Controlled Disassembly and Purification of Functional Viral Subassemblies Using Asymmetrical Flow Field-Flow Fractionation (AF4)


Abstract
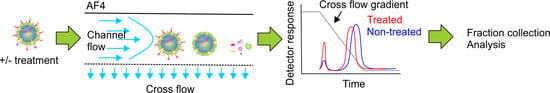
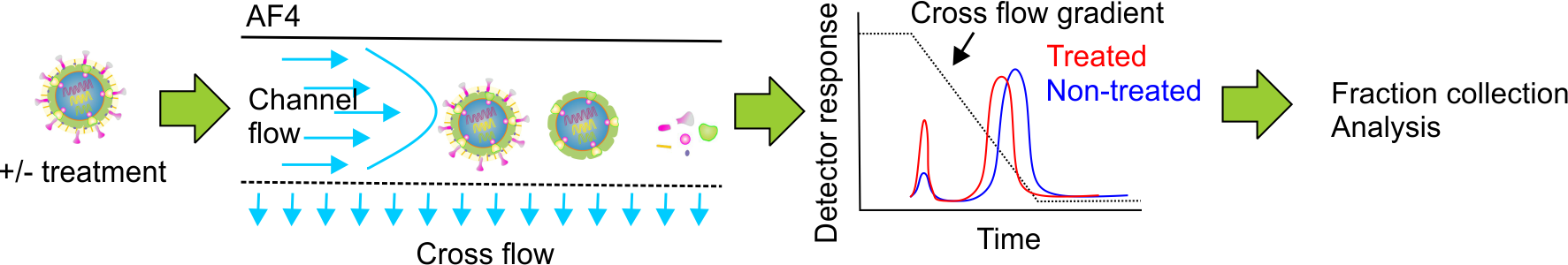{kind=link}
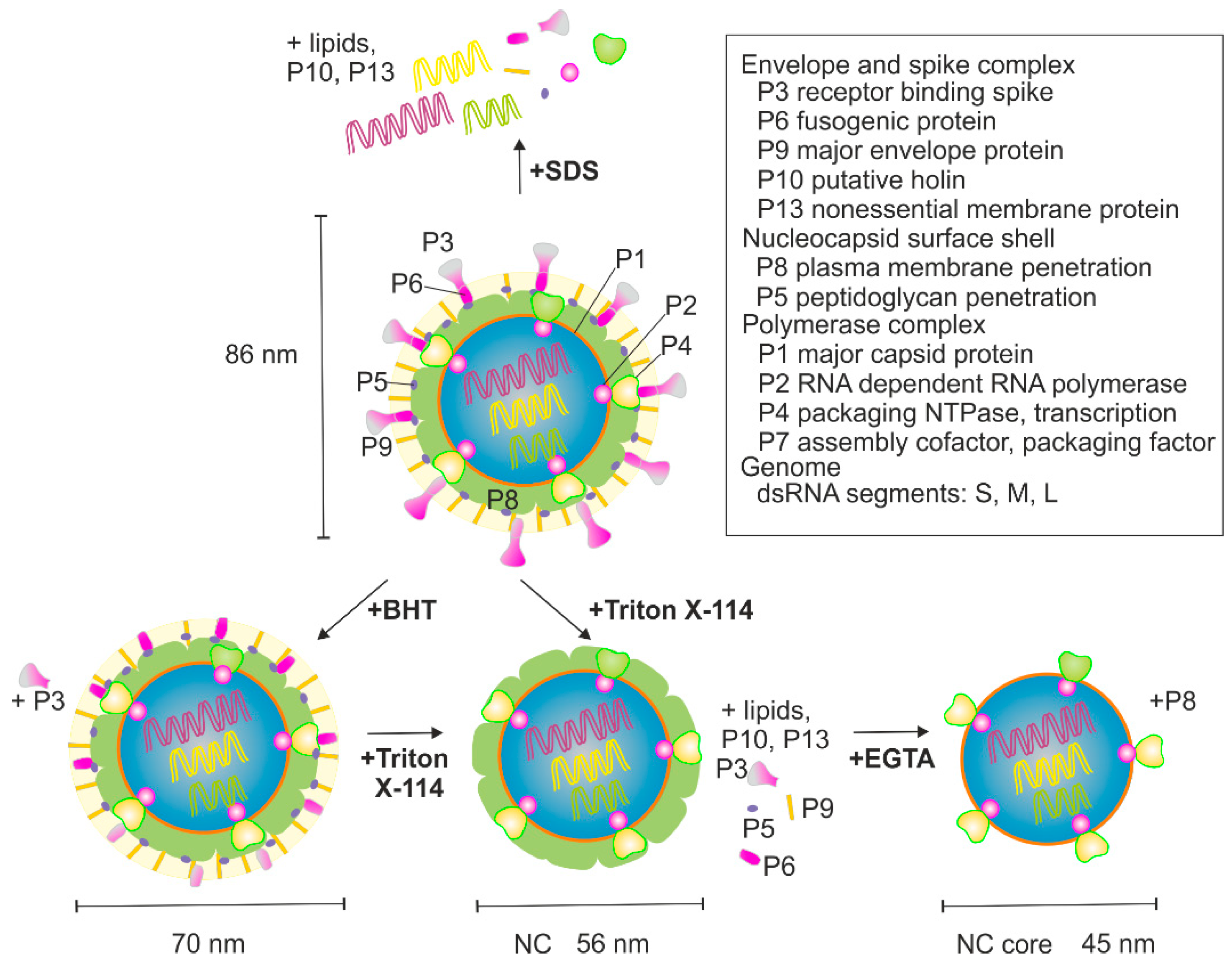{kind=link}
{kind=link}
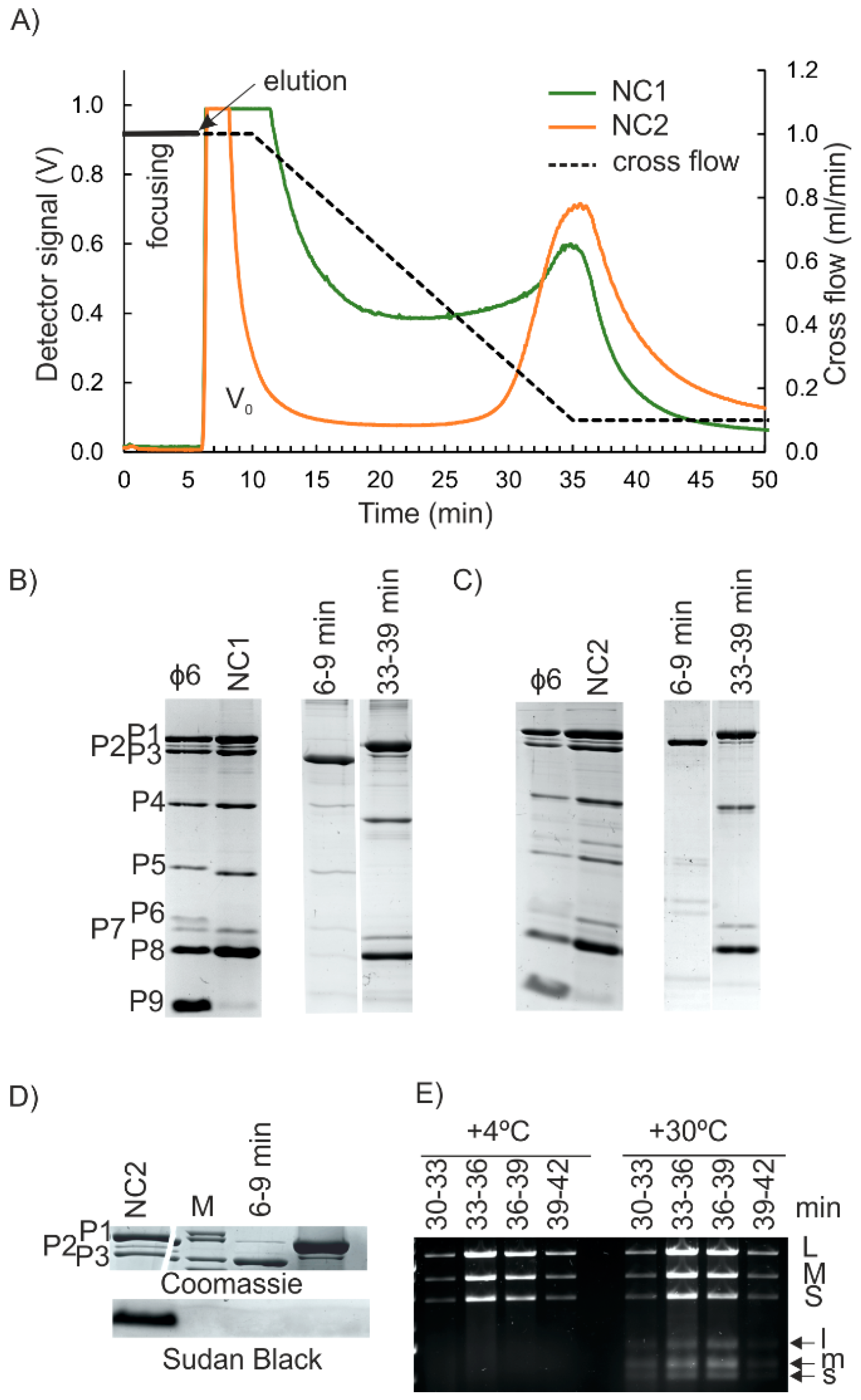{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Virus Purification and Treatments
2.2. AF4 Instrumentation and Operation
2.3. Analyses of Biological Activity, Purity and Yield
3. Results and Discussion
3.1. BHT- and SDS-Treatment of φ6
3.2. Nucleocapsid (NC) Isolation
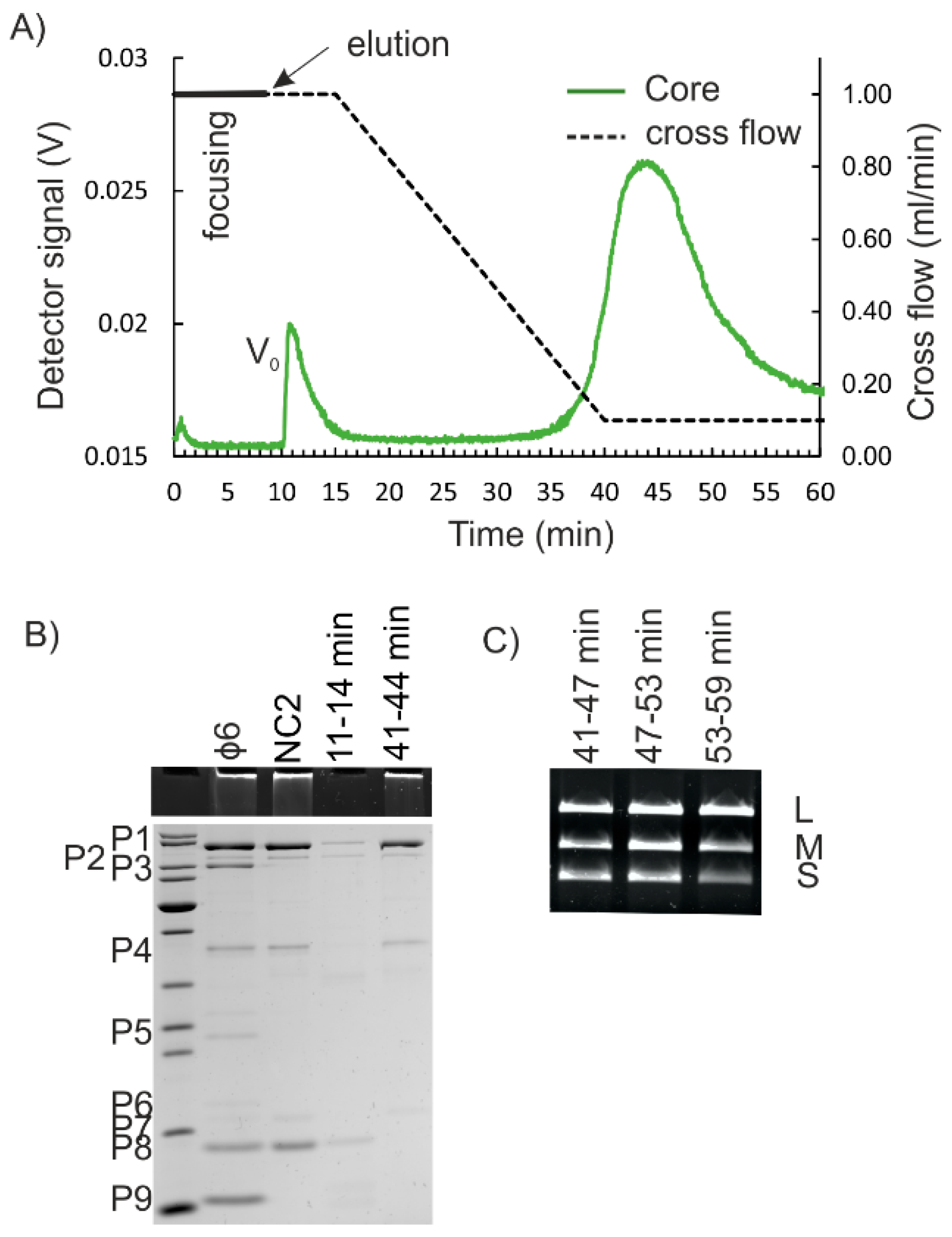
3.3. Isolation of the NC Core
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Atanasova, N.S.; Sencilo, A.; Pietilä, M.K.; Roine, E.; Oksanen, H.M.; Bamford, D.H. Comparison of lipid-containing bacterial and archaeal viruses. Adv. Virus Res. 2015, 92, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Poranen, M.M.; Bamford, D.H. Assembly of large icosahedral double-stranded RNA viruses. Adv. Exp. Med. Biol. 2012, 726, 379–402. [Google Scholar] [CrossRef] [PubMed]
- Vidaver, A.K.; Koski, R.; van Etten, J.L. Bacteriophage φ6: A lipid-containing virus of Pseudomonas phaseolicola. J. Virol. 1973, 11, 799–805. [Google Scholar] [PubMed]
- Day, L.A.; Mindich, L. The molecular weight of bacteriophage φ6 and its nucleocapsid. Virology 1980, 103, 376–385. [Google Scholar] [CrossRef]
- Lampi, M.; Oksanen, H.M.; Meier, F.; Moldenhauer, E.; Poranen, M.M.; Bamford, D.H.; Eskelin, K. Asymmetrical flow field-flow fractionation in purification of an enveloped bacteriophage φ6. J. Chromatogr. B 2018, 1095, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Jäälinoja, H.T.; Huiskonen, J.T.; Butcher, S.J. Electron cryomicroscopy comparison of the architectures of the enveloped bacteriophages φ6 and φ8. Structure 2007, 15, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Wanda, P.; Cupp, J.; Snipes, W.; Deith, A.; Rucinsky, T.; Polish, L.; Sands, J. Inactivation of the enveloped bacteriophage φ6 by butylated hydroxytoluene and butylated hydroxyanisole. Antimicrob. Agents Chemother. 1976, 10, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Mäntynen, S.; Sundberg, L.R.; Poranen, M.M. Recognition of six additional cystoviruses: Pseudomonas virus φ6 is no longer the sole species of the family Cystoviridae. Arch. Virol. 2018, 163, 1117–1124. [Google Scholar] [CrossRef] [PubMed]
- Pirttimaa, M.J.; Bamford, D.H. RNA secondary structures of the bacteriophage φ6 packaging regions. RNA 2000, 6, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, P.; Qiao, X.; Strassman, J.; Frilander, M.; Mindich, L. Identification of the packaging regions within the genomic RNA segments of bacteriophage φ6. Virology 1994, 200, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, P.; Strassman, J.; Qiao, X.; Frilander, M.; Frucht, A.; Mindich, L. In vitro packaging and replication of individual genomic segments of bacteriophage φ6 RNA. J. Virol. 1992, 66, 2611–2616. [Google Scholar] [PubMed]
- Frilander, M.; Bamford, D.H. In vitro packaging of the single-stranded RNA genomic precursors of the segmented double-stranded RNA bacteriophage φ6: The three segments modulate each other’s packaging efficiency. J. Mol. Biol. 1995, 246, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Dulin, D.; Vilfan, I.D.; Berghuis, B.A.; Hage, S.; Bamford, D.H.; Poranen, M.M.; Depken, M.; Dekker, N.H. Elongation-competent pauses govern the fidelity of a viral RNA-dependent RNA polymerase. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.; Poranen, M.M.; Bamford, D.H.; Stuart, D.I.; Grimes, J.M. Noncatalytic ions direct the RNA-dependent RNA polymerase of bacterial double-stranded RNA virus φ6 from de novo initiation to elongation. J. Virol. 2012, 86, 2837–2849. [Google Scholar] [CrossRef] [PubMed]
- Makeyev, E.V.; Bamford, D.H. Replicase activity of purified recombinant protein P2 of double-stranded RNA bacteriophage φ6. EMBO J. 2000, 19, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Butcher, S.J.; Grimes, J.M.; Makeyev, E.V.; Bamford, D.H.; Stuart, D.I. A mechanism for initiating RNA-dependent RNA polymerization. Nature 2001, 410, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Butcher, S.J.; Dokland, T.; Ojala, P.M.; Bamford, D.H.; Fuller, S.D. Intermediates in the assembly pathway of the double-stranded RNA virus φ6. EMBO J. 1997, 16, 4477–4487. [Google Scholar] [CrossRef] [PubMed]
- Huiskonen, J.T.; de Haas, F.; Bubeck, D.; Bamford, D.H.; Fuller, S.D.; Butcher, S.J. Structure of the bacteriophage φ6 nucleocapsid suggests a mechanism for sequential RNA packaging. Structure 2006, 14, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; El Omari, K.; Sun, X.; Ilca, S.L.; Kotecha, A.; Stuart, D.I.; Poranen, M.M.; Huiskonen, J.T. Double-stranded RNA virus outer shell assembly by bona fide domain-swapping. Nat. Commun. 2017, 8, 14814. [Google Scholar] [CrossRef] [PubMed]
- Poranen, M.M.; Paatero, A.O.; Tuma, R.; Bamford, D.H. Self-assembly of a viral molecular machine from purified protein and RNA constituents. Mol. Cell 2001, 7, 845–854. [Google Scholar] [CrossRef]
- Sun, X.; Pirttimaa, M.J.; Bamford, D.H.; Poranen, M.M. Rescue of maturation off-pathway products in the assembly of Pseudomonas phage φ6. J. Virol. 2013, 87, 13279–13286. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, J.F.; Tzagoloff, A.; Levine, D.; Mindich, L. Proteins of bacteriophage φ6. J. Virol. 1975, 16, 685–695. [Google Scholar] [PubMed]
- Bamford, D.H.; Romantschuk, M.; Somerharju, P.J. Membrane fusion in prokaryotes: Bacteriophage φ6 membrane fuses with the Pseudomonas syringae outer membrane. EMBO J. 1987, 6, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Ojala, P.M.; Bamford, D.H. Generation of infectious nucleocapsids by in vitro assembly of the shell protein on to the polymerase complex of the dsRNA bacteriophage φ 6. J. Mol. Biol. 1991, 218, 569–581. [Google Scholar] [CrossRef]
- Ilca, S.L.; Kotecha, A.; Sun, X.; Poranen, M.M.; Stuart, D.I.; Huiskonen, J.T. Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat. Commun. 2015, 6, 8843. [Google Scholar] [CrossRef] [PubMed]
- Nemecek, D.; Qiao, J.; Mindich, L.; Steven, A.C.; Heymann, J.B. Packaging accessory protein P7 and polymerase P2 have mutually occluding binding sites inside the bacteriophage φ6 procapsid. J. Virol. 2012, 86, 11616–11624. [Google Scholar] [CrossRef] [PubMed]
- Giddings, J.C.; Yang, F.; Myers, M.N. Flow-field-flow fractionation: A versatile new separation method. Science 1976, 193, 1244–1245. [Google Scholar] [CrossRef] [PubMed]
- Wahlund, K.G.; Giddings, J.C. Properties of an asymmetrical flow field-flow fractionation channel having one permeable wall. Anal. Chem. 1987, 59, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Ratanathanawongs, S.K.; Williams, D.L. Field-flow fractionation of proteins, polysaccharides, synthetic polymers, and supramolecular assemblies. J. Sep. Sci. 2006, 29, 1720–1732. [Google Scholar] [CrossRef]
- Giddings, J.C. Field-Flow Fractionation. Chem. Eng. News Arch. 1988, 66, 34–45. [Google Scholar] [CrossRef]
- Giddings, J.C.; Ratanathanawongs, S.K.; Moon, M.H. Field-flow fractionation: A versatile technology for particle characterization in the size range 10−3 to 102 micrometers. KONA Powder Part. J. 1991, 9, 200–217. [Google Scholar] [CrossRef]
- Gigault, J.; Pettibone, J.M.; Schmitt, C.; Hackley, V.A. Rational strategy for characterization of nanoscale particles by asymmetric-flow field flow fractionation: A tutorial. Anal. Chim. Acta 2014, 809, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Messaud, F.A.; Sanderson, R.D.; Runyon, J.R.; Otte, T.; Pasch, H.; Williams, S.K.R. An overview on field-flow fractionation techniques and their applications in the separation and characterization of polymers. Prog. Polym. Sci. 2009, 34, 351–368. [Google Scholar] [CrossRef]
- Roda, B.; Zattoni, A.; Reschiglian, P.; Moon, M.H.; Mirasoli, M.; Michelini, E.; Roda, A. Field-flow fractionation in bioanalysis: A review of recent trends. Anal. Chim. Acta 2009, 635, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Wahlund, K.-G. Flow field-flow fractionation: Critical overview. J. Chromatogr. A 2013, 1287, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Bamford, D.H.; Ojala, P.M.; Frilander, M.; Walin, L.; Bamford, J.K. Isolation, purification, and function of assembly intermediates and subviral particles of bacteriophages PRD1 and φ6. Methods Mol. Genet. 1995, 6, 455–474. [Google Scholar]
- Ojala, P.M.; Paatero, A.O.; Bamford, D.H. NTP binding induces conformational changes in the double-stranded RNA bacteriophage φ6 subviral particles. Virology 1994, 205, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Eskelin, K.; Lampi, M.; Meier, F.; Moldenhauer, E.; Bamford, D.H.; Oksanen, H.M. Asymmetric flow field flow fractionation methods for virus purification. J. Chromatogr. A 2016, 1469, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Eskelin, K.; Lampi, M.; Meier, F.; Moldenhauer, E.; Bamford, D.H.; Oksanen, H.M. Halophilic viruses with varying biochemical and biophysical properties are amenable to purification with asymmetrical flow field-flow fractionation. Extremophiles 2017, 21, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Olkkonen, V.M.; Bamford, D.H. Quantitation of the adsorption and penetration stages of bacteriophage φ6 infection. Virology 1989, 171, 229–238. [Google Scholar] [CrossRef]
- Ojala, P.M.; Bamford, D.H. In vitro transcription of the double-stranded RNA bacteriophage φ6 is influenced by purine NTPs and calcium. Virology 1995, 207, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Pagratis, N.; Revel, H.R. Detection of bacteriophage φ6 minus-strand RNA and novel mRNA isoconformers synthesized in vivo and in vitro, by strand-separating agarose gels. Virology 1990, 177, 273–280. [Google Scholar] [CrossRef]
- Poranen, M.M.; Daugelavičius, R.; Ojala, P.M.; Hess, M.W.; Bamford, D.H. A novel virus-host cell membrane interaction. Membrane voltage-dependent endocytic-like entry of bacteriophage straight φ6 nucleocapsid. J. Cell Biol. 1999, 147, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Ojala, P.M.; Romantschuk, M.; Bamford, D.H. Purified φ6 nucleocapsids are capable of productive infection of host cells with partially disrupted outer membranes. Virology 1990, 178, 364–372. [Google Scholar] [CrossRef]
- Wei, Z.; Mcevoy, M.; Razinkov, V.; Polozova, A.; Li, E.; Casas-Finet, J.; Tous, G.I.; Balu, P.; Pan, A.A.; Mehta, H. Biophysical characterization of influenza virus subpopulations using field flow fractionation and multiangle light scattering: Correlation of particle counts, size distribution and infectivity. J. Virol. Methods 2007, 144, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Bamford, D.H.; Bamford, J.K.H.; Thomas, G.J. Structural studies of the enveloped dsRNA bacteriophage φ6 of Pseudomonas syringae by Raman spectroscopy: I. The virion and its membrane envelope. J. Mol. Biol. 1993, 230, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.; Lane, L.; Gonzalez, C.; Partridge, J.; Vidaver, A. Comparative properties of bacteriophage φ6 and φ6 nucleocapsid. J. Virol. 1976, 18, 652–658. [Google Scholar]
- Stitt, B.L.; Mindich, L. Morphogenesis of bacteriophage φ6: A presumptive viral membrane precursor. Virology 1983, 127, 446–458. [Google Scholar] [CrossRef]
- Hantula, J.; Bamford, D.H. Chemical crosslinking of bacteriophage φ6 nucleocapsid proteins. Virology 1988, 165, 482–488. [Google Scholar] [CrossRef]
- Usala, S.J.; Brownstein, B.H.; Haselkorn, R. Displacement of parental RNA strands during in vitro transcription by bacteriophage φ6 nucleocapsids. Cell 1980, 19, 855–862. [Google Scholar] [CrossRef]
- Van Etten, J.L.; Burbank, D.E.; Cuppels, D.A.; Lane, L.C.; Vidaver, A.K. Semiconservative synthesis of single-stranded RNA by bacteriophage φ6 RNA polymerase. J. Virol. 1980, 33, 769–773. [Google Scholar] [PubMed]
- Chen, Y.; Zhang, Y.; Zhou, Y.; Luo, J.; Su, Z. Asymmetrical flow field-flow fractionation coupled with multi-angle laser light scattering for stability comparison of virus-like particles in different solution environments. Vaccine 2016, 34, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Chuan, Y.P.; Fan, Y.Y.; Lua, L.H.; Middelberg, A.P. Virus assembly occurs following a pH-or Ca2+-triggered switch in the thermodynamic attraction between structural protein capsomeres. J. R. Soc. Interface 2010, 7, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Bousse, T.; Shore, D.A.; Goldsmith, C.S.; Hossain, M.J.; Jang, Y.; Davis, C.T.; Donis, R.O.; Stevens, J. Quantitation of influenza virus using field flow fractionation and multi-angle light scattering for quantifying influenza A particles. J. Virol. Methods 2013, 193, 589–596. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eskelin, K.; Poranen, M.M. Controlled Disassembly and Purification of Functional Viral Subassemblies Using Asymmetrical Flow Field-Flow Fractionation (AF4). Viruses 2018, 10, 579. https://doi.org/10.3390/v10110579
Eskelin K, Poranen MM. Controlled Disassembly and Purification of Functional Viral Subassemblies Using Asymmetrical Flow Field-Flow Fractionation (AF4). Viruses. 2018; 10(11):579. https://doi.org/10.3390/v10110579
Chicago/Turabian StyleEskelin, Katri, and Minna M. Poranen. 2018. "Controlled Disassembly and Purification of Functional Viral Subassemblies Using Asymmetrical Flow Field-Flow Fractionation (AF4)" Viruses 10, no. 11: 579. https://doi.org/10.3390/v10110579
APA StyleEskelin, K., & Poranen, M. M. (2018). Controlled Disassembly and Purification of Functional Viral Subassemblies Using Asymmetrical Flow Field-Flow Fractionation (AF4). Viruses, 10(11), 579. https://doi.org/10.3390/v10110579