Photodynamic Inactivation of Herpes Simplex Viruses

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
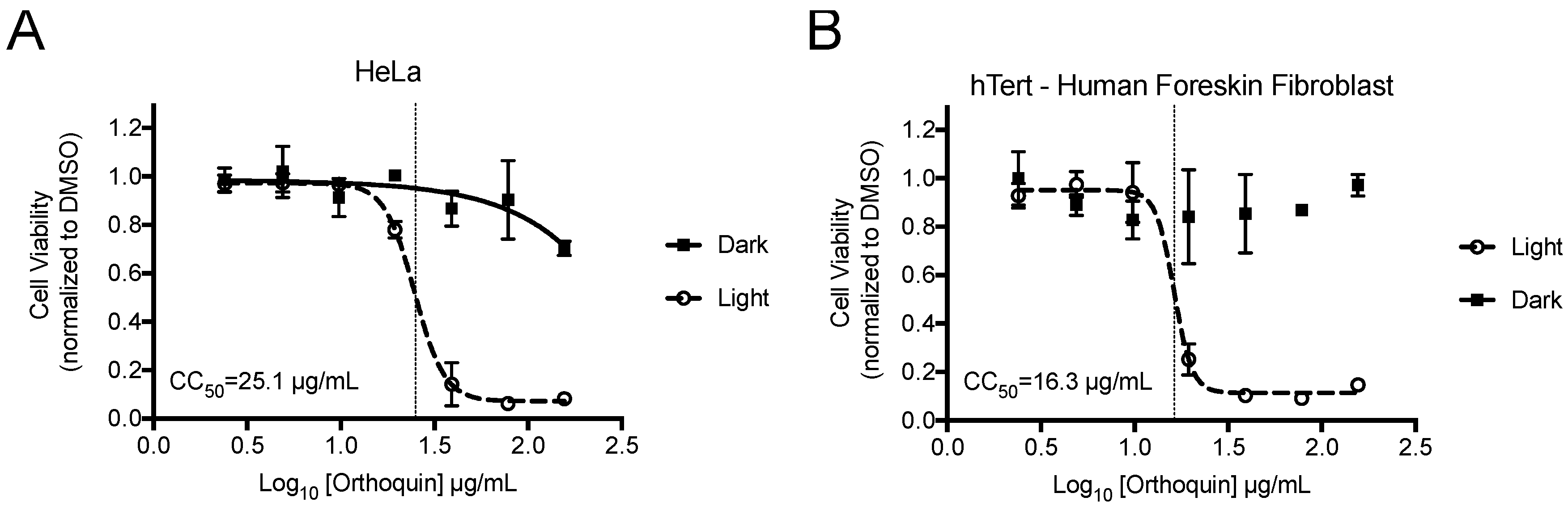
2.1. Characterization of Orthoquin™
2.2. Cells, Viruses, and Reagents
2.3. Cytotoxicity Assay
2.4. Photodynamic Inactivation of Viral Inoculum
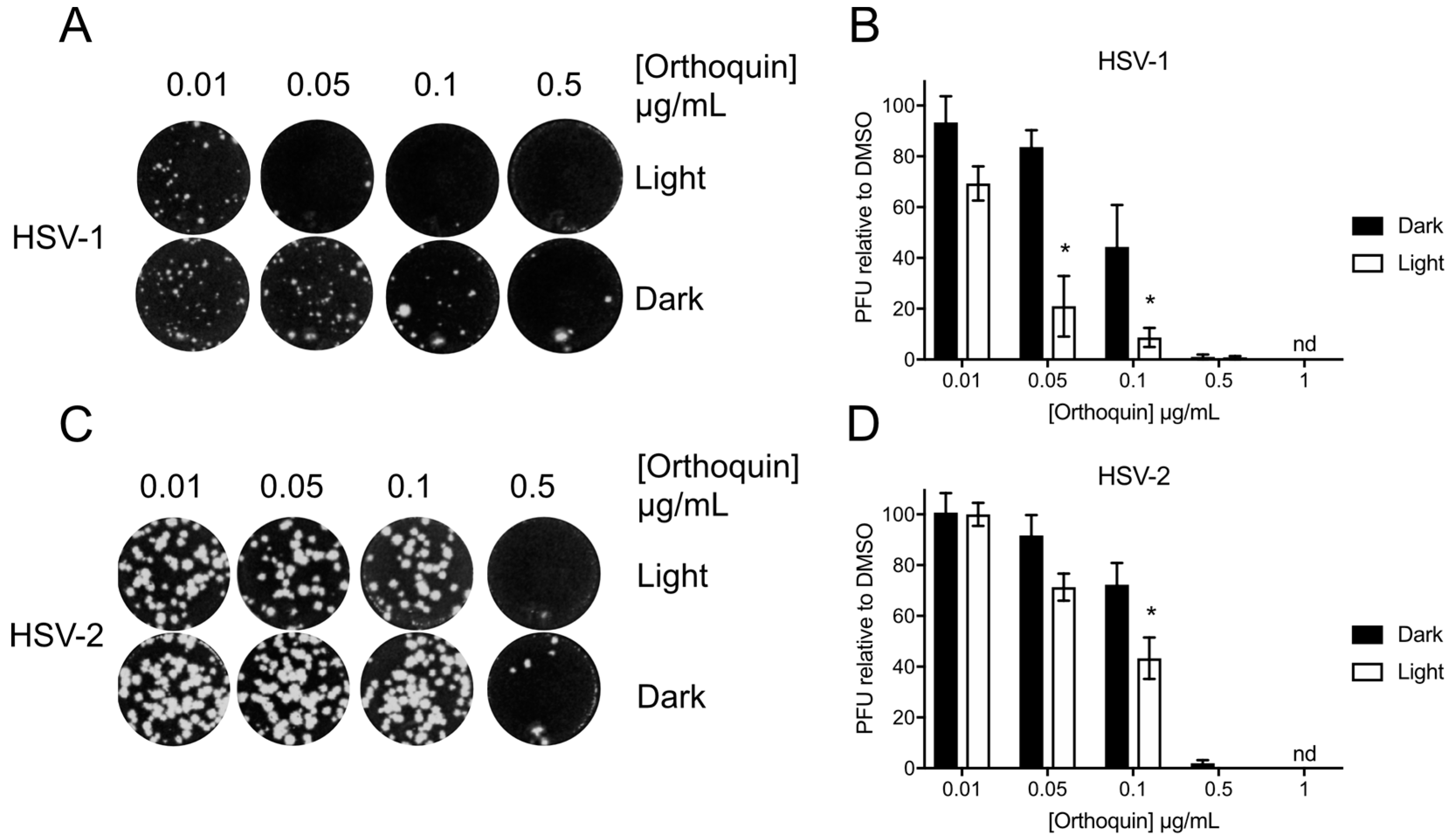
2.5. Plaque Assays
2.5.1. HSV-1 and HSV-2 Plaque Assays
2.5.2. VSV Plaque Assay
2.5.3. AdV Plaque Assay
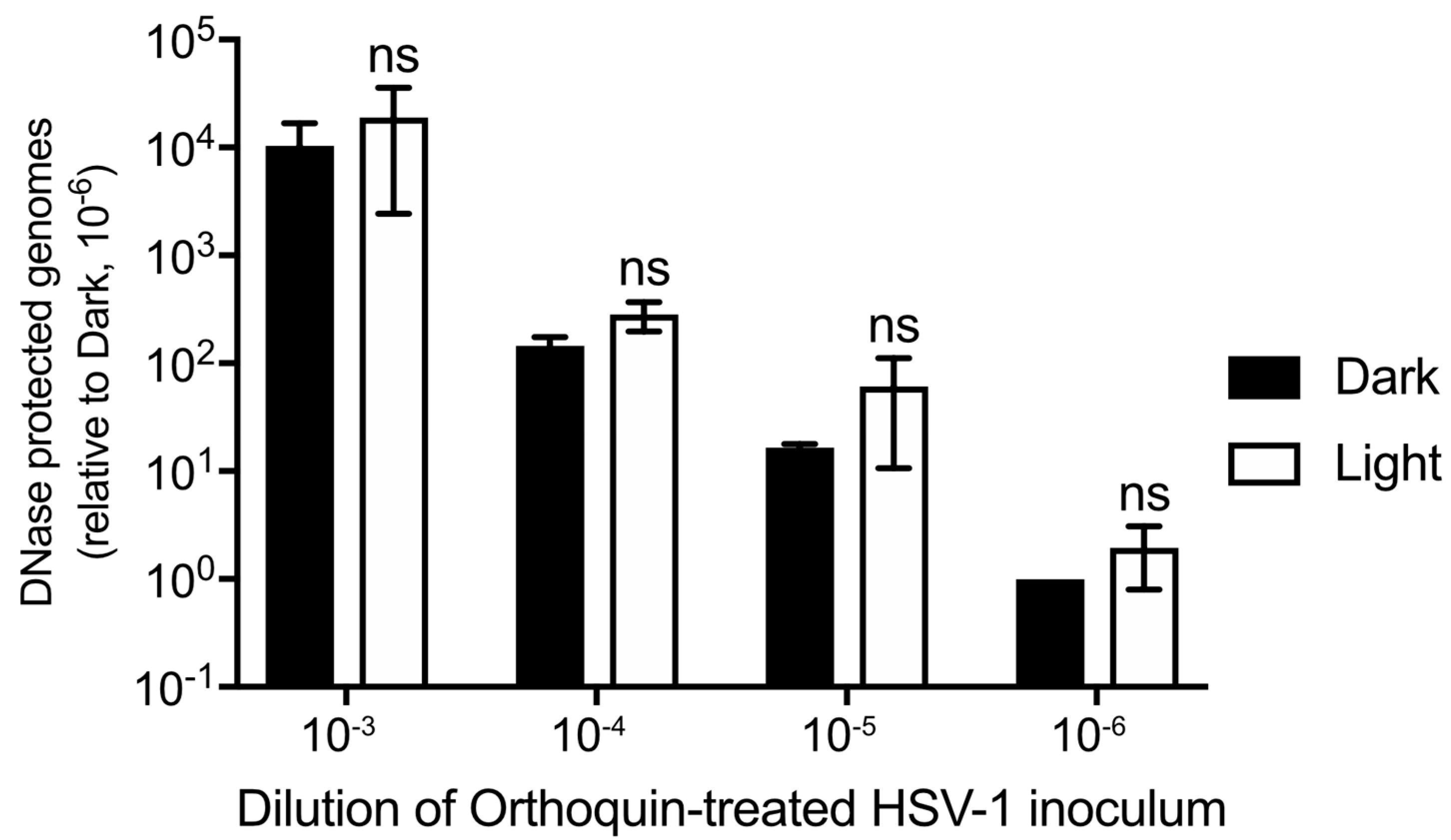
2.6. DNase-Protection Assays and qPCR
2.7. Immunoblotting
2.8. Statistics
3. Results
3.1. Description and Preparation of Orthoquin
3.2. Measurement of Phototoxic Threshold of Orthoquin on HeLa and hTert-BJ Cells
3.3. Differential Baseline Photosensitivity of HSV-1 and HSV-2
3.4. Orthoquin Exhibits Light-Dependent Antiviral Activity against HSV-1 and HSV-2
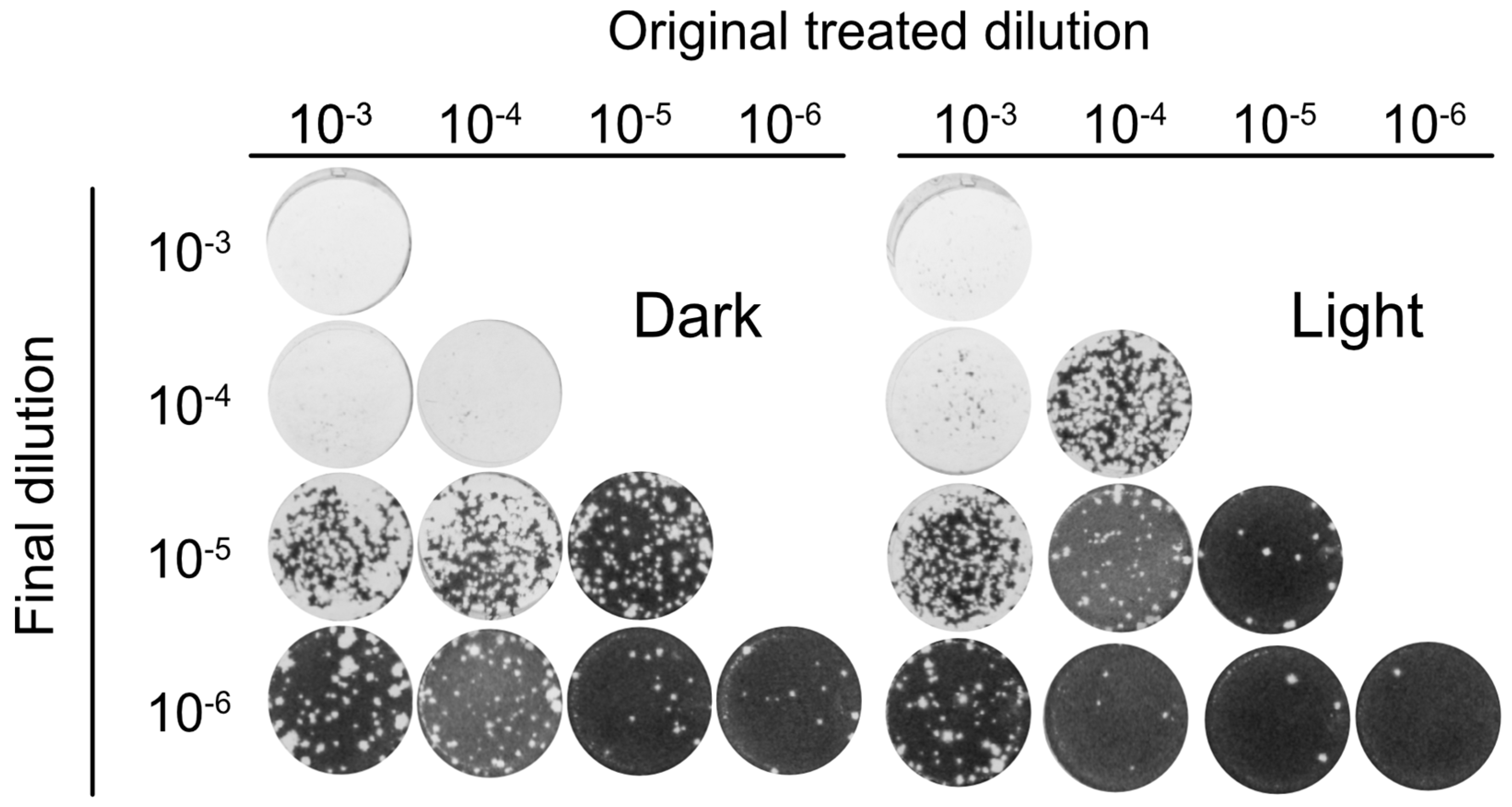
3.5. Orthoquin PDI Inactivates HSV-1 across a 100-Fold Range of Inoculum Titer
3.6. Orthoquin PDI Depends on Direct Contact with HSV-1 Virions
3.7. Orthoquin PDI Does Not Release HSV-1 Genomic DNA from Virions
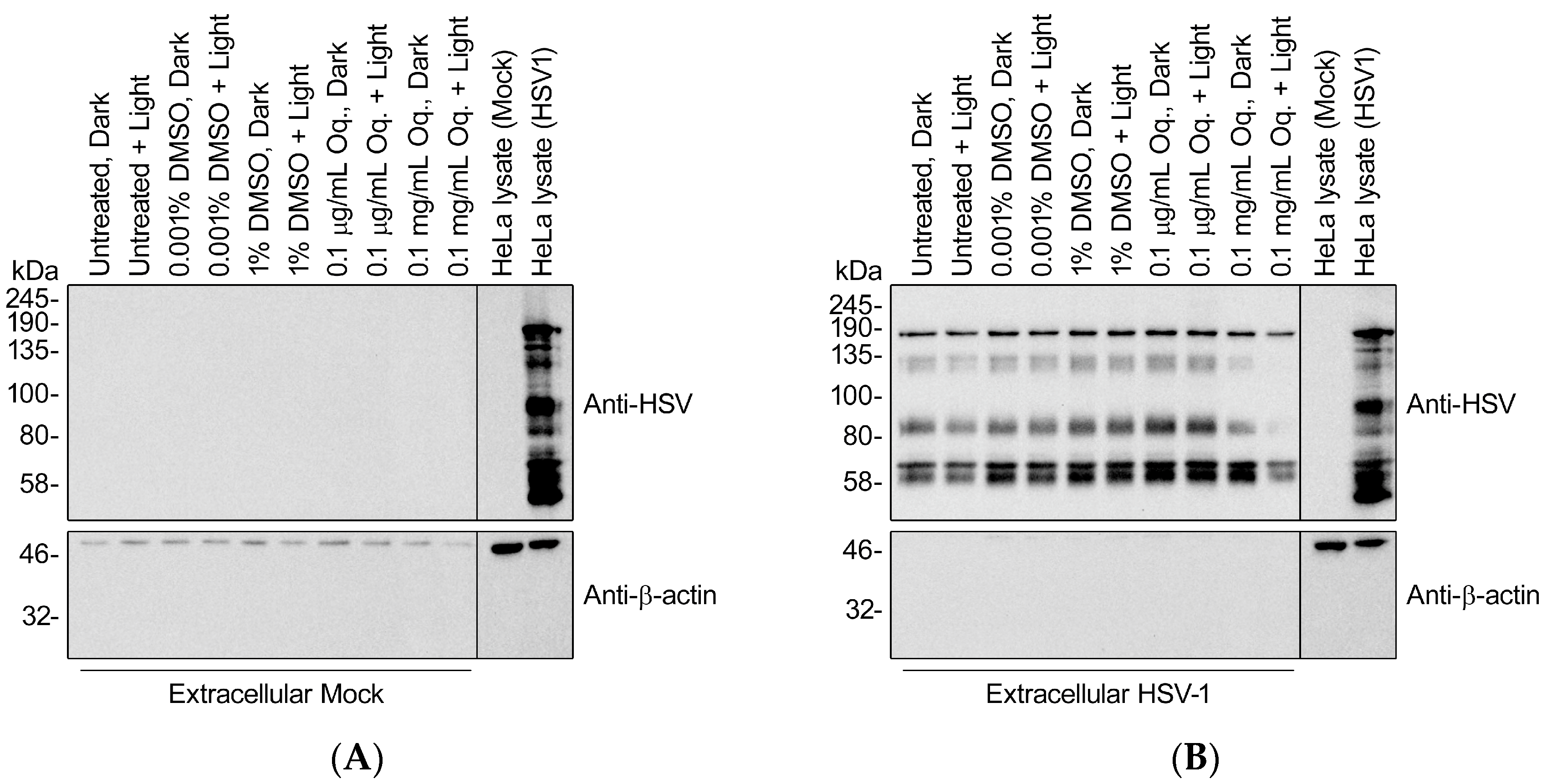
3.8. High Doses of Orthoquin Inhibit Detection of HSV-1 Structural Proteins
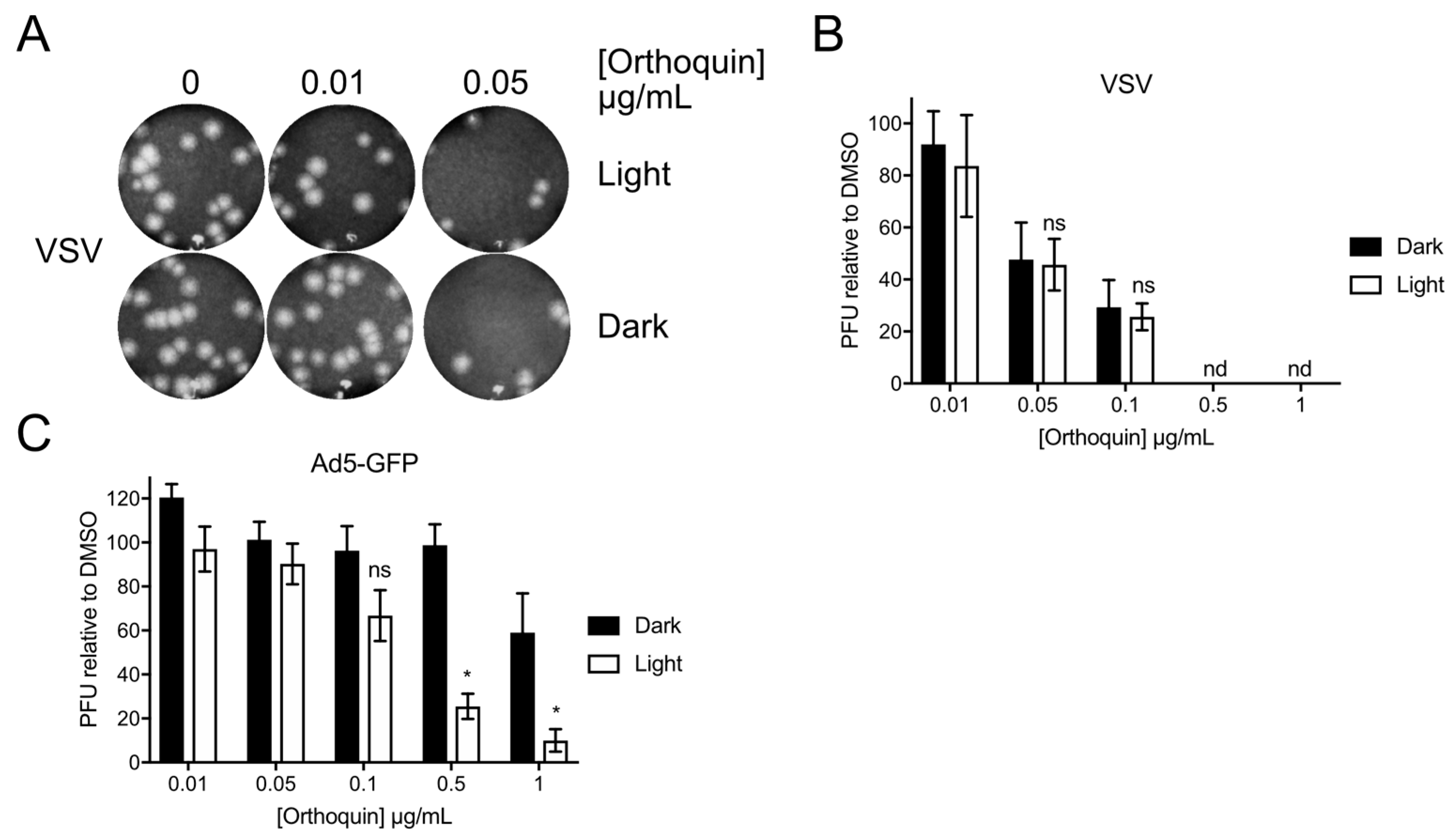
3.9. Orthoquin PDI of Vesicular Stomatitis Virus and Adenovirus
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell. Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R. Persistent and recurring viral infections: The human herpesviruses. Curr. Probl. Pediatr. Adolesc. Health Care 2009, 39, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Crumpacker, C.S. Mechanism of action of foscarnet against viral polymerases. Am. J. Med. 1992, 92, 3S–7S. [Google Scholar] [CrossRef]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin. Microbiol. Rev. 2003, 16, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Resistance of herpes simplex viruses to nucleoside analogues: Mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 2011, 55, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Boivin, G. Antiviral drug resistance in herpesviruses other than cytomegalovirus. Rev. Med. Virol. 2014, 24, 186–218. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.H.; Marcelletti, J.R.; Pope, L.E.; Khalil, M.H.; Katz, L.R.; McFadden, R. n-Docosanol: Broad Spectrum Anti-Viral Activity against Lipid-enveloped Virusesa. Ann. N. Y. Acad. Sci. 1994, 724, 472–488. [Google Scholar] [CrossRef] [PubMed]
- Pope, L.E.; Marcelletti, J.F.; Katz, L.R.; Lin, J.Y.; Katz, D.H.; Parish, M.L.; Spear, P.G. The anti-herpes simplex virus activity of n-docosanol includes inhibition of the viral entry process. Antivir. Res. 1998, 40, 85–94. [Google Scholar] [CrossRef]
- Huang, L.; Dai, T.; Hamblin, M.R. Antimicrobial photodynamic inactivation and photodynamic therapy for infections. Methods Mol. Biol. 2010, 635, 155–173. [Google Scholar] [PubMed]
- Henderson, B.W.; Dougherty, T.J. How does photodynamic therapy work? Photochem. Photobiol. 1992, 55, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Bonnett, R. Chemical Aspects of Photodynamic Therapy; CRC Press: London, UK, 2014. [Google Scholar]
- Van Straten, D.; Mashayekhi, V.; de Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Wormald, R.; Evans, J.; Smeeth, L.; Henshaw, K. Photodynamic therapy for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2005. [Google Scholar] [CrossRef]
- Costa, L.; Faustino, M.A.; Neves, M.G.; Cunha, A.; Almeida, A. Photodynamic inactivation of mammalian viruses and bacteriophages. Viruses 2012, 4, 1034–1074. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—Photosensitizers, photochemistry and cellular localization. Photodiagn. Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef]
- McFarland, S. Novel Polygonum Cuspidatum Extracts and Their Use as Photodynamic Inactivating Agents. U.S. Patent US20170095521A1, 6 April 2017. [Google Scholar]
- Matrosovich, M.; Matrosovich, T.; Garten, W.; Klenk, H.D. New low-viscosity overlay medium for viral plaque assays. Virol. J. 2006, 3, 63. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Loret, S.; Guay, G.; Lippé, R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 2008, 82, 8605–8618. [Google Scholar] [CrossRef] [PubMed]
- State Pharmacopoeia Commission of the PRC. Pharmacopoeia of the People’s Republic of China 2005; BC Decker, Inc.: Hamilton, ON, Canada, 2008. [Google Scholar]
- Smetana, Z.; Ben-Hur, E.; Mendelson, E.; Salzberg, S.; Wagner, P.; Malik, Z. Herpes simplex virus proteins are damaged following photodynamic inactivation with phthalocyanines. J. Photochem. Photobiol. B Biol. 1998, 44, 77–83. [Google Scholar] [CrossRef]
- Smetana, Z.; Mendelson, E.; Manor, J.; van Lier, J.E.; Ben-Hur, E.; Salzberg, S.; Malik, Z. Photodynamic inactivation of herpes viruses with phthalocyanine derivatives. J. Photochem. Photobiol. B Biol. 1994, 22, 37–43. [Google Scholar] [CrossRef]
- Müller-Breitkreutz, K.; Mohr, H.; Briviba, K.; Sies, H. Inactivation of viruses by chemically and photochemically generated singlet molecular oxygen. J. Photochem. Photobiol. B Biol. 1995, 30, 63–70. [Google Scholar] [CrossRef]
- Campagna, M.; Rivas, C. Antiviral activity of resveratrol. Biochem. Soc. Trans. 2010, 38, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Abba, Y.; Hassim, H.; Hamzah, H.; Noordin, M.M. Antiviral Activity of Resveratrol against Human and Animal Viruses. Adv. Virol. 2015, 2015, 184241. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.A.; Hudson, J.B.; Towers, G.H. Antiviral activities of anthraquinones, bianthrones and hypericin derivatives from lichens. Experientia 1996, 52, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.L.; Huffman, J.H.; Morris, J.L.; Wood, S.G.; Hughes, B.G.; Sidwell, R.W. Evaluation of the antiviral activity of anthraquinones, anthrones and anthraquinone derivatives against human cytomegalovirus. Antivir. Res. 1992, 17, 63–77. [Google Scholar] [CrossRef]
- Dang, S.; Zhang, Z.; Chen, Y.; Zhang, X.; Wang, B.; Yuan, L.; Cheng, Y. Inhibition of the replication of hepatitis B virus in vitro by emodin. Med. Sci. Monit. 2006, 12, BR302–BR306. [Google Scholar]
- Lin, C.J.; Lin, H.J.; Chen, T.H.; Hsu, Y.-A.; Liu, C.-S.; Hwang, G.-Y.; Wan, L. Polygonum cuspidatum and its active components inhibit replication of the influenza virus through toll-like receptor 9-induced interferon beta expression. PLoS ONE 2015, 10, e0117602. [Google Scholar]
- Schinazi, R.F.; Chu, C.K.; Babu, J.R.; Oswald, B.J.; Saalmann, V.; Cannon, D.L.; Eriksson, B.F.H.; Nasr, M. Anthraquinones as a new class of antiviral agents against human immunodeficiency virus. Antivir. Res. 1990, 13, 265–272. [Google Scholar] [CrossRef]
- Konoshima, T.; Kozuka, M.; Koyama, J.; Okatani, T.; Tagahara, K.; Tokuda, H. Studies on inhibitors of skin tumor promotion, VI. Inhibitory effects of quinones on Epstein-Barr virus activation. J. Nat. Prod. 1989, 52, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Semple, S.J.; Pyke, S.M.; Reynolds, G.D.; Flower, R.L. In vitro antiviral activity of the anthraquinone chrysophanic acid against poliovirus. Antivir. Res. 2001, 49, 169–178. [Google Scholar] [CrossRef]
- Ho, T.Y.; Wu, S.L.; Chen, J.C.; Li, C.C.; Hsiang, C.Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 2007, 74, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, C.Y.; Ho, T.Y. Emodin is a novel alkaline nuclease inhibitor that suppresses herpes simplex virus type 1 yields in cell cultures. Br. J. Pharmacol. 2008, 155, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.; Lee, Y.H.; Lee, C.H.; Lee, S.K. Emodin, an anthraquinone derivative isolated from the rhizomes of Rheum palmatum, selectively inhibits the activity of casein kinase II as a competitive inhibitor. Planta Med. 1999, 65, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.P.; Roy, P. Cellular Casein Kinase 2 and Protein Phosphatase 2A Modulate Replication Site Assembly of Bluetongue Virus. J. Biol. Chem. 2016, 291, 14566–14574. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.E.; Agaisse, H. Casein kinase 2 regulates vaccinia virus actin tail formation. Virology 2012, 423, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Bayless, A.M.; Goddard, E.T.; Davido, D.J. CK2 inhibitors increase the sensitivity of HSV-1 to interferon-β. Antivir. Res. 2011, 91, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Pari, G.S. Interaction of human cytomegalovirus pUL84 with casein kinase 2 is required for oriLyt-dependent DNA replication. J. Virol. 2009, 83, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Alves, D.S.; Pérez-Fons, L.; Estepa, A.; Micol, V. Membrane-related effects underlying the biological activity of the anthraquinones emodin and barbaloin. Biochem. Pharmacol. 2004, 68, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.; Roth, E.; Mazur, Y.; Silman, I. Targeted cross-linking of a molten globule form of acetylcholinesterase by the virucidal agent hypericin. Biochemistry 1999, 38, 11401–11405. [Google Scholar] [CrossRef] [PubMed]
- Sydiskis, R.J.; Owen, D.G.; Lohr, J.L.; Rosler, K.H.; Blomster, R.N. Inactivation of enveloped viruses by anthraquinones extracted from plants. Antimicrob. Agents Chemother. 1991, 35, 2463–2466. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Wu, C.-F.; Hsiao, N.-W.; Chang, C.-Y.; Li, S.-W.; Wan, L.; Lin, Y.-J.; Lin, W.-Y. Aloe-emodin is an interferon-inducing agent with antiviral activity against Japanese encephalitis virus and enterovirus 71. Int. J. Antimicrob. Agents 2008, 32, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Danve-Szatanek, C.; Aymard, M.; Thouvenot, D.; Morfin, F.; Agius, G.; Bertin, I.; Billaudel, S.; Chanzy, B.; Coste-Burel, M.; Finkielsztejn, L.; et al. Surveillance network for herpes simplex virus resistance to antiviral drugs: 3-Year follow-up. J. Clin. Microbiol. 2004, 42, 242–249. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monjo, A.L.-A.; Pringle, E.S.; Thornbury, M.; Duguay, B.A.; Monro, S.M.A.; Hetu, M.; Knight, D.; Cameron, C.G.; McFarland, S.A.; McCormick, C. Photodynamic Inactivation of Herpes Simplex Viruses. Viruses 2018, 10, 532. https://doi.org/10.3390/v10100532
Monjo AL-A, Pringle ES, Thornbury M, Duguay BA, Monro SMA, Hetu M, Knight D, Cameron CG, McFarland SA, McCormick C. Photodynamic Inactivation of Herpes Simplex Viruses. Viruses. 2018; 10(10):532. https://doi.org/10.3390/v10100532
Chicago/Turabian StyleMonjo, Andrea L.-A., Eric S. Pringle, Mackenzie Thornbury, Brett A. Duguay, Susan M. A. Monro, Marc Hetu, Danika Knight, Colin G. Cameron, Sherri A. McFarland, and Craig McCormick. 2018. "Photodynamic Inactivation of Herpes Simplex Viruses" Viruses 10, no. 10: 532. https://doi.org/10.3390/v10100532
APA StyleMonjo, A. L.-A., Pringle, E. S., Thornbury, M., Duguay, B. A., Monro, S. M. A., Hetu, M., Knight, D., Cameron, C. G., McFarland, S. A., & McCormick, C. (2018). Photodynamic Inactivation of Herpes Simplex Viruses. Viruses, 10(10), 532. https://doi.org/10.3390/v10100532