Interactions between Bacteriophage, Bacteria, and the Mammalian Immune System

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Human Phageome
3. Phages Effects on the Bacterial - Mammalian Host Interface
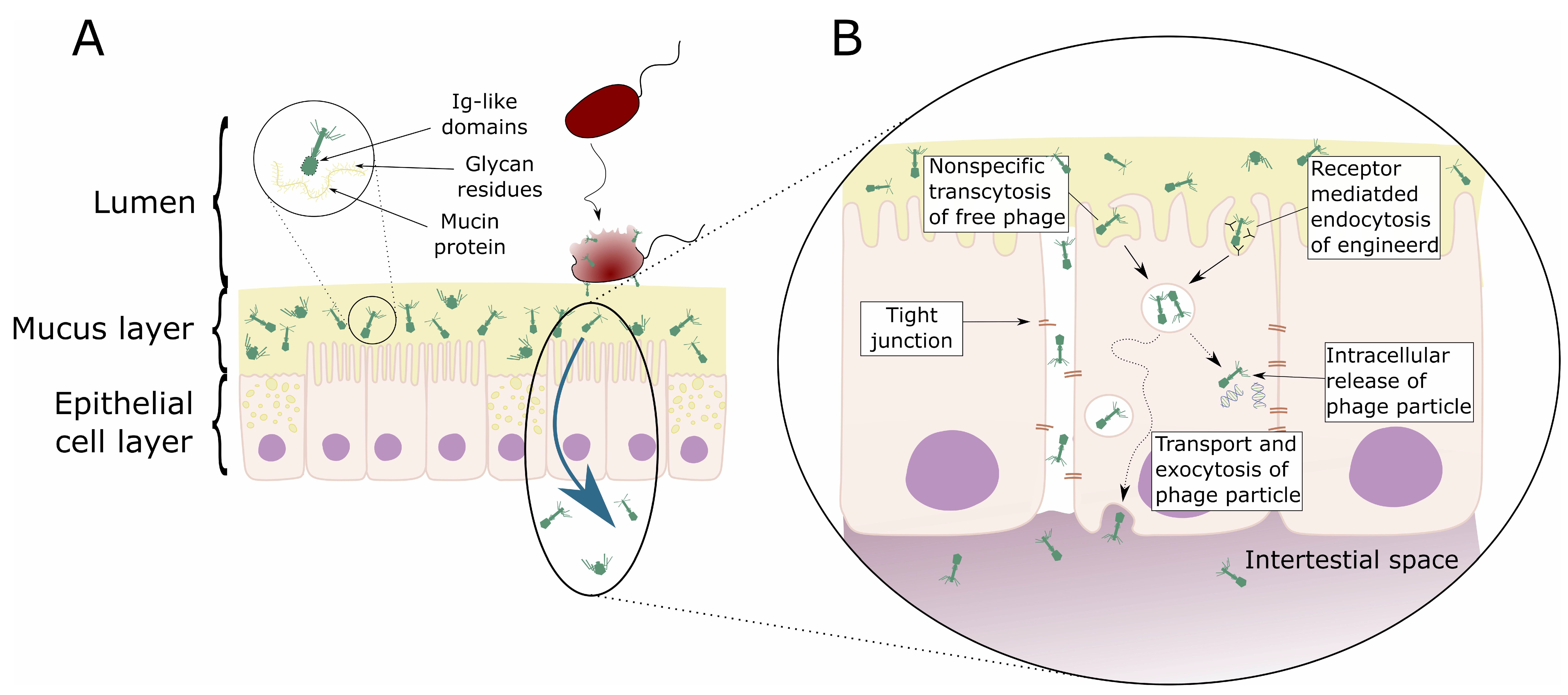
3.1. Phages and Mucosal Tissues
3.2. Phage Transcytosis
4. Cell Perfusion and Access, Interaction with Intracellular Immune Response
5. Phage Innate Immune Response
5.1. Phage Phagocytosis
5.2. Phage Induced Phagocytosis of Bacteria
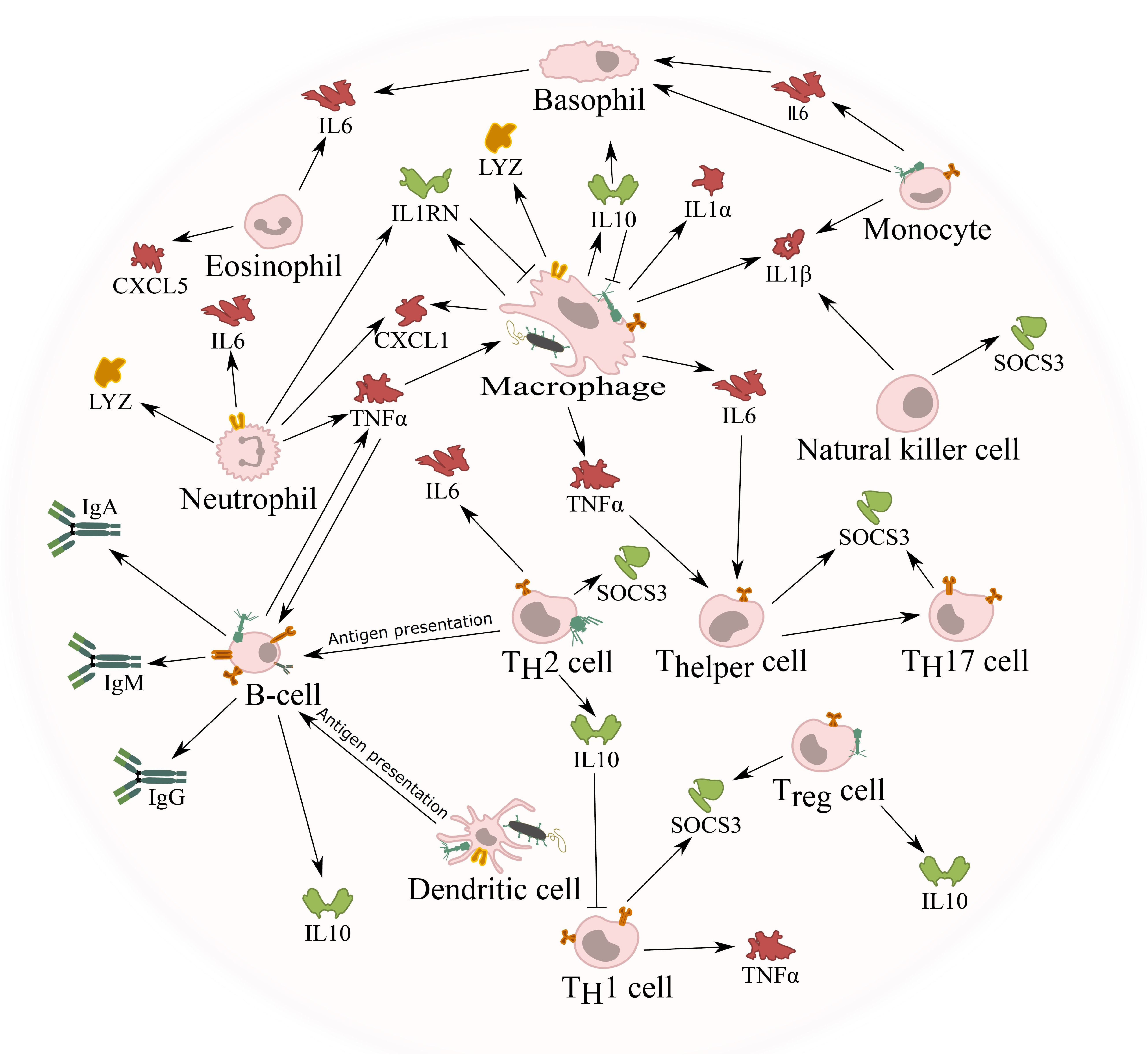
5.3. Cytokine Response against Phages
5.4. Phage Adaptive Immune Response
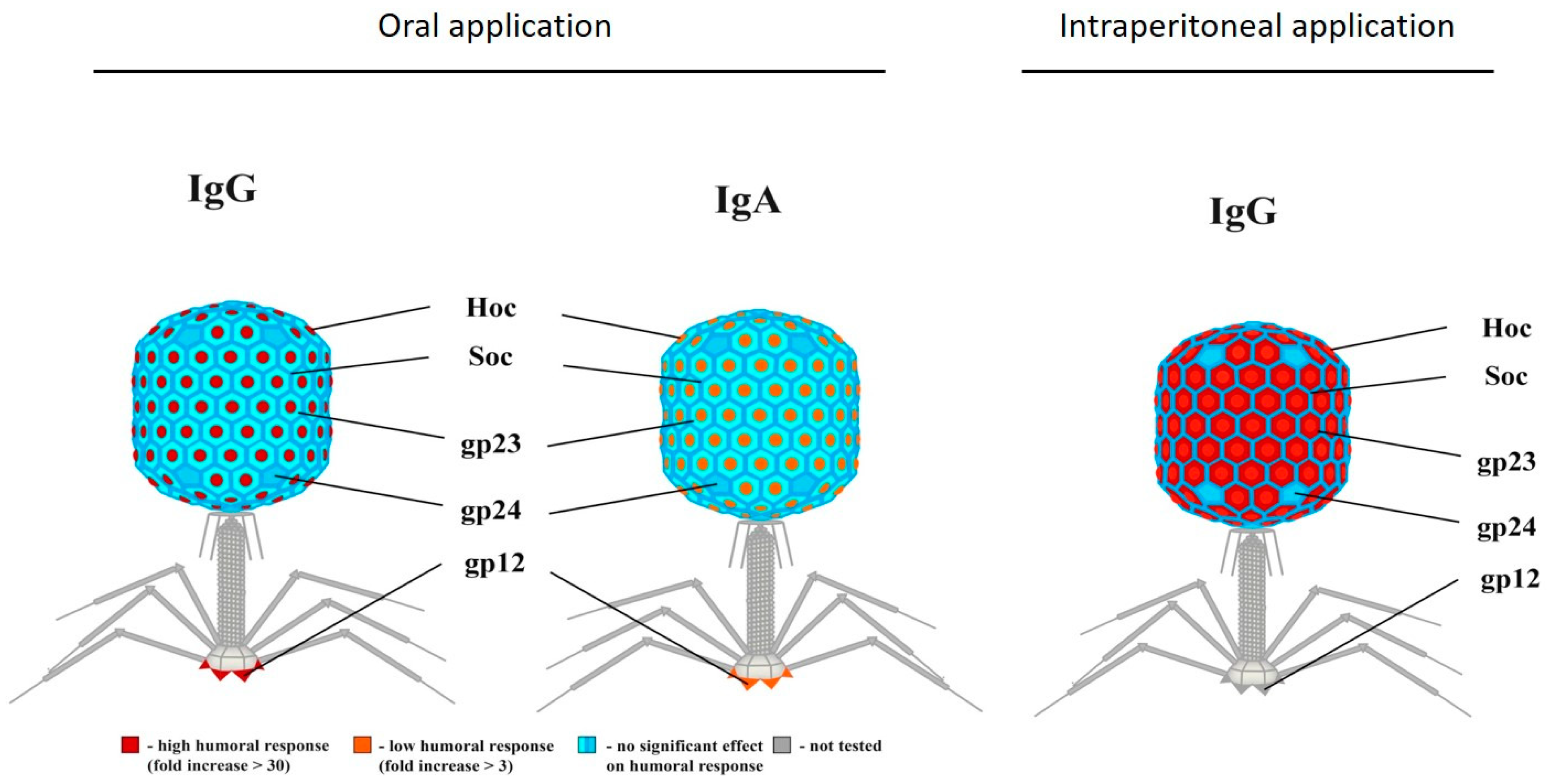
Anti-Phage Antibody Production
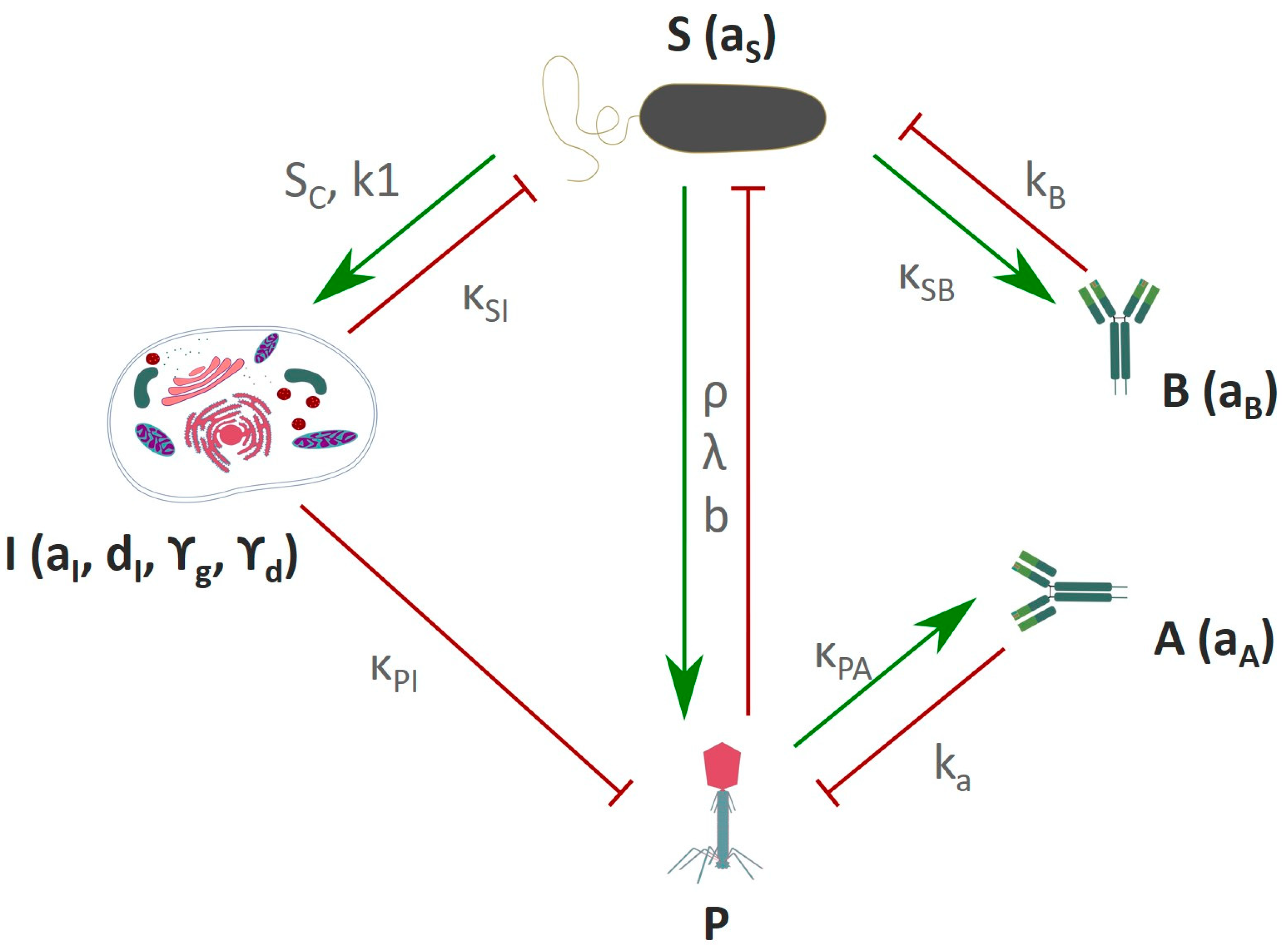
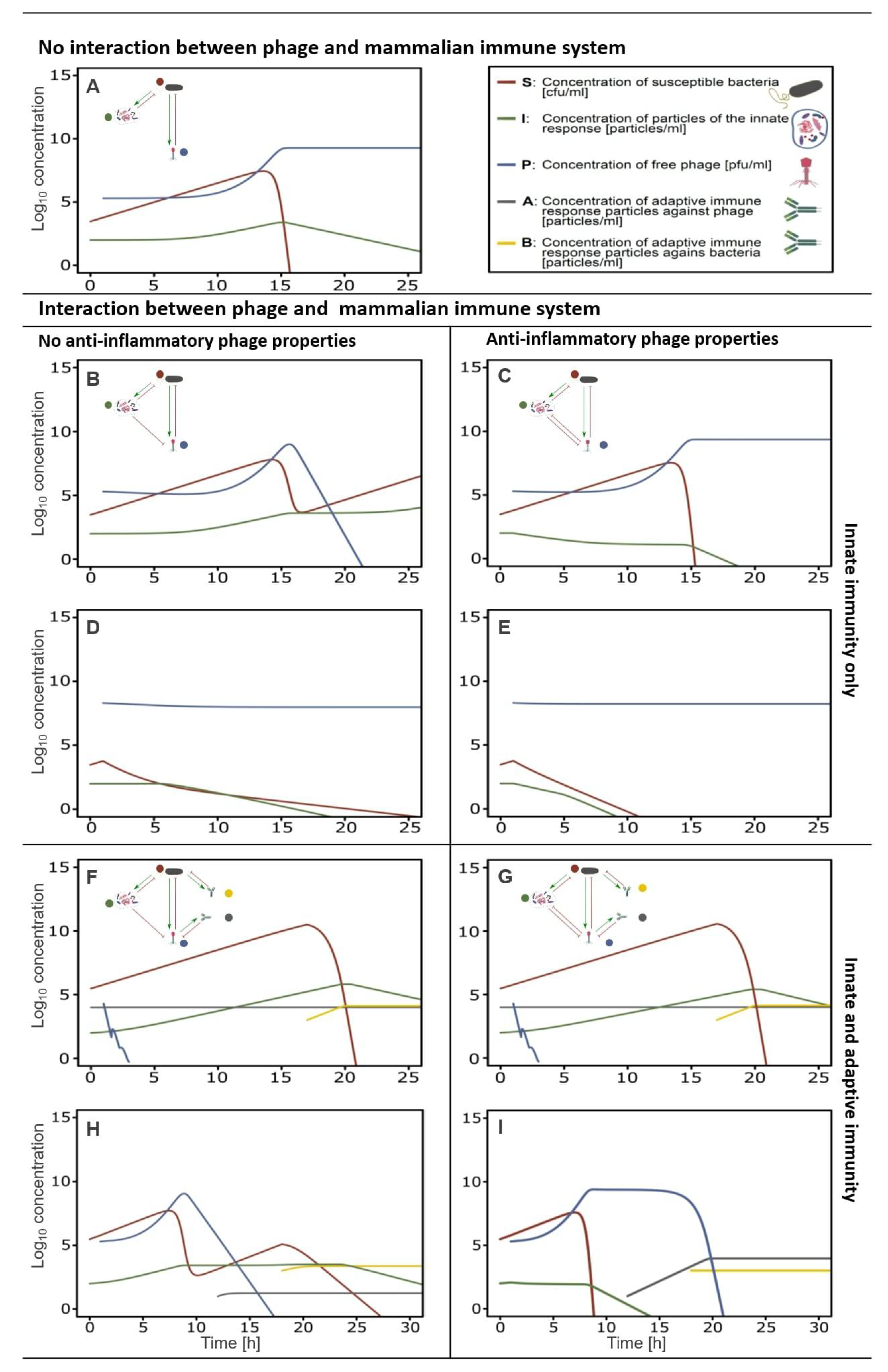
5.5. In-Silico Modeling of the Immune Response Towards Phages
6. Anti-Inflammatory Phage Properties Affect the Outcome of Phage Therapy
7. Relevance of Phage-Mammalian Host Immune Responses
8. Conclusions and Areas of Future Investigation
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Duerkop, B.A.; Hooper, L.V. Resident viruses and their interactions with the immune system. Nat. Immunol. 2013, 14, 654–659. [Google Scholar] [CrossRef]
- Proctor, L.; Sechi, S.; Di Giacomo, N.; Fettweis, J.; Jefferson, K.; Strauss, J., III; Rubens, C.; Brooks, J.; Girerd, P.; Huang, B.; et al. The Integrative Human Microbiome Project: Dynamic Analysis of Microbiome-Host Omics Profiles during Periods of Human Health and Disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Park, M.; Kong, H.H.; Segre, J.A. Temporal Stability of the Human Skin Microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Foulongne, V.; Sauvage, V.; Hebert, C.; Dereure, O.; Cheval, J.; Gouilh, M.A.; Pariente, K.; Segondy, M.; Burguière, A.; Manuguerra, J.C.; et al. Human skin Microbiota: High diversity of DNA viruses identified on the human skin by high throughput sequencing. PLoS ONE 2012, 7, e38499. [Google Scholar] [CrossRef]
- Pride, D.T.; Salzman, J.; Haynes, M.; Rohwer, F.; Davis-Long, C.; White, R.A.; Loomer, P.; Armitage, G.C.; Relman, D.A. Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J. 2012, 6, 915–926. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Zhao, F. Phage-bacteria interaction network in human oral microbiome. Environ. Microbiol. 2016, 18, 2143–2158. [Google Scholar] [CrossRef]
- Edlund, A.; Santiago-Rodriguez, T.M.; Boehm, T.K.; Pride, D.T. Bacteriophage and their potential roles in the human oral cavity. J. Oral Microbiol. 2015, 7, 1–12. [Google Scholar] [CrossRef]
- Dickson, R.P.; Huffnagle, G.B. The Lung Microbiome: New Principles for Respiratory Bacteriology in Health and Disease. PLoS Pathog. 2015, 11, e1004923. [Google Scholar] [CrossRef]
- Lim, Y.W.; Schmieder, R.; Haynes, M.; Willner, D.; Furlan, M.; Youle, M.; Abbott, K.; Edwards, R.; Evangelista, J.; Conrad, D.; et al. Metagenomics and metatranscriptomics: Windows on CF-associated viral and microbial communities. J. Cyst. Fibros. 2013, 12, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Willner, D.; Haynes, M.R.; Furlan, M.; Hanson, N.; Kirby, B.; Lim, Y.W.; Rainey, P.B.; Schmieder, R.; Youle, M.; Conrad, D.; et al. Case studies of the spatial heterogeneity of DNA viruses in the cystic fibrosis lung. Am. J. Respir. Cell Mol. Biol. 2012, 46, 127–131. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Ly, M.; Bonilla, N.; Pride, D.T. The human urine virome in association with urinary tract infections. Front. Microbiol. 2015, 6, 14. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. 2004, 68. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Method for discovering novel DNA viruses in blood using viral particle selection and shotgun sequencing. Biotechniques 2005, 39, 729–736. [Google Scholar] [CrossRef]
- Dinakaran, V.; Rathinavel, A.; Pushpanathan, M.; Sivakumar, R.; Gunasekaran, P.; Rajendhran, J. Elevated levels of circulating DNA in cardiovascular disease patients: Metagenomic profiling of microbiome in the circulation. PLoS ONE 2014, 9, e105221. [Google Scholar] [CrossRef] [PubMed]
- Li, S.K.; Leung, R.K.-K.; Guo, H.X.; Wei, J.F.; Wang, J.H.; Kwong, K.T.; Lee, S.S.; Zhang, C.; Tsui, S.K.-W. Detection and identification of plasma bacterial and viral elements in HIV/AIDS patients in comparison to healthy adults. Clin. Microbiol. Infect. 2012, 18, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Xie, C.; Kirkness, E.; Biggs, W.; Wong, E.; Turpaz, Y.; Bloom, K.; Delwart, E.; Nelson, K.E.; Venter, J.C.; et al. The blood DNA virome in 8000 humans. PLoS Pathog. 2017, 13, e1006292. [Google Scholar] [CrossRef] [PubMed]
- Van Belleghem, J.D.; Clement, F.; Merabishvili, M.; Lavigne, R.; Vaneechoutte, M. Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci. Rep. 2017, 7, 8004. [Google Scholar] [CrossRef] [PubMed]
- Majewska, J.; Beta, W.; Lecion, D.; Hodyra-Stefaniak, K.; Kłopot, A.; Kaźmierczak, Z.; Miernikiewicz, P.; Piotrowicz, A.; Ciekot, J.; Owczarek, B.; et al. Oral application of T4 phage induces weak antibody production in the gut and in the blood. Viruses 2015, 7, 4783–4799. [Google Scholar] [CrossRef] [PubMed]
- Miernikiewicz, P.; Dąbrowska, K.; Piotrowicz, A.; Owczarek, B.; Wojas-Turek, J.; Kicielińska, J.; Rossowska, J.; Pajtasz-Piasecka, E.; Hodyra, K.; Macegoniuk, K.; et al. T4 phage and its head surface proteins do not stimulate inflammatory mediator production. PLoS ONE 2013, 8, e71036. [Google Scholar] [CrossRef] [PubMed]
- Hodyra-Stefaniak, K.; Miernikiewicz, P.; Drapała, J.; Drab, M.; Jonczyk-Matysiak, E.; Lecion, D.; Kazmierczak, Z.; Beta, W.; Majewska, J.; Harhala, M.; et al. Mammalian Host-Versus-Phage immune response determines phage fate in vivo. Sci. Rep. 2015, 5, 3–8. [Google Scholar] [CrossRef]
- Handley, S.A.; Thackray, L.B.; Zhao, G.; Presti, R.; Miller, A.D.; Droit, L.; Abbink, P.; Maxfield, L.F.; Kambal, A.; Duan, E.; et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 2012, 151, 253–266. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, L.; Breitbart, M.; Mobberley, J.; Long, A.; Haynes, M.; Rohwer, F.; Paul, J.H. Metagenomic analysis of lysogeny in Tampa Bay: Implications for prophage gene expression. PLoS ONE 2008, 3, e3263. [Google Scholar] [CrossRef]
- Scarpellini, E.; Ianiro, G.; Attili, F.; Bassanelli, C.; De Santis, A.; Gasbarrini, A. The human gut microbiota and virome: Potential therapeutic implications. Dig. Liver Dis. 2015, 47, 1007–1012. [Google Scholar] [CrossRef]
- Kim, M.-S.; Park, E.-J.; Roh, S.W.; Bae, J.-W. Diversity and abundance of single-stranded DNA viruses in human feces. Appl. Environ. Microbiol. 2011, 77, 8062–8070. [Google Scholar] [CrossRef]
- Reyes, A.; Wu, M.; McNulty, N.P.; Rohwer, F.L.; Gordon, J.I. Gnotobiotic mouse model of phage–bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. USA 2013, 110, 20236–20241. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Haynes, M.; Kelley, S.; Angly, F.; Edwards, R.A.; Felts, B.; Mahaffy, J.M.; Mueller, J.; Nulton, J.; Rayhawk, S.; et al. Viral diversity and dynamics in an infant gut. Res. Microbiol. 2008, 159, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.B.; Rodriguez-Brito, B.; Pašić, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009, 7, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Enault, F.; Briet, A.; Bouteille, L.; Roux, S.; Sullivan, M.B.; Petit, M.-A. Phages rarely encode antibiotic resistance genes: A cautionary tale for virome analyses. ISME J. 2016, 053025. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Brandt, J.M.; Verma, D.J.; Lindberg, A.A. Molecular characterization of the O-acetyl transferase gene of converting bacteriophage SF6 that adds group antigen 6 to Shigella flexneri. Mol. Microbiol. 1991, 5, 71–75. [Google Scholar] [CrossRef]
- Pieraerts, C.; Martin, V.; Jichlinski, P.; Nardelli-Haefliger, D.; Derré, L. Detection of functional antigen-specific T cells from urine of non-muscle invasive bladder cancer patients. Oncoimmunology 2012, 1, 694–698. [Google Scholar] [CrossRef]
- Brüssow, H. Bacteriophage-host interaction: From splendid isolation into a messy reality. Curr. Opin. Microbiol. 2013, 16, 500–506. [Google Scholar] [CrossRef]
- Davies, M.R.; Broadbent, S.E.; Harris, S.R.; Thomson, N.R.; van der Woude, M.W. Horizontally Acquired Glycosyltransferase Operons Drive Salmonellae Lipopolysaccharide Diversity. PLoS Genet. 2013, 9, e1003568. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Frutos R de, L.; Manel, N.; Yoshinaga, K.; Rifkin, D.B.; Sartor, R.B.; Finlay, B.B.; Littman, D.R. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 2008, 4, 337–349. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Cui, H.L.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Przybylski, M.; Borysowski, J.; Jakubowska-Zahorska, R.; Weber-Dąbrowska, B.; Górski, A. T4 bacteriophage-mediated inhibition of adsorption and replication of human adenovirus in vitro. Future Microbiol. 2015, 10, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, L.G.; Saez-Rodriguez, J.; Cosgrove, B.D.; Lauffenburger, D.A.; Sorger, P.K. Networks Inferred from Biochemical Data Reveal Profound Differences in Toll-like Receptor and Inflammatory Signaling between Normal and Transformed Hepatocytes. Mol. Cell Proteom. 2010, 9, 1849–1865. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Beutler, B. Intracellular Toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Cai, Z.; Sanchez, A.; Zhang, T.; Wen, S.; Wang, J.; Yang, J.; Fu, S.; Zhang, D. A novel toll-like receptor that recognizes vesicular stomatitis virus. J. Biol. Chem. 2011, 286, 4517–4524. [Google Scholar] [CrossRef] [PubMed]
- Focà, A.; Liberto, M.C.; Quirino, A.; Marascio, N.; Zicca, E.; Pavia, G. Gut inflammation and immunity: What is the role of the human gut virome? Med. Inflamm. 2015, 2015, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Foxman, E.F.; Iwasaki, A. Genome-virome interactions: Examining the role of common viral infections in complex disease. Nat. Rev. Microbiol. 2011, 9, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Farrar, M.A.; Schreiber, R.D. The Molecular Cell Biology of Interferon-gamma and its Receptor. Annu. Rev. Immunol. 1993, 11, 571–611. [Google Scholar] [CrossRef] [PubMed]
- Linden, S.K.; Sutton, P.; Karlsson, N.G.; Korolik, V.; McGuckin, M.A. Mucins in the mucosal barrier to infection. Mucosal. Immunol. 2008, 1, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C.; et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. PNAS 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, J.L. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 2005, 307, 1955–1959. [Google Scholar] [CrossRef] [PubMed]
- Schluter, J.; Foster, K.R. The evolution of mutualism in gut microbiota via host epithelial selection. PLoS Biol. 2012, 10, e1001424. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Xu, J.; Falk, P.G.; Midtvedt, T.; Gordon, J.I. A molecular sensor that allows a gut commensal to control its nutrient foundation in a competitive ecosystem. Proc. Natl. Acad. Sci. USA 1999, 96, 9833–9838. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.C.; Chiang, H.C.; Gordon, J.I. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 2008, 4, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.K.; Lan, F.; Kristensen, C.S.; Hobolth, P.; Molin, S.; Krogfelt, K.A. Spatial distribution of Escherichia coli in the mouse large intestine inferred from rRNA in situ hybridization. Infect. Immun. 1994, 62, 5191–5194. [Google Scholar] [PubMed]
- Koropatkin, N.M.; Cameron, E.A.; Martens, E.C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012, 10, 323–335. [Google Scholar] [CrossRef]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjöberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–83. [Google Scholar] [CrossRef]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The Antibacterial Lectin RegIII Promotes the Spatial Segregation of Microbiota and Host in the Intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef]
- Gill, D.J.; Tham, K.M.; Chia, J.; Wang, S.C.; Steentoft, C.; Clausen, H.; Bard-Chapeau, E.A.; Bard, F.A. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc. Natl. Acad. Sci. USA 2013, 110, E3152–E3161. [Google Scholar] [CrossRef]
- Jentoft, N. Why are proteins O-glycosylated? Trends Biochem. Sci. 1990, 15, 291–294. [Google Scholar] [CrossRef]
- Schulz, B.L.; Sloane, A.J.; Robinson, L.J.; Prasad, S.S.; Lindner, R.A.; Robinson, M.; Bye, P.T.; Nielson, D.W.; Harry, J.L.; Packer, N.H.; et al. Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology 2007, 17, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef]
- Nguyen-Kim, H.; Bettarel, Y.; Bouvier, T.; Bouvier, C.; Doan-Nhu, H.; Nguyen-Ngoc, L.; Nguyen-Thanh, T.; Tran-Quang, H.; Brune, J. Coral mucus is a hot spot for viral infections. Appl. Environ. Microbiol. 2015, 81, 5773–5783. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Kim, H.; Bouvier, T.; Bouvier, C.; Doan-Nhu, H.; Nguyen-Ngoc, L.; Rochelle-Newall, E.; Baudoux, A.C.; Desnues, C.; Reynaud, S.; Ferrier-Pages, C.; et al. High occurrence of viruses in the mucus layer of scleractinian corals. Environ. Microbiol. Rep. 2014, 6, 675–682. [Google Scholar] [CrossRef]
- Bork, P.; Holm, L.; Sander, C. The Immunoglobulin Fold. Structural classification, sequence patterns and common core. J. Mol. Biol. 1994, 242, 309–320. [Google Scholar] [CrossRef]
- Halaby, D.M.; Mornon, J.P.E. The immunoglobulin superfamily: An insight on its tissular, species, and functional diversity. J. Mol. Evol. 1998, 46, 389–400. [Google Scholar] [CrossRef]
- Fraser, J.S.; Maxwell, K.L.; Davidson, A.R. Immunoglobulin-like domains on bacteriophage: Weapons of modest damage? Curr. Opin. Microbiol. 2007, 10, 382–387. [Google Scholar] [CrossRef]
- Fraser, J.S.; Yu, Z.; Maxwell, K.L.; Davidson, A.R. Ig-like domains on bacteriophages: A tale of promiscuity and deceit. J. Mol. Biol. 2006, 359, 496–507. [Google Scholar] [CrossRef]
- McMahon, S.A.; Miller, J.L.; Lawton, J.A.; Kerkow, D.E.; Hodes, A.; Marti-Renom, M.A.; Doulatov, S.; Narayanan, E.; Sali, A.; Miller, J.F.; et al. The C-type lectin fold as an evolutionary solution for massive sequence variation. Nat. Struct. Mol. Biol. 2005, 12, 886–892. [Google Scholar] [CrossRef]
- Barr, J.J.; Auro, R.; Sam-Soon, N.; Kassegne, S.; Peters, G.; Bonilla, N.; Hatay, M.; Mourtada, S.; Bailey, B.; Youle, M.; et al. Subdiffusive motion of bacteriophage in mucosal surfaces increases the frequency of bacterial encounters. Proc. Natl. Acad. Sci. USA 2015, 112, 13675–13680. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J. A bacteriophages journey through the human body. Immunol. Rev. 2017, 279, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Bille, E.; Meyer, J.; Jamet, A.; Euphrasie, D.; Barnier, J.P.; Brissac, T.; Larsen, A.; Pelissier, P.; Nassif, X. A virulence-associated filamentous bacteriophage of Neisseria meningitidis increases host-cell colonisation. PLoS Pathog. 2017, 13, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Guarner, F.; Malagelada, J.R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Wiest, R.; Garcia-Tsao, G. Bacterial translocation (BT) in cirrhosis. Hepatology 2005, 41, 422–433. [Google Scholar] [CrossRef]
- Costantini, T.W.; Putnam, J.G.; Sawada, R.; Baird, A.; Loomis, W.H.; Eliceiri, B.P.; Bansal, V.; Coimbra, R. Targeting the gut barrier: Identification of a homing peptide sequence for delivery into the injured intestinal epithelial cell. Surgery 2009, 146, 206–212. [Google Scholar] [CrossRef]
- Ivanenkov, V.V.; Felici, F.; Menon, A.G. Uptake and intracellular fate of phage display vectors in mammalian cells. Biochim. Biophys. Acta Mol. Cell Res. 1999, 1448, 450–462. [Google Scholar] [CrossRef]
- Keller, R.; Engley, F.B. Fate of Bacteriophage Particles Introduced into Mice by Various Routes. Exp. Biol. Med. 1958, 98, 577–580. [Google Scholar] [CrossRef]
- Wolochow, H.; Hildebrand, G.J.; Lamanna, C. Translocation of microorganisms across the intestinal wall of the rat: Effect of microbial size and concentration. J. Infect. Dis. 1966, 116, 523–528. [Google Scholar] [CrossRef]
- Reynaud, A.; Cloastre, L.; Bernard, J.; Laveran, H.; Ackermann, H.W.; Licois, D.; Joly, B. Characteristics and diffusion in the rabbit of a phage for Escherichia coli 0103. Attempts to use this phage for therapy. Vet. Microbiol. 1992, 30, 203–212. [Google Scholar] [CrossRef]
- Jaiswal, A.; Koley, H.; Mitra, S.; Saha, D.R.; Sarkar, B. Comparative analysis of different oral approaches to treat Vibrio cholerae infection in adult mice. Int. J. Med. Microbiol. 2014, 304, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.W.; Shin, T.H.; Kim, J.H.; Shin, S.P.; Han, J.E.; Heo, G.J.; De Zoysa, M.; Shin, G.W.; Chai, J.Y.; Park, S.C. Bacteriophage therapy of a Vibrio parahaemolyticus infection caused by a multiple-antibiotic-resistant O3:K6 pandemic clinical strain. J. Infect. Dis. 2014, 210, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Duerr, D.M.; White, S.J.; Schluesener, H.J. Identification of peptide sequences that induce the transport of phage across the gastrointestinal mucosal barrier. J. Virol. Methods 2004, 116, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Bruttin, A.; Brüssow, H. Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Denou, E.; Bruttin, A.; Barretto, C.; Ngom-Bru, C.; Brüssow, H.; Zuber, S. T4 phages against Escherichia coli diarrhea: Potential and problems. Virology 2009, 388, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.; Sereno, R.; Nicolau, A.; Azeredo, J. The influence of the mode of administration in the dissemination of three coliphages in chickens. Poult. Sci. 2009, 88, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Letarova, M.; Strelkova, D.; Nevolina, S.; Letarov, A. A test for the “physiological phagemia” hypothesis—Natural intestinal coliphages do not penetrate to the blood in horses. Folia Microbiol. (Praha) 2012, 57, 81–83. [Google Scholar] [CrossRef]
- McCallin, S.; Alam Sarker, S.; Barretto, C.; Sultana, S.; Berger, B.; Huq, S.; Krause, L.; Bibiloni, R.; Schmitt, B.; Reuteler, G.; et al. Safety analysis of a Russian phage cocktail: From MetaGenomic analysis to oral application in healthy human subjects. Virology 2013, 443, 187–196. [Google Scholar] [CrossRef]
- Górski, A.; Ważna, E.; Weber-Dąbrowska, B.; Dąbrowska, K.; Switała-Jelen, K.; Miedzybrodzki, R.; Wazna, E.; Weber-Da̧browska, B.; Da̧browska, K.; Świtała-Jeleń, K.; et al. Bacteriophage translocation. FEMS Immunol. Med. Microbiol. 2006, 46, 313–319. [Google Scholar] [CrossRef]
- Weiss, M.; Denou, E.; Bruttin, A.; Serra-Moreno, R.; Dillmann, M.L.; Brüssow, H. In vivo replication of T4 and T7 bacteriophages in germ-free mice colonized with Escherichia coli. Virology 2009, 393, 16–23. [Google Scholar] [CrossRef]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, K.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage Transcytosis Provides a Mechanism To Cross Epithelial Cell Layers. MBio 2017, 8, e01874-17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, L.; Wei, R.; Gao, Q.; He, T.; Xu, C.; Liu, X.; Wang, R. Intracellular Staphylococcus aureus control by virulent bacteriophages within MAC-T bovine mammary epithelial cells. Antimicrob. Agents Chemother. 2017, 61, AAC.01990-16. [Google Scholar] [CrossRef] [PubMed]
- Sharp, R. Bacteriophages: Biology and history. J. Chem. Technol. Biotechnol. 2011, 667–672. [Google Scholar] [CrossRef]
- Di Giovine, M.; Salone, B.; Martina, Y.; Amati, V.; Zambruno, G.; Cundari, E.; Failla, C.M.; Saggio, I. Binding properties, cell delivery, and gene transfer of adenoviral penton base displaying bacteriophage. Virology 2001, 282, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Boulifard, D.A.; Labouvie, E. Gene Transfer into Mammalian Cells Using Targeted Filamentous Bacteriophage. Int. J. Psychol. 2009, 19, 1–20. [Google Scholar] [CrossRef]
- Poul, M.A.; Marks, J.D. Targeted gene delivery to mammalian cells by filamentous bacteriophage. J. Mol. Biol. 1999, 288, 203–211. [Google Scholar] [CrossRef]
- Lengeling, A.; Mahajan, A.; Gally, D. Bacteriophages as pathogens and immune modulators? MBio 2013. [Google Scholar] [CrossRef]
- Lehti, T.A.; Pajunen, M.I.; Skog, M.S.; Finne, J. Internalization of a polysialic acid-binding Escherichia coli bacteriophage into eukaryotic neuroblastoma cells. Nat. Commun. 2017, 8, 1915. [Google Scholar] [CrossRef]
- Tam, J.C.H.; Jacques, D.A. Intracellular immunity: Finding the enemy within-how cells recognize and respond to intracellular pathogens. J. Leukoc. Biol. 2014, 96, 233–244. [Google Scholar] [CrossRef]
- Barfoot, R.; Denham, S.; Gyure, L.A.; Hall, J.G.; Hobbs, S.M.; Jackson, L.E.; Robertson, D. Some properties of dendritic macrophages from peripheral lymph. Immunology 1989, 68, 233–239. [Google Scholar]
- Wenger, S.L.; Turner, J.H.; Petricciani, J.C. The cytogenetic, proliferative and viability effects of four bacteriophages on human lymphocytes. In Vitro 1978, 14, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Aronow, R.; Danon, D. Electron microscopy of in vitro endocytosis of T2 phage by cells from rabbit peritoneal exudate. J. Exp. Med. 1964, 120, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Nungester, W.J.; Watrous, R.M. Accumulation of Bacteriophage in Spleen and Liver Following Its Intravenous Inoculation. Exp. Biol. Med. 1934, 31, 901–905. [Google Scholar] [CrossRef]
- Geier, M.R.; Trigg, M.E.; Merril, C.R. Fate of bacteriophage lamba in non-immune germ-free mice. Nature 1973, 246, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, S.; Ghosh, S.N. Localization of cholera bacteriophage after intravenous injection. Ann. Biochem. Exp. Med. 1962, 22, 73–76. [Google Scholar] [PubMed]
- Inchley, C.J. The activity of mouse Kupffer cells following intravenous injection of T4 bacteriophage. Clin Exp. Immunol 1969, 5, 173–187. [Google Scholar]
- Cerveny, K.E.; DePaola, A.; Duckworth, D.H.; Gulig, P.A. Phage therapy of local and systemic disease caused by Vibrio vulnificus in iron-dextran-treated mice. Infect. Immun. 2002, 70, 6251–6262. [Google Scholar] [CrossRef]
- Tiwari, B.R.; Kim, S.; Rahman, M.; Kim, J. Antibacterial efficacy of lytic Pseudomonas bacteriophage in normal and neutropenic mice models. J. Microbiol. 2011, 49, 994–999. [Google Scholar] [CrossRef]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Santo, P.D.; Weitz, J.S.; Debarbieux, L.; Unit, I.I. Immunophage synergy is essential for eradicating pathogens that provoke acute respiratory infections. Cell Host Microbe 2017, 22, 38–47. [Google Scholar] [CrossRef]
- Pincus, N.B.; Reckhow, J.D.; Saleem, D.; Jammeh, M.L.; Datta, S.K.; Myles, I.A. Strain specific phage treatment for Staphylococcus aureus infection is influenced by host immunity and site of infection. PLoS ONE 2015, 10, e0124280. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.Y.; Weitz, J.S. Modeling the synergistic elimination of bacteria by phage and the innate immune system. J. Theor. Biol. 2017, 429, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Weber-Dąbrowska, B.; Zimecki, M.; Mulczyk, M. Effective phage therapy is associated with normalization of cytokine production by blood cell cultures. Arch. Immunol. Ther. Exp. 2000, 48, 31–37. [Google Scholar]
- Zhang, L.; Hou, X.; Sun, L.; He, T.; Wei, R.; Pang, M.; Wang, R. Staphylococcus aureus Bacteriophage Suppresses LPS-Induced Inflammation in MAC-T Bovine Mammary Epithelial Cells. Front. Microbiol. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Park, K.; Cha, K.E.; Myung, H. Observation of inflammatory responses in mice orally fed with bacteriophage T7. J. Appl. Microbiol. 2014, 117, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Harjai, K.; Chhibber, S. Bacteriophage-aided intracellular killing of engulfed methicillin-resistant Staphylococcus aureus (MRSA) by murine macrophages. Appl. Microbiol. Biotechnol. 2014, 98, 4653–4661. [Google Scholar] [CrossRef] [PubMed]
- Górski, A.; Międzybrodzki, R.; Borysowski, J.; Dąbrowska, K.; Wierzbicki, P.; Ohams, M.; Korczak-Kowalska, G.; Olszowska-Zaremba, N.; Łusiak-Szelachowska, M.; Kłak, M.; et al. Phage as a modulator of immune responses: Practical implications for phage therapy. Adv. Virus Res. 2012, 83, 41–71. [Google Scholar] [CrossRef]
- Jończyk-Matysiak, E.; Łusiak-Szelachowska, M.; Kłak, M.; Bubak, B.; Międzybrodzki, R.; Weber-Dąbrowska, B.; Zaczek, M.; Fortuna, W.; Rogóz, P.; Letkiewicz, S.; et al. The effect of bacteriophage preparations on intracellular killing of bacteria by phagocytes. J. Immunol. Res. 2015, 2015. [Google Scholar] [CrossRef]
- Przerwa, A.; Zimecki, M.; Świtała-Jeleń, K.; Dąbrowska, K.; Krawczyk, E.; Łuczak, M.; Weber-Dąbrowska, B.; Syper, D.; Miȩdzybrodzki, R.; Górski, A. Effects of bacteriophages on free radical production and phagocytic functions. Med. Microbiol. Immunol. 2006, 195, 143–150. [Google Scholar] [CrossRef]
- Miedzybrodzki, R.; Switala-Jelen, K.; Fortuna, W.; Weber-Dąbrowska, B.; Przerwa, A.; Lusiak-Szelachowska, M.; Dąbrowska, K.; Kurzepa, A.; Boratynski, J.; Syper, D.; et al. Bacteriophage preparation inhibition of reactive oxygen species generation by endotoxin-stimulated polymorphonuclear leukocytes. Virus Res. 2008, 131, 233–242. [Google Scholar] [CrossRef]
- Miernikiewicz, P.; Klopot, A.; Soluch, R.; Szkuta, P.; Keska, W.; Hodyra-Stefaniak, K.; Konopka, A.; Nowak, M.; Lecion, D.; Kazmierczak, Z.; et al. T4 phage tail Adhesin Gp12 counteracts LPS-induced inflammation In Vivo. Front. Microbiol. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Górski, A.; Nowaczyk, M.; Weber-Dabrowska, B.; Kniotek, M.; Boratynski, J.; Ahmed, A.; Dabrowska, K.; Wierzbicki, P.; Switala-Jelen, K.; Opolski, A. New insights into the possible role of bacteriophages in transplantation. Transplant. Proc. 2003, 35, 2372–2373. [Google Scholar] [CrossRef]
- Górski, A.; Miedzybrodzki, R.; Weber-Dabrowska, B.; Fortuna, W.; Letkiewicz, S.; Rogóz, P.; Jończyk-Matysiak, E.; Dabrowska, K.; Majewska, J.; Borysowski, J. Phage therapy: Combating infections with potential for evolving from merely a treatment for complications to targeting diseases. Front. Microbiol. 2016, 7, 1515. [Google Scholar] [CrossRef] [PubMed]
- Kamme, C. Antibodies against staphylococcal bacteriophages in human sera. Acta Pathol. Microbiol. Scand. B Microbiol. Immunol. 1973, 81, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W.; Huggins, M.B.; Shaw, K.M. Factors influencing the survival and multiplication of bacteriophages in calves and in their environment. J. Gen. Microbiol. 1987, 133, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Ka mierczak, Z.; Letarov, A.; Górski, A. Immunogenicity studies of Proteins Forming the T4 Phage Head Surface. J. Virol. 2014, 88, 12551–12557. [Google Scholar] [CrossRef] [PubMed]
- Fogelman, I.; Davey, V.; Ochs, H.D.; Elashoff, M.; Feinberg, M.B.; Mican, J.; Siegel, J.P.; Sneller, M.; Lane, H.C. Evaluation of CD4+ T cell function In vivo in HIV-infected patients as measured by bacteriophage phiX174 immunization. J. Infect. Dis. 2000, 182, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.D.; Davis, S.D.; Wedgwood, R.J. Immunologic responses to bacteriophage phi-X 174 in immunodeficiency diseases. J. Clin. Investig. 1971, 50, 2559–2568. [Google Scholar] [CrossRef]
- Rubinstein, A.; Mizrachi, Y.; Bernstein, L.; Shliozberg, J.; Golodner, M.; Liu, G.Q.; Ochs, H.D. Progressive specific immune attrition after primary, secondary and tertiary immunizations with bacteriophage phi X174 in asymptomatic HIV-1 infected patients. AIDS 2000, 14, F55–F62. [Google Scholar] [CrossRef]
- Shearer, W.T.; Lugg, D.J.; Rosenblatt, H.M.; Nickolls, P.M.; Sharp, R.M.; Reuben, J.M.; Ochs, H.D. Antibody responses to bacteriophage φX-174 in human subjects exposed to the Antarctic winter-over model of spaceflight. J. Allergy Clin. Immunol. 2001, 107, 160–164. [Google Scholar] [CrossRef]
- Dąbrowska, K.; Switala-Jelen, K.; Opolski, A.; Weber-Dąbrowska, B.; Górski, A. Bacteriophage penetration in vertebrates. J. Appl. Microbiol. 2005, 98, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Jerne, N.K. The presence in normal serum of specific antibody against bacteriophage T4 and its increase during the earliest stages of immunization. J. Immunol. 1956, 76, 209–216. [Google Scholar] [PubMed]
- Jerne, N.K. Bacteriophage inactivation by antiphage serum diluted in distilled water. Nature 1952, 169, 117–118. [Google Scholar] [CrossRef] [PubMed]
- Żaczek, M.; Łusiak-Szelachowska, M.; Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Owczarek, B.; Kopciuch, A.; Fortuna, W.; Rogóż, P.; Górski, A.; et al. Antibody production in response to Staphylococcal MS-1 phage cocktail in patients undergoing phage therapy. Front. Microbiol. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Żaczek, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Kłak, M.; Fortuna, W.; Letkiewicz, S.; Rogóż, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; et al. Phage Neutralization by Sera of Patients Receiving Phage Therapy. Viral Immunol. 2014, 27, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.J.H.; Jansen, V.A.A. Pharmacokinetic principles of bacteriophage therapy. Clin. Pharmacokinet. 2003, 42, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Payne, R.J.; Jansen, V.A. Understanding bacteriophage therapy as a density-dependent kinetic process. J. Theor. Biol. 2001, 208, 37–48. [Google Scholar] [CrossRef]
- Payne, R.J.H.; Jansen, V.A.A. Phage therapy: The peculiar kinetics of self-replicating pharmaceuticals. Clin. Pharmacol. Ther. 2000, 68, 225–230. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Cairns, B.J.; Timms, A.R.; Jansen, V.A.A.; Connerton, I.F.; Payne, R.J.H. Quantitative Models of In Vitro Bacteriophage–Host Dynamics and Their Application to Phage Therapy. PLoS Pathog. 2009, 5, e1000253. [Google Scholar] [CrossRef] [PubMed]
- Kasman, L.M.; Kasman, A.; Westwater, C.; Dolan, J.; Schmidt, M.G.; Norris, J.S. Overcoming the phage replication threshold: A mathematical model with implications for phage therapy. J. Virol. 2002, 76, 5557–5564. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.; Thomas-Abedon, C. Phage therapy pharmacology. Curr. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef] [PubMed]
- Ho, K. Bacteriophage Therapy for Bacterial Infections: Rekindling a Memory from the Pre-Antibiotics Era. Perspect. Biol. Med. 2001, 44, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Aleshkin, A.V.; Ershova, O.N.; Volozhantsev, N.V.; Svetoch, E.A.; Popova, A.V.; Rubalskii, E.O.; Borzilov, A.I.; Aleshkin, V.A.; Afanas’ev, S.S.; Karaulov, A.V.; et al. Phagebiotics in treatment and prophylaxis of healthcare-associated infections. Bacteriophage 2016, 6, e1251379. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, K. Bacteriophage therapies re-enter clinical trials. Nat. Rev. Drug Discov. 2015, 14, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Mattey, M.; Spencer, J. Bacteriophage therapy--cooked goose or phoenix rising? Curr. Opin. Biotechnol. 2008, 19, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Debarbieux, L.; Leduc, D.; Maura, D.; Morello, E.; Criscuolo, A.; Grossi, O.; Balloy, V.; Touqui, L. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J. Infect. Dis. 2010, 201, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Secor, P.R.; Michaels, L.A.; Smigiel, K.S.; Rohani, M.G.; Jennings, L.K.; Hisert, K.B.; Arrigoni, A.; Braun, K.R.; Birkland, T.P.; Lai, Y.; et al. Filamentous bacteriophage produced by Pseudomonas aeruginosa alters the inflammatory response and promotes noninvasive infection in vivo. Infect. Immun. 2017, 85, e00648-16. [Google Scholar] [CrossRef]
- Janeway, C.A.; Medzhitov, R. Innate Immune Recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Eriksson, F.; Tsagozis, P.; Lundberg, K.; Parsa, R.; Mangsbo, S.M.; Persson, M.A.A.; Harris, R.A.; Pisa, P.; Culp, W.D.; Massey, R.; et al. Tumor-specific bacteriophages induce tumor destruction through activation of tumor-associated macrophages. J. Immunol. 2009, 182, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, F.; Culp, W.D.; Massey, R.; Egevad, L.; Garland, D.; Persson, M.A.A.A.; Pisa, P. Tumor specific phage particles promote tumor regression in a mouse melanoma model. Cancer Immunol. Immunother. 2007, 56, 677–687. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Belleghem, J.D.; Dąbrowska, K.; Vaneechoutte, M.; Barr, J.J.; Bollyky, P.L. Interactions between Bacteriophage, Bacteria, and the Mammalian Immune System. Viruses 2019, 11, 10. https://doi.org/10.3390/v11010010
Van Belleghem JD, Dąbrowska K, Vaneechoutte M, Barr JJ, Bollyky PL. Interactions between Bacteriophage, Bacteria, and the Mammalian Immune System. Viruses. 2019; 11(1):10. https://doi.org/10.3390/v11010010
Chicago/Turabian StyleVan Belleghem, Jonas D., Krystyna Dąbrowska, Mario Vaneechoutte, Jeremy J. Barr, and Paul L. Bollyky. 2019. "Interactions between Bacteriophage, Bacteria, and the Mammalian Immune System" Viruses 11, no. 1: 10. https://doi.org/10.3390/v11010010
APA StyleVan Belleghem, J. D., Dąbrowska, K., Vaneechoutte, M., Barr, J. J., & Bollyky, P. L. (2019). Interactions between Bacteriophage, Bacteria, and the Mammalian Immune System. Viruses, 11(1), 10. https://doi.org/10.3390/v11010010