Antibiotic Therapy Using Phage Depolymerases: Robustness Across a Range of Conditions

Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Cell Culture
2.2. Coliphage Strains and Culture
2.3. Plasmids, Protein Expression and Purification
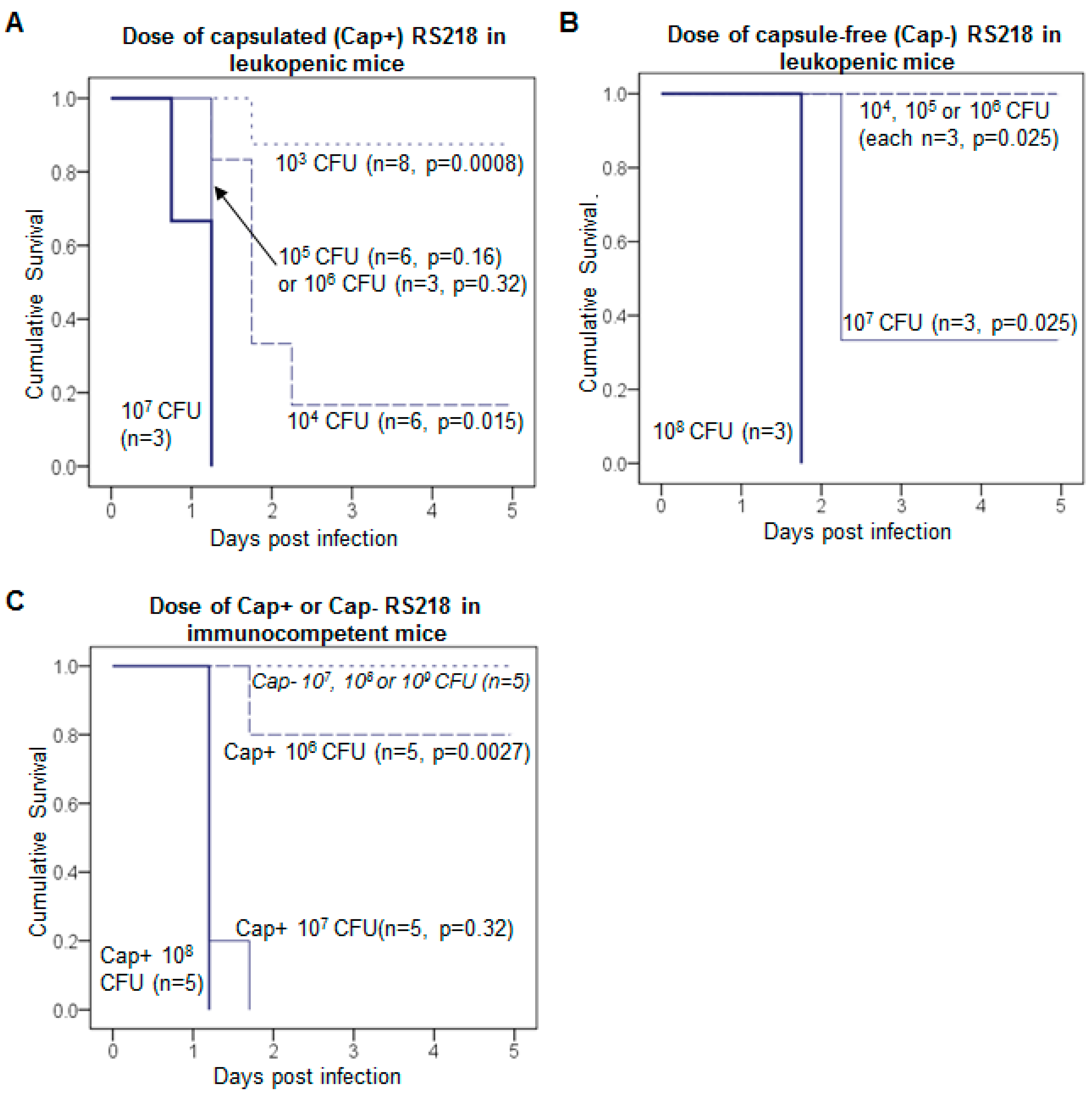
2.4. Mouse Infection Model
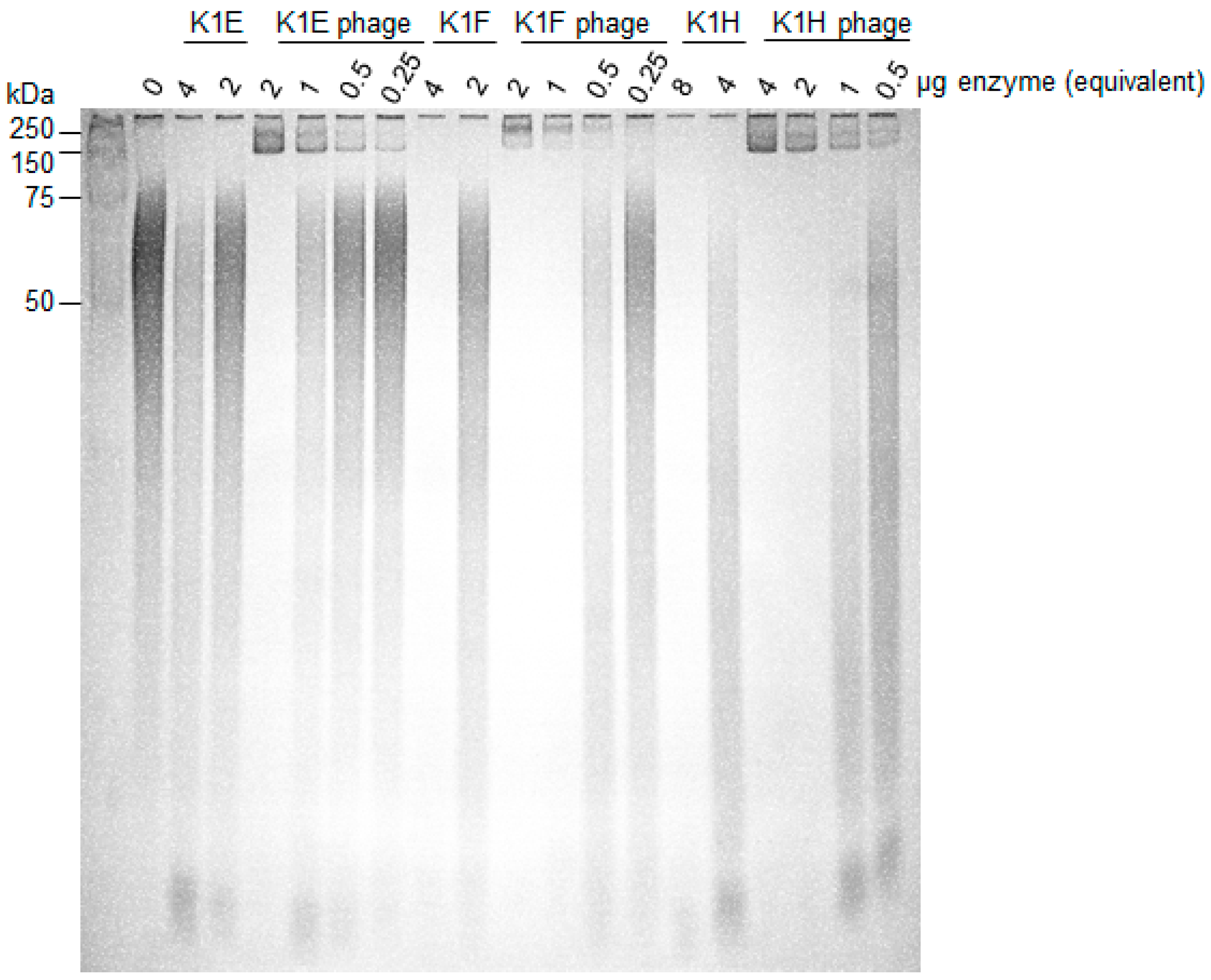
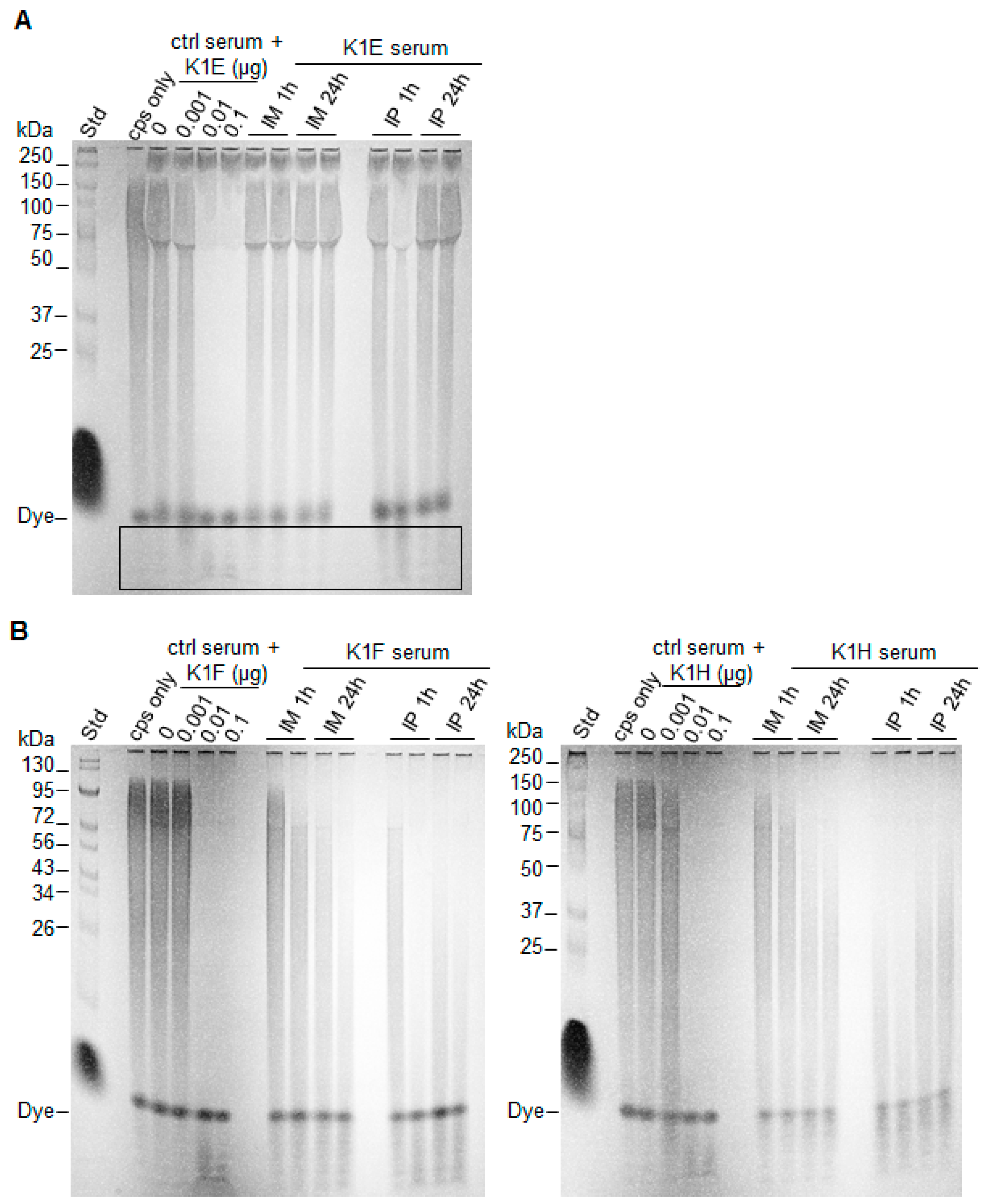
2.5. Capsule Isolation and Degradation Assay
2.6. Resistance Competition Assay (RCA)
3. Results
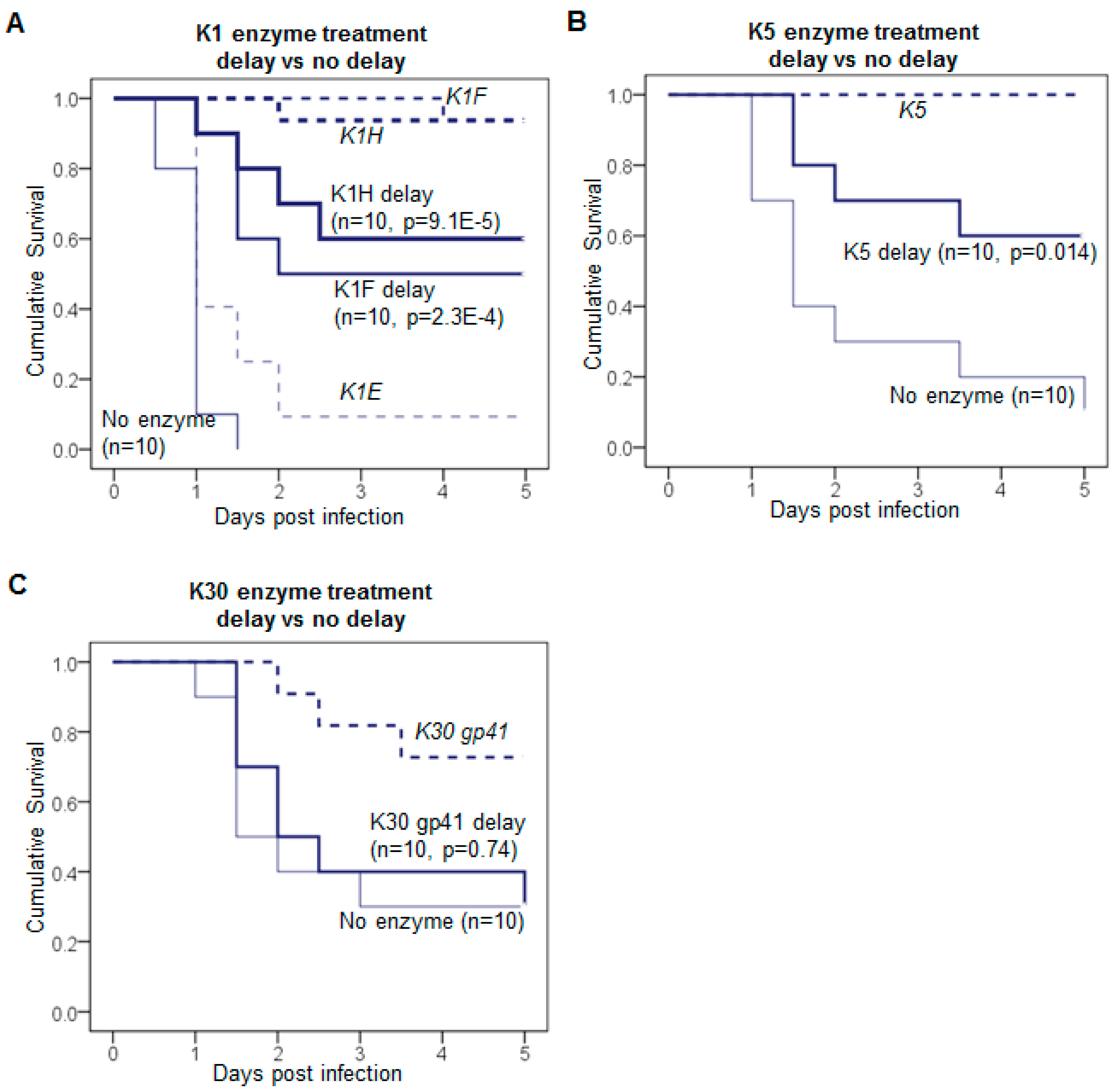
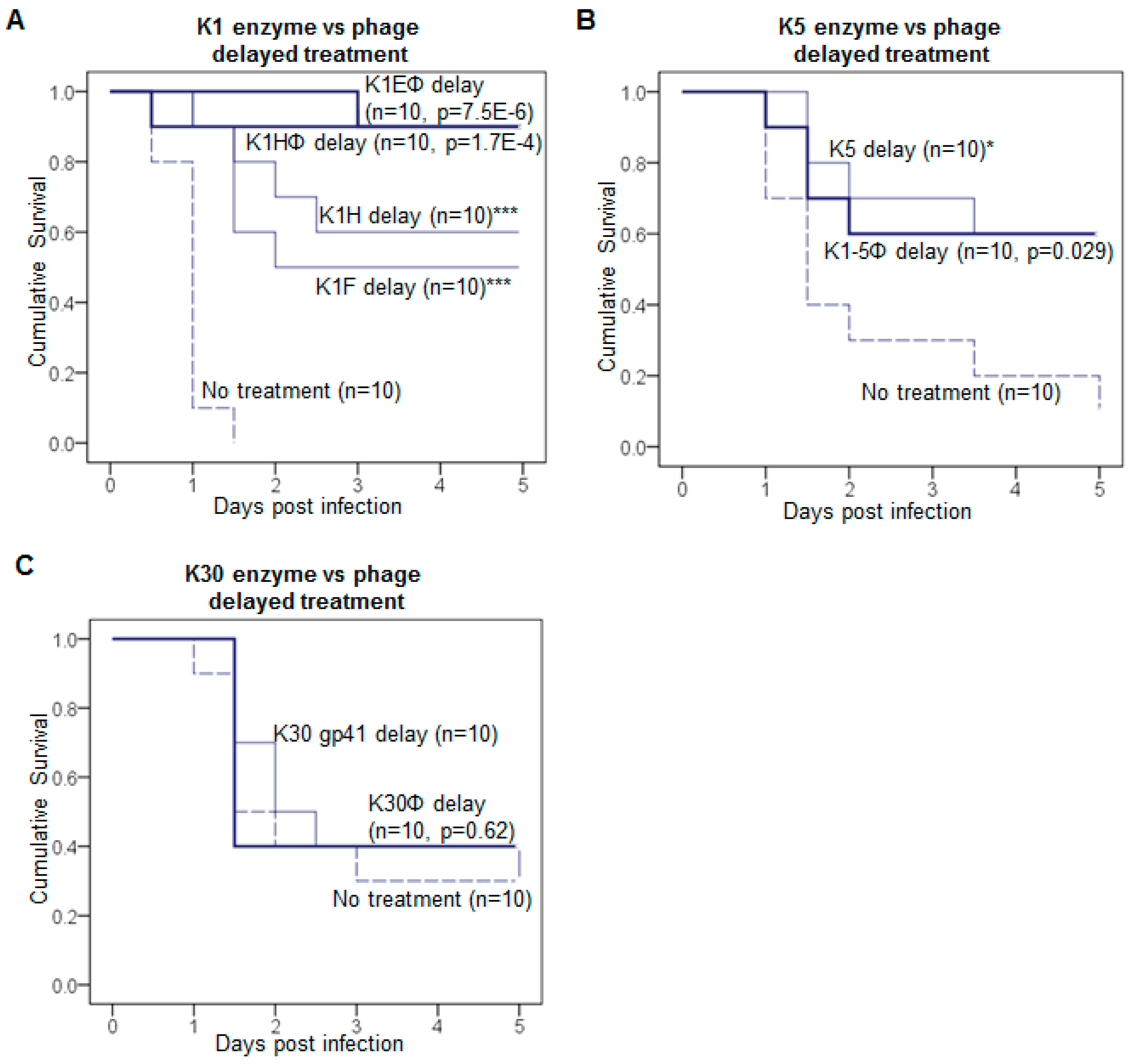
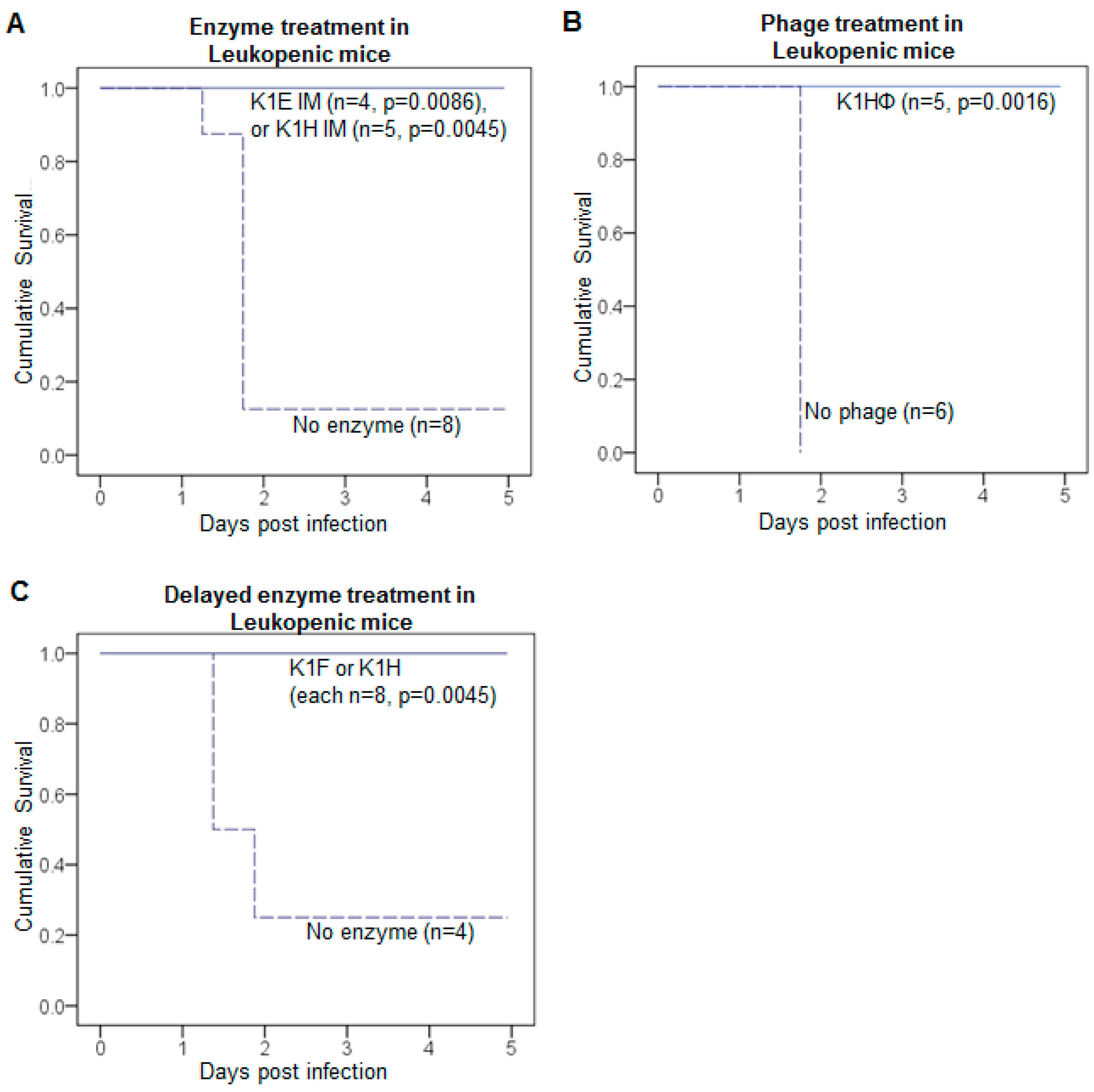
3.1. Delayed Treatment Reduces Efficacy
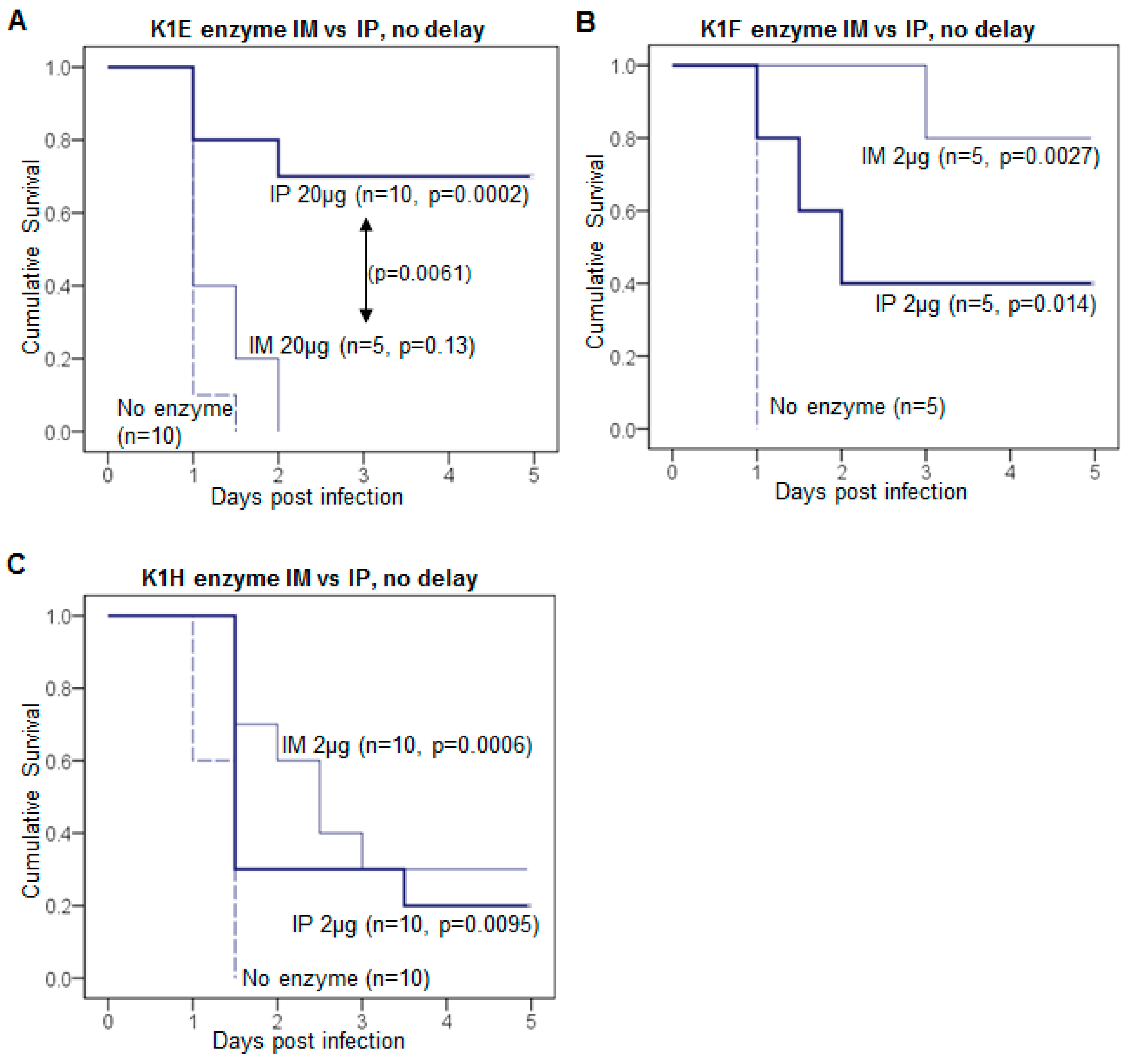
3.2. Efficacy Varies with Route of Administration and Enzyme
3.3. K1: Treatment Is Successful with Leukopenia
3.4. Measuring the Dynamical Impact of Treatment: Resistance Competition Assay
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B. Bacteriophages and phage-derived proteins—Application approaches. Curr. Med. Chem. 2015, 22, 1757–1773. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.J.; Koonjan, S.; Nilsson, A.S. Enhancing whole phage therapy and their derived antimicrobial enzymes through complex formulation. Pharmaceuticals (Basel) 2018, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Donovan, D.M. Antimicrobial bacteriophage-derived proteins and therapeutic applications. Bacteriophage 2015, 5, e1062590. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; García, P.; Mullany, P.; Aminov, R. Editorial: Phage Therapy: Past, Present and Future. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Vandenheuvel, D.; Lavigne, R.; Brussow, H. Bacteriophage Therapy: Advances in Formulation Strategies and Human Clinical Trials. Annu. Rev. Virol. 2015, 2, 599–618. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, D.D.; Wolcott, R.D.; Kuskowski, M.A.; Wolcott, B.M.; Ward, L.S.; Sulakvelidze, A. Bacteriophage therapy of venous leg ulcers in humans: Results of a phase I safety trial. J. Wound Care 2009, 18, 237–238, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.A.; Sultana, S.; Reuteler, G.; Moine, D.; Descombes, P.; Charton, F.; Bourdin, G.; McCallin, S.; Ngom-Bru, C.; Neville, T.; et al. Oral Phage Therapy of Acute Bacterial Diarrhea with Two Coliphage Preparations: A Randomized Trial in Children from Bangladesh. EBioMedicine 2016, 4, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2018. [Google Scholar] [CrossRef]
- Lin, H.; Paff, M.L.; Molineux, I.J.; Bull, J.J. Therapeutic Application of Phage Capsule Depolymerases against K1, K5, and K30 Capsulated E. coli in Mice. Front. Microbiol. 2017, 8, 2257. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Lavigne, R. Breaking barriers: Expansion of the use of endolysins as novel antibacterials against Gram-negative bacteria. Future Microbiol. 2015, 10, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; Sao-Jose, C.; Azeredo, J. Phage-Derived Peptidoglycan Degrading Enzymes: Challenges and Future Prospects for in vivo Therapy. Viruses 2018, 10, 292. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Xia, F.; Jiang, H.; Li, X.; Hu, L.; Gong, P.; Lei, L.; Feng, X.; Sun, C.; Gu, J.; et al. Identification and characterization of HolGH15: The holin of Staphylococcus aureus bacteriophage GH15. J. Gen. Virol. 2016, 97, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Development of Phage Lysins as Novel Therapeutics: A Historical Perspective. Viruses 2018, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.S. The biochemistry and genetics of capsular polysaccharide production in bacteria. Annu. Rev. Microbiol. 1996, 50, 285–315. [Google Scholar] [CrossRef] [PubMed]
- Azeredo, J.; Sutherland, I.W. The use of phages for the removal of infectious biofilms. Curr. Pharm. Biotechnol. 2008, 9, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.; Chahin, L.; Rumbaugh, K. Glycoside Hydrolases Degrade Polymicrobial Bacterial Biofilms in Wounds. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.; Rumbaugh, K.P. Approaches to Dispersing Medical Biofilms. Microorganisms 2017, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, N.; Redpath, M.B.; Luzio, J.P.; Taylor, P.W. Prevention and cure of systemic Escherichia coli K1 infection by modification of the bacterial phenotype. Antimicrob. Agents Chemother. 2004, 48, 1503–1508. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, N.; Redpath, M.B.; Luzio, J.P.; Taylor, P.W. Treatment of experimental Escherichia coli infection with recombinant bacteriophage-derived capsule depolymerase. J. Antimicrob. Chemother. 2005, 56, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Scorpio, A.; Tobery, S.A.; Ribot, W.J.; Friedlander, A.M. Treatment of experimental anthrax with recombinant capsule depolymerase. Antimicrob. Agents Chemother. 2008, 52, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Negus, D.; Vipond, J.; Hatch, G.J.; Rayner, E.L.; Taylor, P.W. Parenteral Administration of Capsule Depolymerase EnvD Prevents Lethal Inhalation Anthrax Infection. Antimicrob. Agents Chemother. 2015, 59, 7687–7692. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Hsieh, P.F.; Huang, Y.T.; Lee, W.C.; Tsai, Y.T.; Su, P.A.; Pan, Y.J.; Hsu, C.R.; Wu, M.C.; Wang, J.T. Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: Implication in typing and treatment. J. Infect. Dis. 2014, 210, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.J.; Lin, T.L.; Lin, Y.T.; Su, P.A.; Chen, C.T.; Hsieh, P.F.; Hsu, C.R.; Chen, C.C.; Hsieh, Y.C.; Wang, J.T. Identification of capsular types in carbapenem-resistant Klebsiella pneumoniae strains by WZC sequencing and implications for capsule depolymerase treatment. Antimicrob. Agents Chemother. 2015, 59, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Majkowska-Skrobek, G.; Latka, A.; Berisio, R.; Maciejewska, B.; Squeglia, F.; Romano, M.; Lavigne, R.; Struve, C.; Drulis-Kawa, Z. Capsule-targeting depolymerase, derived from Klebsiella KP36 phage, as a tool for the development of anti-virulent strategy. Viruses 2016, 8, 324. [Google Scholar] [CrossRef] [PubMed]
- Achtman, M.; Mercer, A.; Kusecek, B.; Pohl, A.; Heuzenroeder, M.; Aaronson, W.; Sutton, A.; Silver, R.P. Six widespread bacterial clones among Escherichia coli K1 isolates. Infect. Immun. 1983, 39, 315–335. [Google Scholar] [PubMed]
- Orskov, I.; Orskov, F.; Jann, B.; Jann, K. Serology, chemistry, and genetics of O and K antigens of Escherichia coli. Bacteriol. Rev. 1977, 41, 667–710. [Google Scholar] [PubMed]
- Vimr, E.R.; Troy, F.A. Regulation of sialic acid metabolism in Escherichia coli: Role of N-acylneuraminate pyruvate-lyase. J. Bacteriol. 1985, 164, 854–860. [Google Scholar] [PubMed]
- Smith, H.W.; Huggins, M.B. The association of the O18, K1 and H7 antigens and the ColV plasmid of a strain of E. coli with virulence and immunogenicity. J. Gen. Microbiol. 1980, 121, 387–400. [Google Scholar] [PubMed]
- Bull, J.J.; Vimr, E.R.; Molineux, I.J. A tale of tails: Sialidase is key to success in a model of phage therapy against K1-capsulated Escherichia coli. Virology 2010, 398, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Scholl, D.; Kieleczawa, J.; Kemp, P.; Rush, J.; Richardson, C.C.; Merril, C.; Adhya, S.; Molineux, I.J. Genomic analysis of bacteriophages SP6 and K1-5, an estranged subgroup of the T7 supergroup. J. Mol. Biol. 2004, 335, 1151–1171. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, C.; Lam, M. Characterisation of coliphage K30, a bacteriophage specific for Escherichia coli capsular serotype K30. FEMS Microbiol. Lett. 1986, 37, 351–355. [Google Scholar] [CrossRef]
- Leiman, P.G.; Battisti, A.J.; Bowman, V.D.; Stummeyer, K.; Muhlenhoff, M.; Gerardy-Schahn, R.; Scholl, D.; Molineux, I.J. The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J. Mol. Biol. 2007, 371, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.J.; Levin, B.R.; DeRouin, T.; Walker, N.; Bloch, C.A. Dynamics of success and failure in phage and antibiotic therapy in experimental infections. BMC Microbiol. 2002, 2, 35. [Google Scholar] [CrossRef]
- Bull, J.J.; Otto, G.; Molineux, I.J. In vivo growth rates are poorly correlated with phage therapy success in a mouse infection model. Antimicrob. Agents Chemother. 2012, 56, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.T.; Neely, J.G.; Paniello, R.C.; Voelker, C.C.; Nussenbaum, B.; Wang, E.W. A practical guide to understanding Kaplan-Meier curves. Otolaryngol. Head Neck Surg. 2010, 143, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 2017, 22, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, S.; Häyrinen, J.; Finne, J. Polyacrylamide gel electrophoresis of the capsular polysaccharides of Escherichia coli K1 and other bacteria. J. Bacteriol. 1988, 170, 2646–2653. [Google Scholar] [CrossRef] [PubMed]
- Muhlenhoff, M.; Stummeyer, K.; Grove, M.; Sauerborn, M.; Gerardy-Schahn, R. Proteolytic processing and oligomerization of bacteriophage-derived endosialidases. J. Biol. Chem. 2003, 278, 12634–12644. [Google Scholar] [CrossRef] [PubMed]
- Gerardy-Schahn, R.; Bethe, A.; Brennecke, T.; Mühlenhoff, M.; Eckhardt, M.; Ziesing, S.; Lottspeich, F.; Frosch, M. Molecular cloning and functional expression of bacteriophage PK1E-encoded endoneuraminidase Endo NE. Mol. Microbiol. 1995, 16, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Luke, D.R.; Brunner, L.J.; Vadiei, K. Bioavailability assessment of cyclosporine in the rat. Influence of route of administration. Drug Metab. Dispos. 1990, 18, 158–162. [Google Scholar] [PubMed]
- Yamamura, Y.; Santa, T.; Kotaki, H.; Uchino, K.; Sawada, Y.; Iga, T. Administration-route dependency of absorption of glycyrrhizin in rats: Intraperitoneal administration dramatically enhanced bioavailability. Biol. Pharm. Bull. 1995, 18, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Kijanka, G.; Prokopowicz, M.; Schellekens, H.; Brinks, V. Influence of aggregation and route of injection on the biodistribution of mouse serum albumin. PLoS ONE 2014, 9, e85281. [Google Scholar] [CrossRef] [PubMed]
- Torres-Barcelo, C. Phage Therapy Faces Evolutionary Challenges. Viruses 2018, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Dennehy, J.J. What Can Phages Tell Us about Host-Pathogen Coevolution? Int. J. Evol. Biol. 2012, 2012, 396165. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.S. Phage therapy—Constraints and possibilities. Ups J. Med. Sci. 2014, 119, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.J.; Vegge, C.S.; Schmerer, M.; Chaudhry, W.N.; Levin, B.R. Phenotypic resistance and the dynamics of bacterial escape from phage control. PLoS ONE 2014, 9, e94690. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W.; Huggins, M.B. Successful treatment of experimental Escherichia coli infections in mice using phage: Its general superiority over antibiotics. J. Gen. Microbiol. 1982, 128, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.; De Sordi, L.; Debarbieux, L. The Diversity of Bacterial Lifestyles Hampers Bacteriophage Tenacity. Viruses 2018, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.J.; Khan Mirzaei, M.; Nilsson, A.S. Adapting Drug Approval Pathways for Bacteriophage-Based Therapeutics. Front. Microbiol. 2016, 7, 1209. [Google Scholar] [CrossRef] [PubMed]
- Hagens, S.; Habel, A.; von Ahsen, U.; von Gabain, A.; Blasi, U. Therapy of experimental pseudomonas infections with a nonreplicating genetically modified phage. Antimicrob. Agents Chemother. 2004, 48, 3817–3822. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Freeman, T.A.; Hilbert, D.W.; Duff, M.; Fuortes, M.; Stapleton, P.P.; Daly, J.M. Lysis-deficient bacteriophage therapy decreases endotoxin and inflammatory mediator release and improves survival in a murine peritonitis model. Surgery 2005, 137, 639–646. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Capsule Depolymerases | Animal Infection Model | References |
|---|---|---|
| EndoE (K1E) | E. coli; neonatal rats | Mushtaq et al., 2004 [21]; 2005 [22] |
| CapD; EnvD | B. anthracis; mice | Scorpio et al., 2008 [23]; Negus et al., 2015 [24] |
| K1-ORF34; K64dep | K. pneumoniae; mice | Lin et al., 2014 [25]; Pan et al., 2015 [26] |
| depoKP36 | K. pneumoniae; moth larvae | Majkowska-Skrobek et al., 2016 [27] |
| K1F, K1H, K5, K30 | E. coli; mice | Lin et al., 2017 [11] |
| Control Mice | Treated Mice 1 | Average RCA |
|---|---|---|
| 0.001, 0.0008, 0.0029 | 0.0052, 0.0049, 0.0047 | 0.30 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, H.; Paff, M.L.; Molineux, I.J.; Bull, J.J. Antibiotic Therapy Using Phage Depolymerases: Robustness Across a Range of Conditions. Viruses 2018, 10, 622. https://doi.org/10.3390/v10110622
Lin H, Paff ML, Molineux IJ, Bull JJ. Antibiotic Therapy Using Phage Depolymerases: Robustness Across a Range of Conditions. Viruses. 2018; 10(11):622. https://doi.org/10.3390/v10110622
Chicago/Turabian StyleLin, Han, Matthew L. Paff, Ian J. Molineux, and James J. Bull. 2018. "Antibiotic Therapy Using Phage Depolymerases: Robustness Across a Range of Conditions" Viruses 10, no. 11: 622. https://doi.org/10.3390/v10110622
APA StyleLin, H., Paff, M. L., Molineux, I. J., & Bull, J. J. (2018). Antibiotic Therapy Using Phage Depolymerases: Robustness Across a Range of Conditions. Viruses, 10(11), 622. https://doi.org/10.3390/v10110622