentiation and Evolution
3. Results
3.1. Prevalence of HfMV1 in H. albidus
3.2. Pairwise Sequence Identity
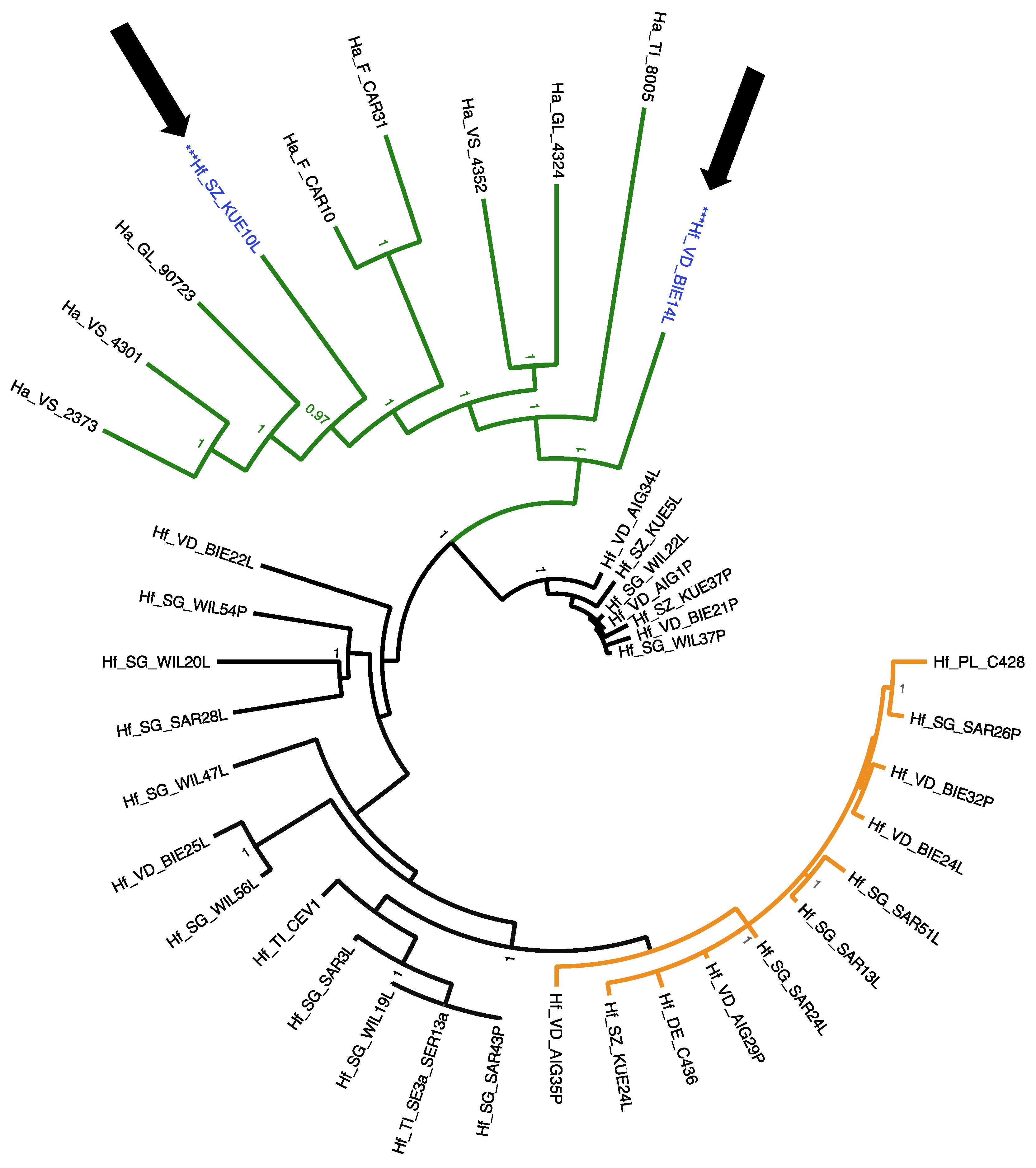
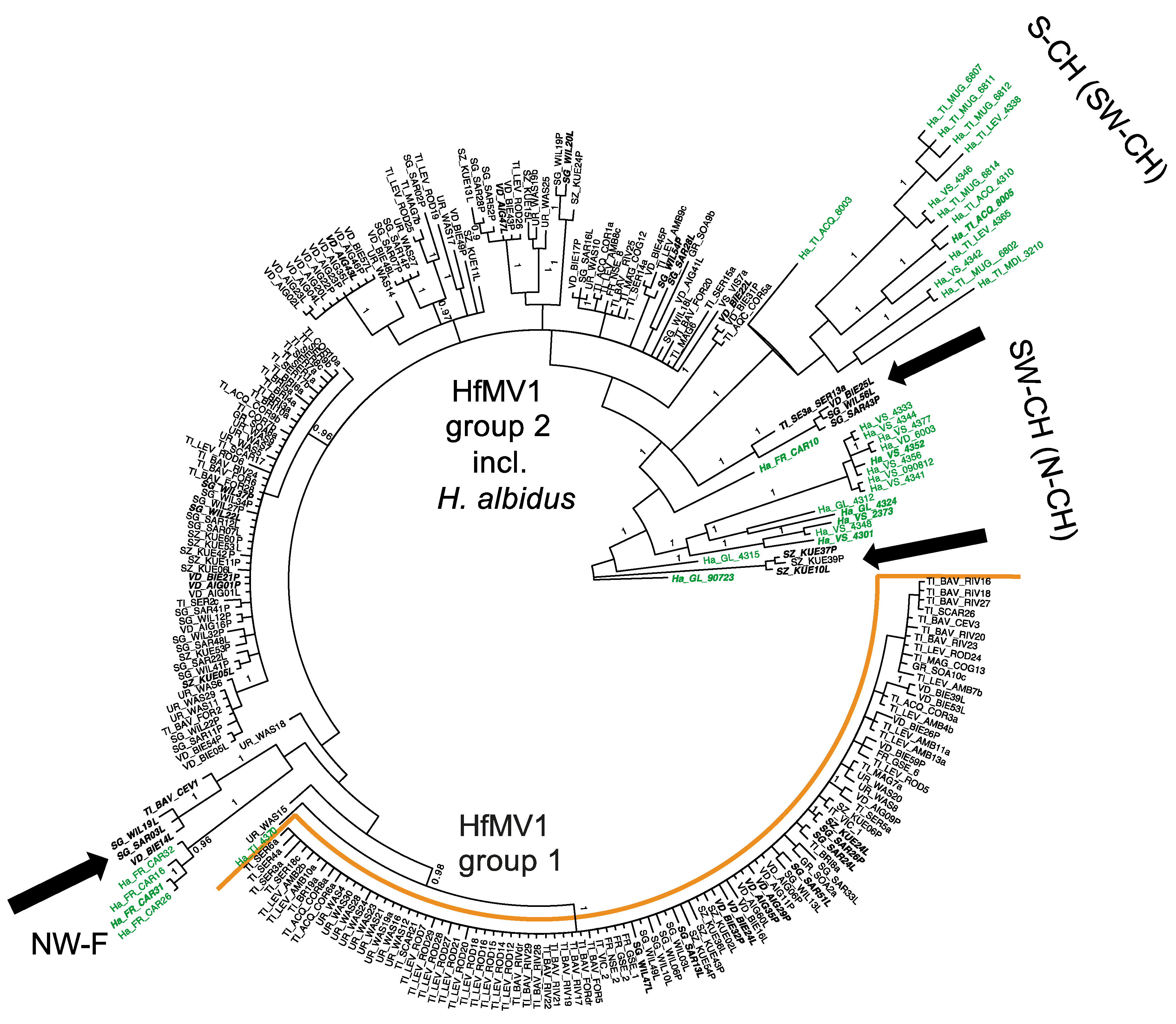
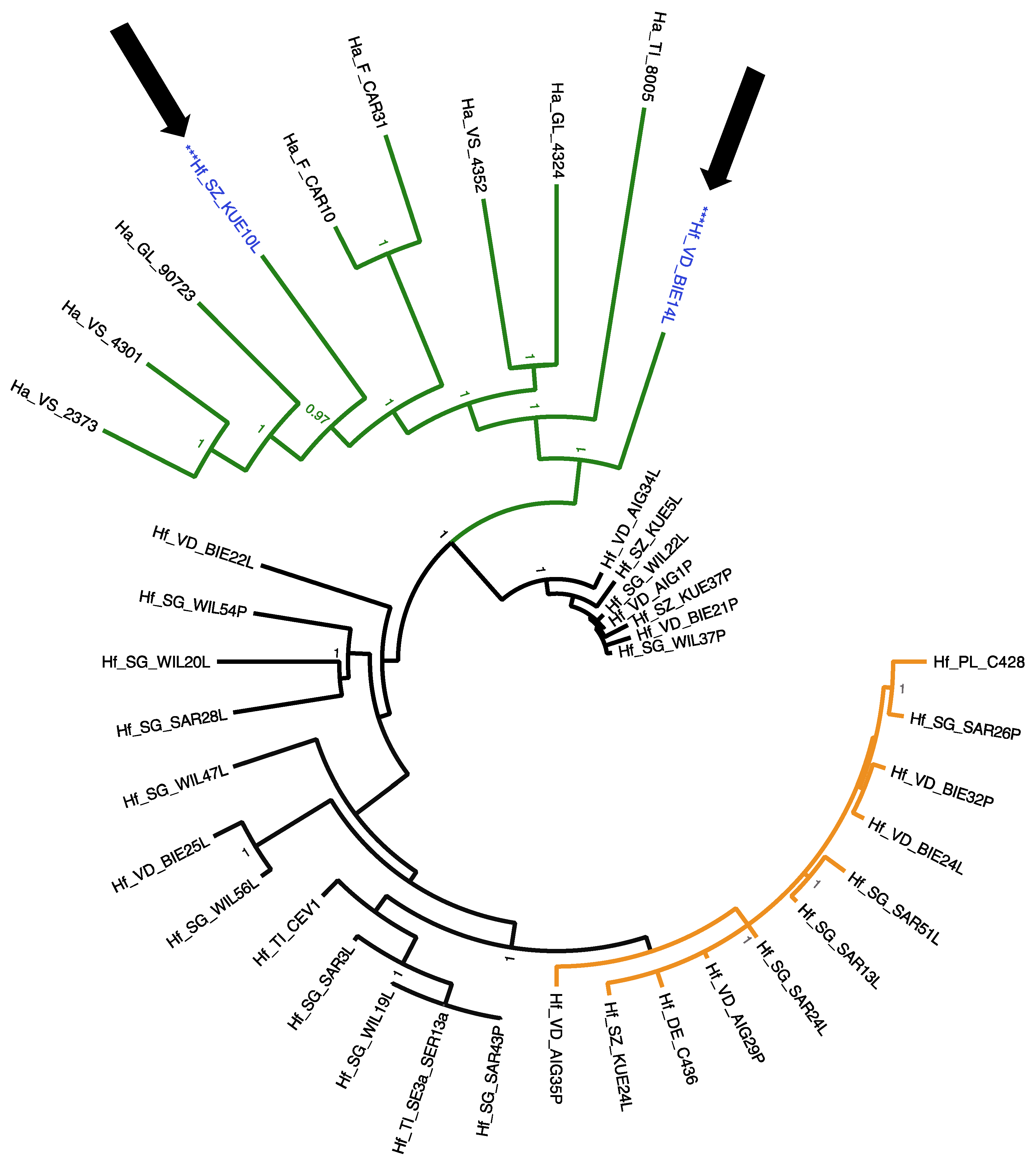
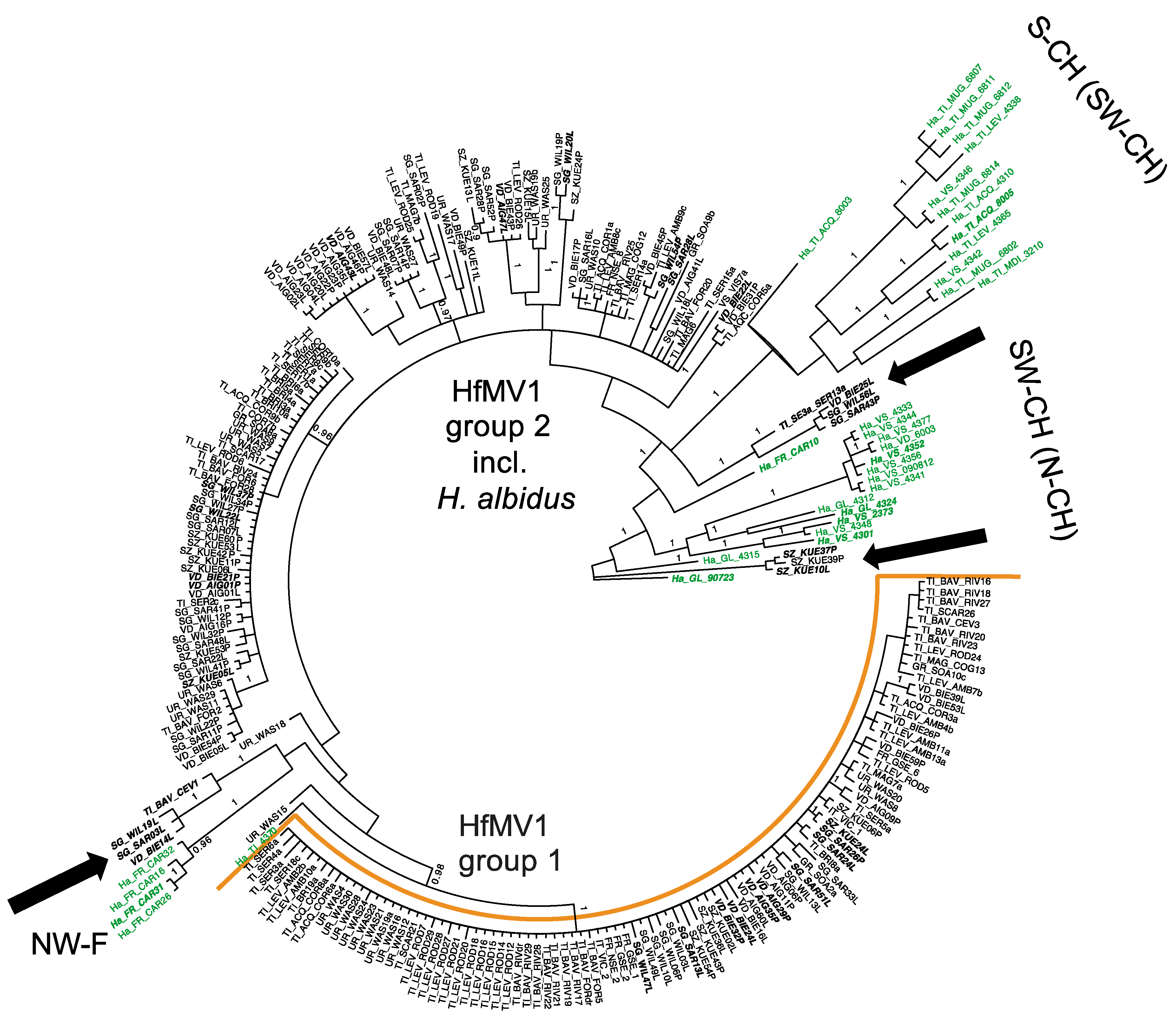
3.3. Phylogenetic Relationships of HfMV1 across Two Different Host Species
3.4. Population Genetic Parameters
3.4.1. Full RdRP Gene
3.4.2. Partial RdRP Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Westphal, M.I.; Browne, M.; MacKinnon, K.; Noble, I. The link between international trade and the global distribution of invasive alien species. Biol. Invasions 2008, 10, 391–398. [Google Scholar] [CrossRef]
- Santini, A.; Ghelardini, L.; De Pace, C.; Desprez-Loustau, M.L.; Capretti, P.; Chandelier, A.; Cech, T.; Chira, D.; Diamandis, S.; Gaitniekis, T.; et al. Biogeographical patterns and determinants of invasion by forest pathogens in Europe. New Phytol. 2013, 197, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Hantula, J.; Mu, M.M.; Uusivuori, J. International plant trade associated risks: Laissez-faire or novel solutions. Environ. Sci. Policy 2014, 37, 158–160. [Google Scholar] [CrossRef]
- Grünwald, N.J.; Garbelotto, M.; Goss, E.M.; Heunges, K.; Prospero, S. Emergence of the sudden oak death pathogen Phytophthora ramorum. Trends Microbiol. 2012, 20, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Rigling, D.; Prospero, S. Cryphonectria parasitica, the causal agent of chestnut blight: Invasion history, population biology and disease control. Mol. Plant Pathol. 2018, 19, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Hoegger, P.J.; Rigling, D.; Holdenrieder, O.; Heiniger, U. Cryphonectria radicalis: Rediscovery of a lost fungus. Mycologia 2002, 94, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Gonthier, P.; Nicolotti, G.; Linzer, R.; Guglielmo, F.; Garbelotto, M. Invasion of European pine stands by a North American forest pathogen and its hybridization with a native interfertile taxon. Mol. Ecol. 2007, 16, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Érsek, T.; Nagy, Z.A. Species hybrids in the genus Phytophthora with emphasis on the alder pathogen Phytophthora alni: A review. Eur. J. Plant Pathol. 2008, 122, 31–39. [Google Scholar] [CrossRef]
- Gross, A.; Han, J.G. Hymenoscyphus fraxineus and two new Hymenoscyphus species identified in Korea. Mycol. Prog. 2015, 14, 1. [Google Scholar] [CrossRef]
- Gross, A.; Zaffarano, P.L.; Duo, A.; Grünig, C.R. Reproductive mode and life-cycle of the ash dieback pathogen Hymenoscyphus pseudoalbidus. Fungal Genet. Biol. 2012, 49, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Baral, H.O.; Bemmann, M. Hymenoscyphus fraxineus vs. Hymenoscyphus albidus—A comparative light microscopic study on the causal agent of European ash dieback and related foliicolous, stroma-forming species. Mycology 2014, 5, 228–290. [Google Scholar] [PubMed]
- Bengtsson, S.B.K.; Vasaitis, R.; Kirisits, T.; Solheim, H.; Stenlid, J. Population structure of Hymenoscyphus pseudoalbidus and its genetic relationship to Hymenoscyphus albidus. Fungal Ecol. 2012, 5, 147–153. [Google Scholar] [CrossRef]
- Burokiene, D.; Prospero, S.; Jung, E.; Marciulyniene, D.; Moosbrugger, K.; Norkute, G.; Rigling, D.; Lygis, V.; Schoebel, C.N. Genetic population structure of the invasive ash dieback pathogen Hymenoscyphus fraxineus in its expanding range. Biol. Invasions 2015, 17, 2743–2756. [Google Scholar] [CrossRef]
- Gross, A.; Hosoya, T.; Queloz, V. Population structure of the invasive forest pathogen Hymenoscyphus pseudoalbidus. Mol. Ecol. 2014, 23, 2943–2960. [Google Scholar] [CrossRef] [PubMed]
- Schoebel, C.N.; Botella, L.; Lygis, V.; Rigling, D. Population genetic analysis of a parasitic mycovirus to infer the invasion history of its fungal host. Mol. Ecol. 2017, 26, 2482–2497. [Google Scholar] [CrossRef] [PubMed]
- Sonstebo, J.H.; Smith, A.V.; Adamson, K.; Drenkhan, R.; Solheim, H.; Hietala, A. Genome-wide population diversity in Hymenoscyphus fraxineus points to an eastern Russian origin of European Ash dieback. bioRxiv 2017, 154492. [Google Scholar] [CrossRef]
- Kirisits, T.; Dämpfle, L.; Kräutler, K. Hymenoscyphus albidus is not associated with an anamorphic stage and displays slower growth than Hymenoscyphus pseudoalbidus on agar media. For. Pathol. 2013, 43, 386–389. [Google Scholar]
- Brasier, C.; King, K.; Kirisits, T.; Orton, E.; Webber, J. High frequency of vegetative incompatibility combined with haploid selfing in the native European ash foliage coloniser Hymenoscyphus albidus. Fungal Ecol. 2017, 28, 11–24. [Google Scholar] [CrossRef]
- McKinney, L.V.; Thomsen, I.M.; Kjær, D.; Bengtsson, S.B.K.; Nielsen, L.R. Rapid invasion by an aggressive pathogenic fungus (Hymenoscyphus pseudoalbidus) replaces a native decomposer (Hymenoscyphus albidus): A case of local cryptic extinction? Fungal Ecol. 2012, 5, 663–669. [Google Scholar] [CrossRef]
- King, K.M.; Webber, J.F. Development of a multiplex PCR assay to discriminate native Hymenoscyphus albidus and introduced Hymenoscyphus fraxineus in Britain and assess their distribution. Fungal Ecol. 2016, 23, 79–85. [Google Scholar] [CrossRef]
- Hietala, A.M.; Timmermann, V.; Barja, I.; Solheim, H. The invasive ash dieback pathogen Hymenoscyphus pseudoalbidus exerts maximal infection pressure prior to the onset of host leaf senescence. Fungal Ecol. 2013, 6, 302–308. [Google Scholar] [CrossRef]
- Kowalski, T.; Holdenrieder, O. Pathogenicity of Chalara fraxinea. For. Pathol. 2009, 39, 1–7. [Google Scholar] [CrossRef]
- Pautasso, M.; Aas, G.; Queloz, V.; Holdenrieder, O. European ash (Fraxinus excelsior) dieback—A conservation biology challenge. Biol. Conserv. 2013, 158, 37–49. [Google Scholar] [CrossRef]
- Orton, E.S.; Brasier, C.M.; Bilham, L.J.; Bansal, A.; Webber, J.F.; Brown, J.K.M. Population structure of the ash dieback pathogen, Hymenoscyphus fraxineus, in relation to its mode of arrival in the UK. Plant Pathol. 2018, 67, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Ghabrial, S.A.; Suzuki, N. Viruses of plant pathogenic fungi. Annu. Rev. Phytopathol. 2009, 47, 353–384. [Google Scholar] [CrossRef] [PubMed]
- Schoebel, C.N.; Zoller, S.; Rigling, D. Detection and genetic characterization of a novel mycovirus in Hymenoscyphus fraxineus, the causal agent of ash dieback. Infect. Genet. Evol. 2014, 28, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H. Molecular Evolution; Sinauer Associates, Inc.: Sunderland, MA, USA, 1997. [Google Scholar]
- Polashock, J.J.; Hillman, B.I. A small mitochondrial double-stranded (ds) RNA element associated with a hypovirulent strain of the chestnut blight fungus and ancestrally related to yeast cytoplasmic T and W dsRNAs. Proc. Natl. Acad. Sci. USA 1994, 91, 8680–8684. [Google Scholar] [CrossRef] [PubMed]
- Polashock, J.; Bedker, P.; Hillman, B. Movement of a small mitochondrial double-stranded RNA element of Cryphonectria parasitica: Ascospore inheritance and implications for mitochondrial recombination. Mol. Gen. Genet. 1997, 256, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Linder-Basso, D.; Hillman, B.I.; Kaneko, S.; Milgroom, M.G. Evidence for interspecies transmission of viruses in natural populations of filamentous fungi in the genus Cryphonectria. Mol. Ecol. 2003, 12, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Vainio, E.J.; Hakanpää, J.; Dai, Y.C.; Hansen, E.; Korhonen, K.; Hantula, J. Species of Heterobasidion host a diverse pool of partitiviruses with global distribution and interspecies transmission. Fungal Biol. 2011, 115, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.; Ikeda, S.S.; Boland, G.J. Interspecific transmission of double-stranded RNA and hypovirulence from Sclerotinia sclerotiorum to S. minor. Phytopathology 2002, 92, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Xu, R.; Boland, G. Hypovirulence-associated double-stranded RNA from Sclerotinia homoeocarpa is conspecific with Ophiostoma novo-ulmi mitovirus 3a-Ld. Phytopathology 2003, 93, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- White, T.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M., Gelfand, D., Sninsky, J., White, T., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Queloz, V.; Grünig, C.R.; Berndt, R.; Berndt, R.; Kowalski, T.; Sieber, T.N.; Holdenrieder, O. Cryptic speciation in Hymenoscyphus albidus. For. Pathol. 2011, 41, 133–142. [Google Scholar] [CrossRef]
- Ranwez, V.; Harispe, S.; Delsuc, F.; Douzery, E.J.P. MACSE: Multiple Alignment of Coding Sequences accounting for frameshifts and stop codons. PLoS ONE 2011, 6, e22594. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Rigling, D.; Hilfiker, S.; Schöbel, C.; Meier, F.; Engesser, R.; Scheidegger, C.; Stofer, S.; Senn-Irlet, B.; Queloz, V. Das Eschentriebsterben: Biologie, Krankheitssymptome und Handlungsempfehlungen; Merkbl. Publisher: Birmensdorf, Switzerland, 2016. [Google Scholar]
- Buck, K.W.; Esteban, R.; Hillman, B.I. Narnaviridae. In Virus Taxonomy: Eighth Report of the International Committee on Taxonomy of Viruses, 1st ed.; Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2005; pp. 751–756. [Google Scholar]
- Botella, L.; Tuomivirta, T.T.; Vervuurt, S.; Diez, J.J.; Hantula, J. Occurrence of two different species of mitoviruses in the European race of Gremmeniella abietina var. abietina, both hosted by the genetically unique Spanish population. Fungal Biol. 2012, 116, 872–882. [Google Scholar] [PubMed]
- Wey, T.; Schlegel, M.; Stroheker, S.; Gross, A. MAT–gene structure and mating behavior of Hymenoscyphus fraxineus and Hymenoscyphus albidus. Fungal Genet. Biol. 2016, 87, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, L.; Li, G.; Jiang, D.; Ghabrial, S.A. Genome characterization of a debilitation-associated mitovirus infecting the phytopathogenic fungus Botrytis cinereal. Virology 2010, 406, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Stenlid, J.; Elfstand, M.; Cleary, M.; Ihrmark, K.; Karlsson, M.; Davydenko, K.; Brandström Durling, M. Genomes of Hymenoscyphus fraxineus and Hymenoscyphus albidus Encode Surprisingly Large Cell Wall Degrading Potential, Balancing Saprotrophic and Necrotrophic Signatures. Baltic For. 2017, 23, 89–106. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
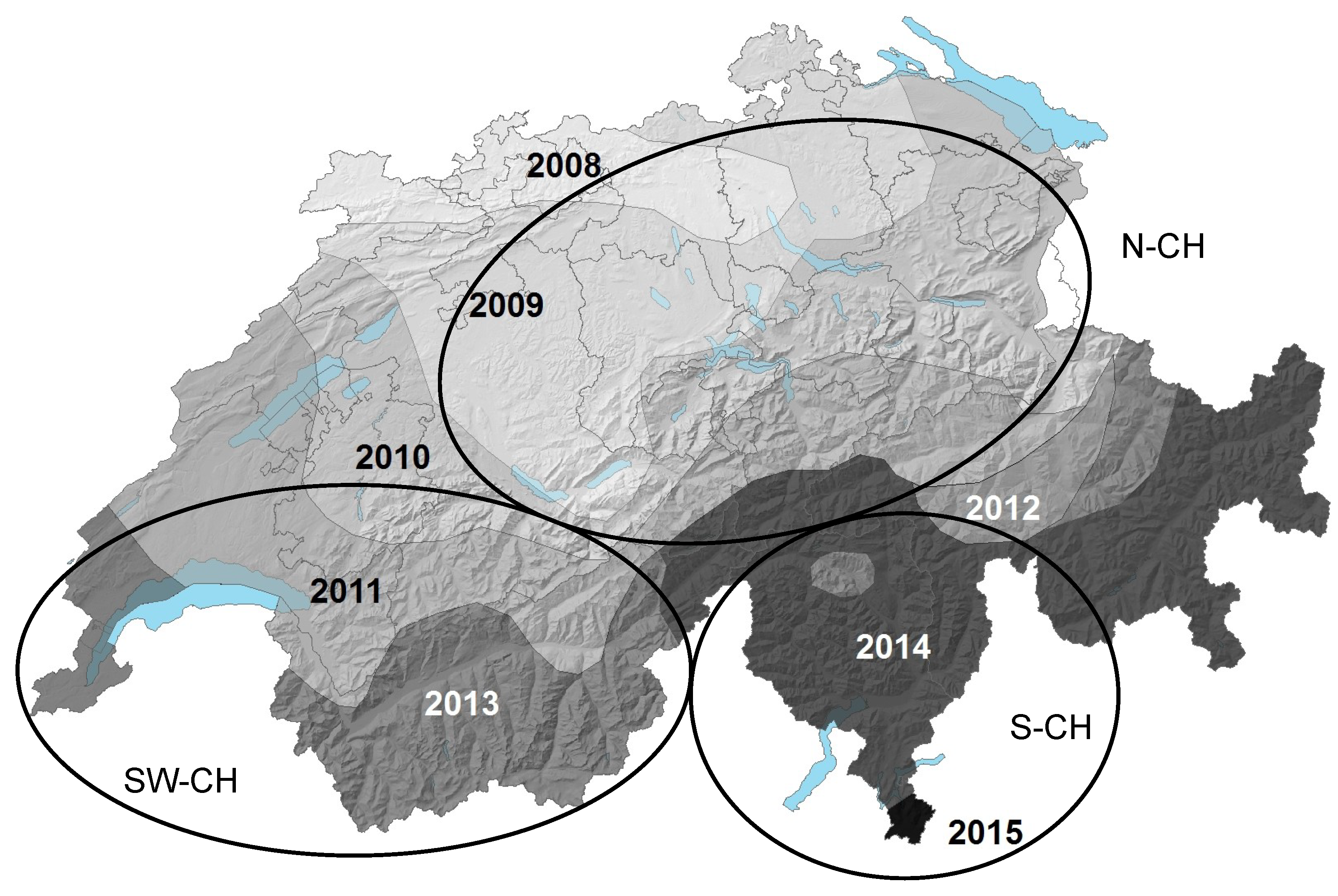
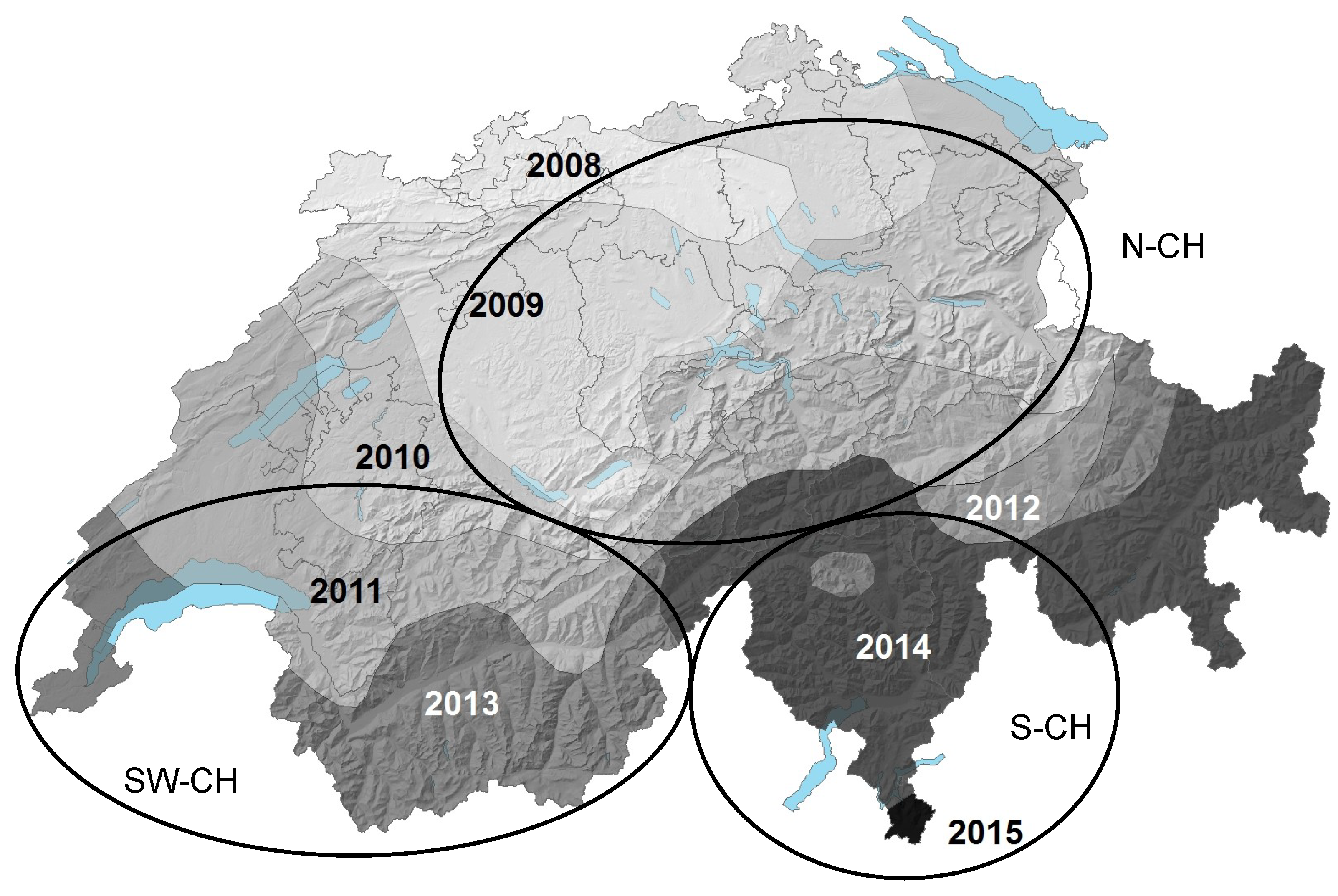
| Host | Region (Abbreviation) | Canton (s)/Region | Number of Samples Screened | Number of Samples with HfMV1 | Sampling Year (s) | Reference for Fungal Isolates |
|---|---|---|---|---|---|---|
| H. albidus | 67 | 34 | ||||
| Switzerland | South-Western Switzerland (SW-CH) | VS, VD | 13 | 13 | 2008, 2009, 2012 | This study |
| Southern Switzerland (S-CH) | TI | 33 | 12 | 2009, 2010, 2012 | [35]; this study | |
| Northern Switzerland (N-CH) | GL, ZH | 7 | 4 | 2009 | [35] | |
| France | North-Western France (NW-F) | Bretagne | 14 | 5 | 2012 | [17]; this study |
| H. fraxineus | ** | 221 | ||||
| Switzerland | South-Western Switzerland (SW-CH) | VS, VD | ** | 41 | 2013, 2016 | [13]; this study |
| Southern-Switzerland * (S-CH) | TI, GR | ** | 90 | 2014, 2013, 2016 | [15]; this study | |
| Northern-Switzerland (N-CH) | SG, SZ, UR | ** | 85 | 2013, 2014 | [13]; this study |
| Population | N | Sequence Length | Net Sites | S | η | h | Hd | π | K |
|---|---|---|---|---|---|---|---|---|---|
| H. fraxineus | 32 | 2151 | 1791 | 219 | 239 | 30 | 1.0 | 0.021 | 38 |
| H. albidus | 8 | 2151 | 2122 | 400 | 464 | 8 | 1.0 | 0.077 | 163 |
| H. fraxineus CH | 30 | 2151 | 1851 | 218 | 238 | 28 | 1.0 | 0.021 | 39 |
| H. albidus CH | 6 | 2151 | 2129 | 360 | 403 | 6 | 1.0 | 0.076 | 163 |
| H. fraxineus & H. albidus total | 40 | 2151 | 1774 | 328 | 380 | 38 | 1.0 | 0.029 | 52 |
| Population | N | Sequence Length | Net Sites | S | η | h | Hd | π | K |
|---|---|---|---|---|---|---|---|---|---|
| H. fraxineus all | 221 | 495 | 348 | 49 | 57 | 48 | 0.79 | 0.007 | 2.35 |
| H. fraxineus CH | 214 | 495 | 349 | 50 | 58 | 49 | 0.80 | 0.007 | 2.41 |
| H. fraxineus S-CH * | 90 | 495 | 359 | 33 | 37 | 28 | 0.80 | 0.008 | 2.98 |
| H. fraxineus SW-CH | 41 | 495 | 485 | 89 | 102 | 27 | 0.96 | 0.039 | 19.21 |
| H. fraxineus N-CH | 85 | 495 | 458 | 73 | 81 | 53 | 0.96 | 0.019 | 8.72 |
| H. fraxineus NW-F | 5 | 495 | 488 | 16 | 16 | 3 | 0.70 | 0.014 | 6.60 |
| H. albidus all | 34 | 495 | 426 | 99 | 129 | 28 | 0.99 | 0.080 | 34.02 |
| H. albidus CH | 29 | 495 | 427 | 94 | 122 | 24 | 0.99 | 0.078 | 33.39 |
| H. albidus S-CH | 12 | 495 | 443 | 79 | 94 | 12 | 1.00 | 0.067 | 29.74 |
| H. albidus SW-CH | 13 | 495 | 467 | 76 | 88 | 10 | 0.96 | 0.052 | 24.03 |
| H. albidus NW-F | 5 | 495 | 494 | 34 | 34 | 4 | 0.90 | 0.026 | 14.40 |
| H. fraxineus & H. albidus total | 255 | 495 | 348 | 71 | 89 | 73 | 0.84 | 0.015 | 5.33 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoebel, C.N.; Prospero, S.; Gross, A.; Rigling, D. Detection of a Conspecific Mycovirus in Two Closely Related Native and Introduced Fungal Hosts and Evidence for Interspecific Virus Transmission. Viruses 2018, 10, 628. https://doi.org/10.3390/v10110628
Schoebel CN, Prospero S, Gross A, Rigling D. Detection of a Conspecific Mycovirus in Two Closely Related Native and Introduced Fungal Hosts and Evidence for Interspecific Virus Transmission. Viruses. 2018; 10(11):628. https://doi.org/10.3390/v10110628
Chicago/Turabian StyleSchoebel, Corine N., Simone Prospero, Andrin Gross, and Daniel Rigling. 2018. "Detection of a Conspecific Mycovirus in Two Closely Related Native and Introduced Fungal Hosts and Evidence for Interspecific Virus Transmission" Viruses 10, no. 11: 628. https://doi.org/10.3390/v10110628
APA StyleSchoebel, C. N., Prospero, S., Gross, A., & Rigling, D. (2018). Detection of a Conspecific Mycovirus in Two Closely Related Native and Introduced Fungal Hosts and Evidence for Interspecific Virus Transmission. Viruses, 10(11), 628. https://doi.org/10.3390/v10110628