
Non-Uniform and Non-Random Binding of Nucleoprotein to Influenza A and B Viral RNA


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. HITS-CLIP and Deep Sequencing Data Analysis
2.3. Pearson Correlation Analysis
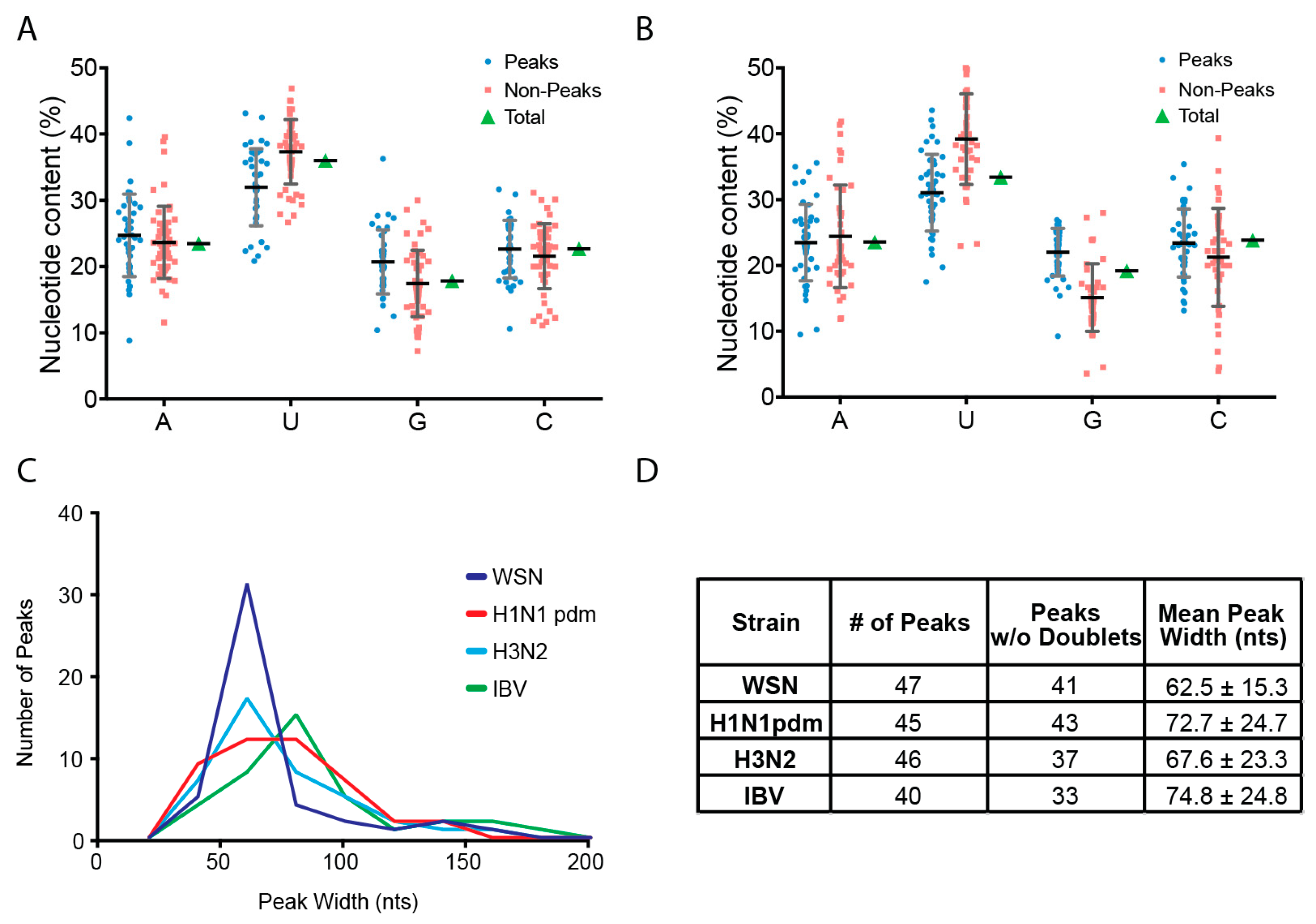
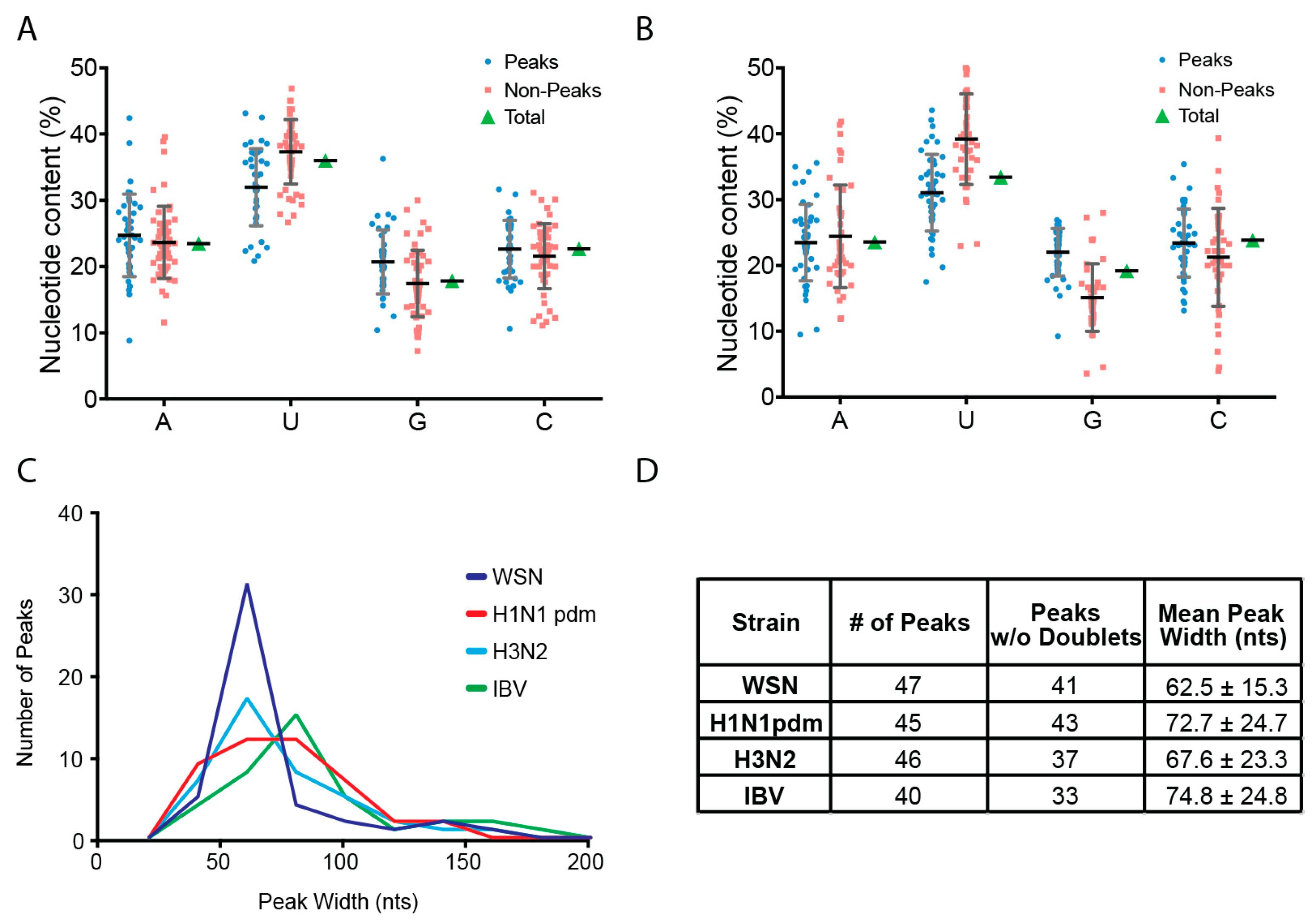
2.4. Peak Analysis
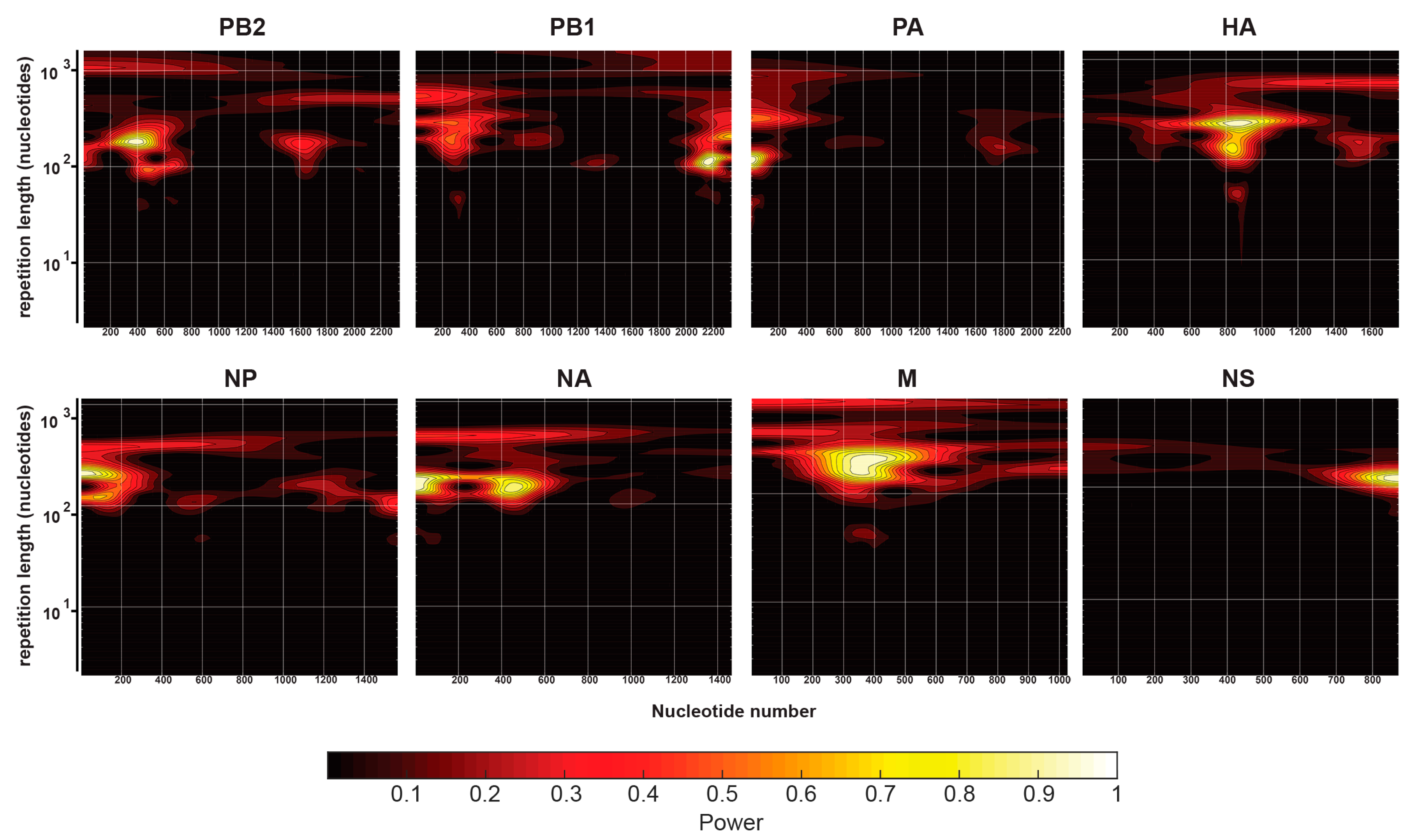
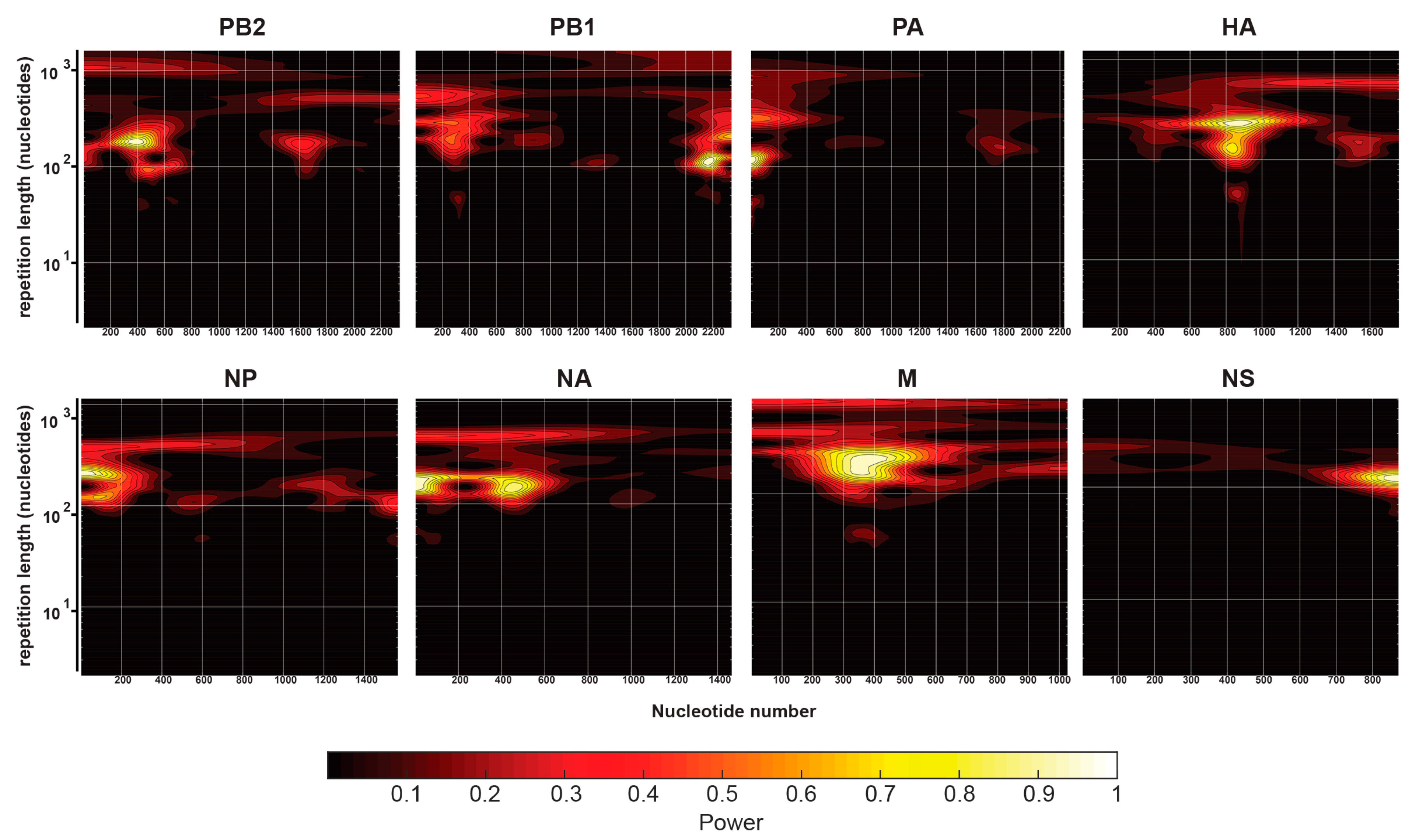
2.5. Continuous Wavelet Transforms
3. Results
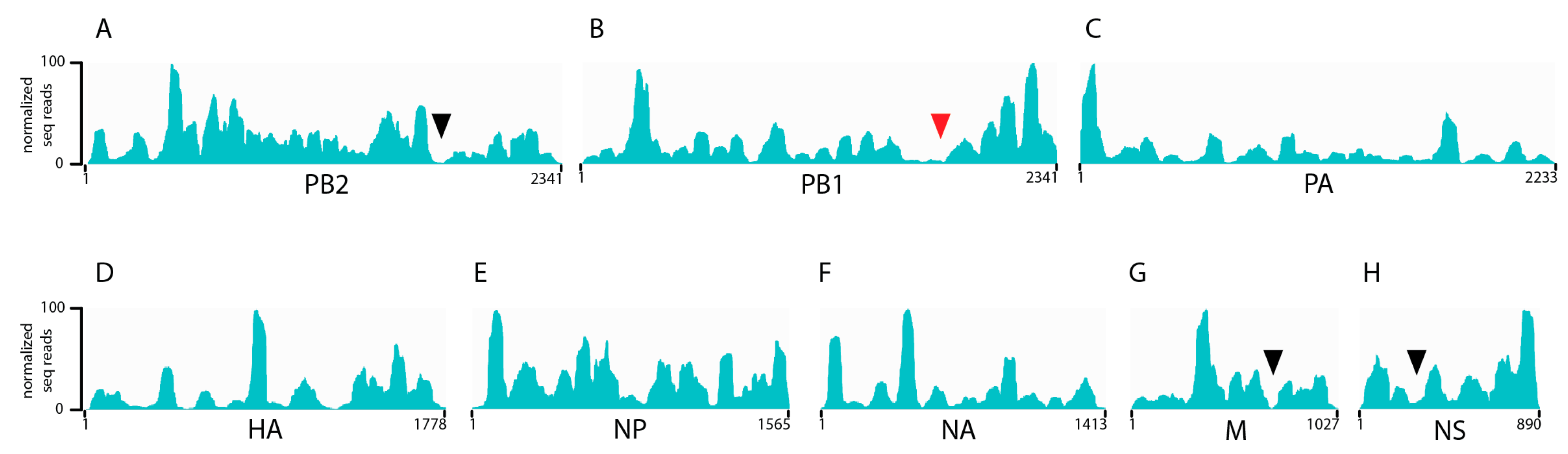
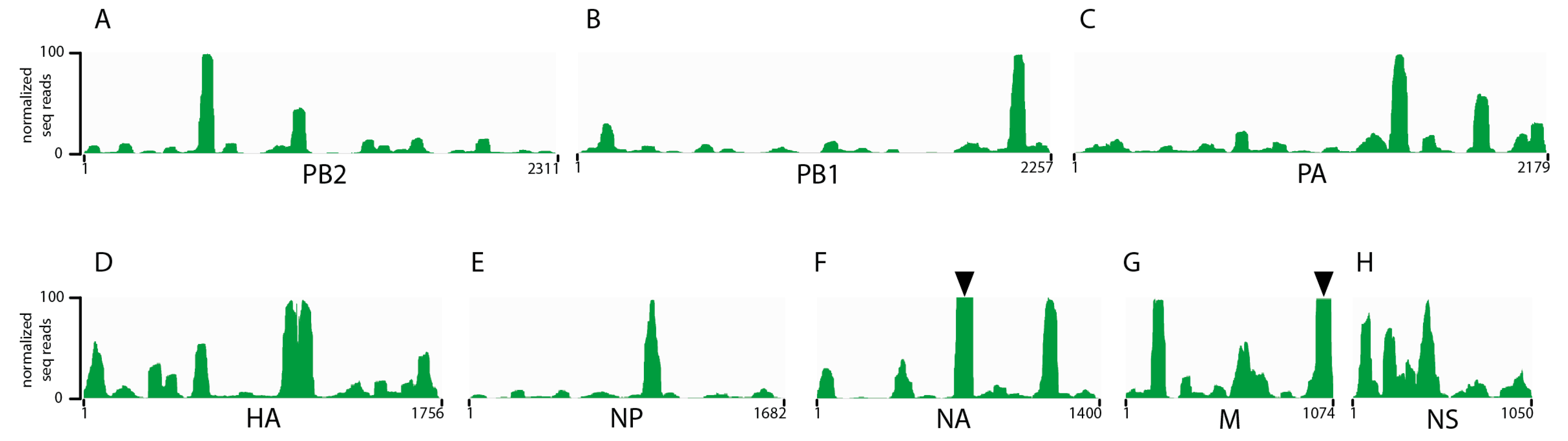
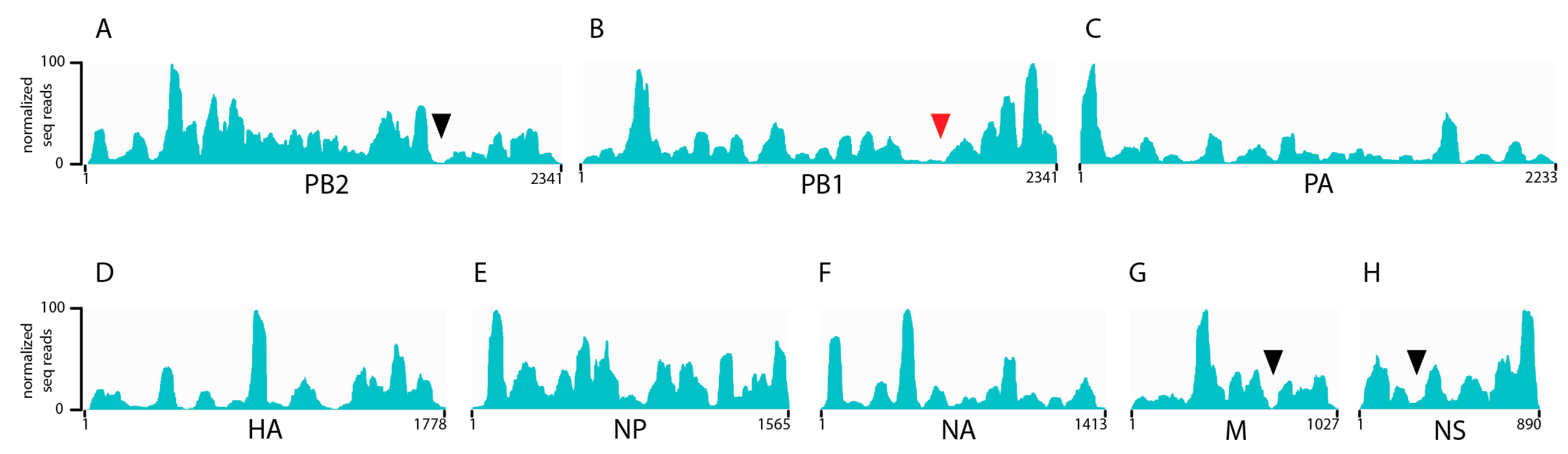
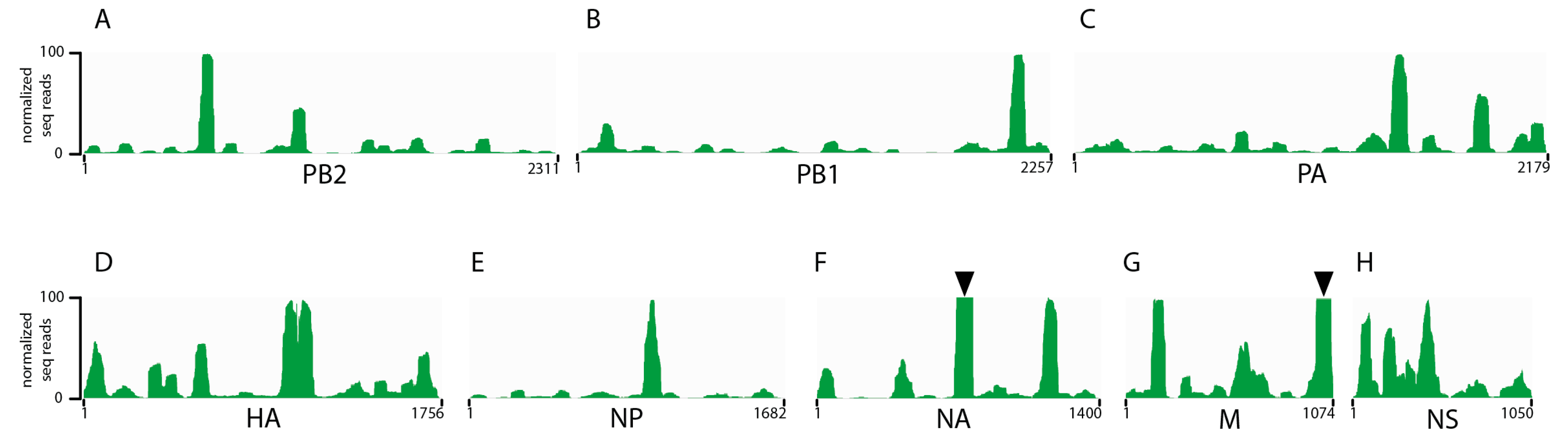
3.1. Seasonal H3N2 and IBV NP-vRNA Binding Profiles
3.2. Characterization of Enriched NP-Binding Regions
3.3. Comparison of IAV and IBV NP-vRNA Binding Profile
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Su, S.; Chaves, S.S.; Perez, A.; D’Mello, T.; Kirley, P.D.; Yousey-Hindes, K.; Farley, M.M.; Harris, M.; Sharangpani, R.; Lynfield, R.; et al. Comparing clinical characteristics between hospitalized adults with laboratory-confirmed influenza A and B virus infection. Clin. Infect. Dis. 2014, 59, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Holmes, E.C. The evolutionary dynamics of human influenza B virus. J. Mol. Evol. 2008, 66, 655–663. [Google Scholar] [CrossRef]
- Hatada, E.; Hasegawa, M.; Mukaigawa, J.; Shimizu, K.; Fukuda, R. Control of influenza virus gene expression: Quantitative analysis of each viral RNA species in infected cells. J. Biochem. 1989, 105, 537–546. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.; Fellner, P.; Newton, C. Influenza virus genome consists of eight distinct RNA species. Proc. Natl. Acad. Sci. USA 1976, 73, 3045–3049. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, S.S.; Wu, Y.; Wawrzusin, P.; Kabat, J.; Broadbent, A.J.; Lamirande, E.W.; Fodor, E.; Altan-Bonnet, N.; Shroff, H.; Subbarao, K. Influenza A virus assembly intermediates fuse in the cytoplasm. PLoS Pathog. 2014, 10, e1003971. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.Y.; Heaton, N.S.; Gao, Q.; Palese, P.; Singer, R.H.; Lionnet, T. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity fish analysis. PLoS Pathog. 2013, 9, e1003358. [Google Scholar] [CrossRef]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; von Kirchbach, J.C.; Gog, J.R.; Digard, P. Genome packaging in influenza A virus. J. Gen. Virol. 2010, 91, 313–328. [Google Scholar] [CrossRef]
- Nakatsu, S.; Sagara, H.; Sakai-Tagawa, Y.; Sugaya, N.; Noda, T.; Kawaoka, Y. Complete and incomplete genome packaging of influenza A and b viruses. MBio 2016, 7, e01248-16. [Google Scholar] [CrossRef]
- Fournier, E.; Moules, V.; Essere, B.; Paillart, J.C.; Sirbat, J.D.; Isel, C.; Cavalier, A.; Rolland, J.P.; Thomas, D.; Lina, B.; et al. A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic Acids Res. 2012, 40, 2197–2209. [Google Scholar] [CrossRef] [Green Version]
- Gavazzi, C.; Isel, C.; Fournier, E.; Moules, V.; Cavalier, A.; Thomas, D.; Lina, B.; Marquet, R. An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: Comparison with a human H3N2 virus. Nucleic Acids Res. 2013, 41, 1241–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noda, T.; Sagara, H.; Yen, A.; Takada, A.; Kida, H.; Cheng, R.H.; Kawaoka, Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006, 439, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Sugita, Y.; Aoyama, K.; Hirase, A.; Kawakami, E.; Miyazawa, A.; Sagara, H.; Kawaoka, Y. Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat. Commun. 2012, 3, 639. [Google Scholar] [CrossRef] [Green Version]
- Gavazzi, C.; Yver, M.; Isel, C.; Smyth, R.P.; Rosa-Calatrava, M.; Lina, B.; Moules, V.; Marquet, R. A functional sequence-specific interaction between influenza A virus genomic RNA segments. Proc. Natl. Acad. Sci. USA 2013, 110, 16604–16609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arranz, R.; Coloma, R.; Chichon, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortin, J.; Martin-Benito, J. The structure of native influenza virion ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef]
- Moeller, A.; Kirchdoerfer, R.N.; Potter, C.S.; Carragher, B.; Wilson, I.A. Organization of the influenza virus replication machinery. Science 2012, 338, 1631–1634. [Google Scholar] [CrossRef] [PubMed]
- Beyleveld, G.; White, K.M.; Ayllon, J.; Shaw, M.L. New-generation screening assays for the detection of anti-influenza compounds targeting viral and host functions. Antivir. Res. 2013, 100, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Coloma, R.; Valpuesta, J.M.; Arranz, R.; Carrascosa, J.L.; Ortin, J.; Martin-Benito, J. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 2009, 5, e1000491. [Google Scholar] [CrossRef]
- Martin-Benito, J.; Area, E.; Ortega, J.; Llorca, O.; Valpuesta, J.M.; Carrascosa, J.L.; Ortin, J. Three-dimensional reconstruction of a recombinant influenza virus ribonucleoprotein particle. EMBO Rep. 2001, 2, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.; Le Sage, V.; Nanni, A.V.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S. Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic Acids Res. 2017, 45, 8968–8977. [Google Scholar] [CrossRef] [Green Version]
- Williams, G.D.; Townsend, D.; Wylie, K.M.; Kim, P.J.; Amarasinghe, G.K.; Kutluay, S.B.; Boon, A.C.M. Nucleotide resolution mapping of influenza A virus nucleoprotein-RNA interactions reveals RNA features required for replication. Nat. Commun. 2018, 9, 465. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Mukaka, M.M. Statistics corner: A guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChiP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Ma, W.; Noble, W.S.; Bailey, T.L. Motif-based analysis of large nucleotide data sets using meme-chip. Nat. Protoc. 2014, 9, 1428–1450. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.P.; Barclay, W.S. The N-terminal extension of the influenza B virus nucleoprotein is not required for nuclear accumulation or the expression and replication of a model RNA. J. Virol. 1998, 72, 5307–5312. [Google Scholar] [PubMed]
- Compans, R.W.; Content, J.; Duesberg, P.H. Structure of the ribonucleoprotein of influenza virus. J. Virol. 1972, 10, 795–800. [Google Scholar]
- Ortega, J.; Martin-Benito, J.; Zurcher, T.; Valpuesta, J.M.; Carrascosa, J.L.; Ortin, J. Ultrastructural and functional analyses of recombinant influenza virus ribonucleoproteins suggest dimerization of nucleoprotein during virus amplification. J. Virol. 2000, 74, 156–163. [Google Scholar] [CrossRef]
- Ng, A.K.; Zhang, H.; Tan, K.; Li, Z.; Liu, J.H.; Chan, P.K.; Li, S.M.; Chan, W.Y.; Au, S.W.; Joachimiak, A.; et al. Structure of the influenza virus a h5n1 nucleoprotein: Implications for RNA binding, oligomerization, and vaccine design. FASEB J. 2008, 22, 3638–3647. [Google Scholar] [CrossRef]
- Ye, Q.; Krug, R.M.; Tao, Y.J. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006, 444, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.K.; Lam, M.K.; Zhang, H.; Liu, J.; Au, S.W.; Chan, P.K.; Wang, J.; Shaw, P.C. Structural basis for RNA binding and homo-oligomer formation by influenza B virus nucleoprotein. J. Virol. 2012, 86, 6758–6767. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Fujii, Y.; Noda, T.; Muramoto, Y.; Watanabe, T.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J. Virol. 2005, 79, 3766–3774. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Goto, H.; Watanabe, T.; Yoshida, T.; Kawaoka, Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA 2003, 100, 2002–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Hong, Y.; Parslow, T.G. Cis-acting packaging signals in the influenza virus pb1, pb2, and pa genomic RNA segments. J. Virol. 2005, 79, 10348–10355. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Huang, T.; Ly, H.; Parslow, T.G.; Liang, Y. Mutational analyses of packaging signals in influenza virus pa, pb1, and pb2 genomic RNA segments. J. Virol. 2008, 82, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Marsh, G.A.; Hatami, R.; Palese, P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J. Virol. 2007, 81, 9727–9736. [Google Scholar] [CrossRef]
- Muramoto, Y.; Takada, A.; Fujii, K.; Noda, T.; Iwatsuki-Horimoto, K.; Watanabe, S.; Horimoto, T.; Kida, H.; Kawaoka, Y. Hierarchy among viral RNA (vRNA) segments in their role in vRNA incorporation into influenza A virions. J. Virol. 2006, 80, 2318–2325. [Google Scholar] [CrossRef]
- Ozawa, M.; Fujii, K.; Muramoto, Y.; Yamada, S.; Yamayoshi, S.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 2007, 81, 30–41. [Google Scholar] [CrossRef]
- Watanabe, T.; Watanabe, S.; Noda, T.; Fujii, Y.; Kawaoka, Y. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J. Virol. 2003, 77, 10575–10583. [Google Scholar] [CrossRef]
- Essere, B.; Yver, M.; Gavazzi, C.; Terrier, O.; Isel, C.; Fournier, E.; Giroux, F.; Textoris, J.; Julien, T.; Socratous, C.; et al. Critical role of segment-specific packaging signals in genetic reassortment of influenza A viruses. Proc. Natl. Acad. Sci. USA 2013, 110, E3840–E3848. [Google Scholar] [CrossRef] [PubMed]
- Fournier, E.; Moules, V.; Essere, B.; Paillart, J.C.; Sirbat, J.D.; Cavalier, A.; Rolland, J.P.; Thomas, D.; Lina, B.; Isel, C.; et al. Interaction network linking the human H3N2 influenza A virus genomic RNA segments. Vaccine 2012, 30, 7359–7367. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, B.; Zheng, T.; Gerber, M.; Printz-Schweigert, A.; Ong, C.; Marquet, R.; Isel, C.; Rockman, S.; Brown, L. Influenza NA and PB1 gene segments interact during the formation of viral progeny: Localization of the binding region within the PB1 gene. Viruses 2016, 8, 238. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Smith, C.B.; Mungall, B.A.; Lindstrom, S.E.; Hall, H.E.; Subbarao, K.; Cox, N.J.; Klimov, A. Intercontinental circulation of human influenza A(H1N2) reassortant viruses during the 2001–2002 influenza season. J. Infect. Dis. 2002, 186, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.F.; Nogales, A.; Finch, C.; Tuffy, K.M.; Domm, W.; Perez, D.R.; Topham, D.J.; Martinez-Sobrido, L. Influenza A and B virus intertypic reassortment through compatible viral packaging signals. J. Virol. 2014, 88, 10778–10791. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hatta, M.; Watanabe, S.; Neumann, G.; Kawaoka, Y. Compatibility among polymerase subunit proteins is a restricting factor in reassortment between equine H7N7 and human H3N2 influenza viruses. J. Virol. 2008, 82, 11880–11888. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Lowen, A.C. Implications of segment mismatch for influenza A virus evolution. J. Gen. Virol. 2018, 99, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The Viruses and Their Replication, 6th ed.; Virology, F., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; p. 2456. [Google Scholar]
- Labaronne, A.; Swale, C.; Monod, A.; Schoehn, G.; Crepin, T.; Ruigrok, R.W. Binding of RNA by the nucleoproteins of influenza viruses A and B. Viruses 2016, 8, 247. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Segments | Virus Comparison | ||||||
|---|---|---|---|---|---|---|---|
| H1N1pdm | H3N2 | IBV | |||||
| Pearson | p-Value | Pearson | p-Value | Pearson | p-Value | ||
| Combined eight segments | WSN | 0.42 | <0.00001 | 0.29 | <0.00001 | −0.01 | 0.33816 |
| H1N1pdm | 0.33 | <0.00001 | 0.04 | <0.00001 | |||
| H3N2 | −0.02 | 0.00618 | |||||
| Segment 1 (PB2) | WSN | 0.40 | <0.00001 | 0.33 | <0.00001 | −0.07 | 0.00124 |
| H1N1pdm | 0.20 | <0.00001 | 0.05 | 0.02013 | |||
| H3N2 | 0.35 | <0.00001 | |||||
| Segment 2 (PB1) | WSN | 0.08 | 0.00023 | 0.22 | <0.00001 | 0.14 | <0.00001 |
| H1N1pdm | 0.68 | <0.00001 | 0.01 | 0.73457 | |||
| H3N2 | 0.20 | <0.00001 | |||||
| Segment 3 (PA) | WSN | 0.66 | <0.00001 | 0.65 | <0.00001 | −0.07 | 0.00061 |
| H1N1pdm | 0.67 | <0.00001 | −0.04 | 0.03624 | |||
| H3N2 | −0.15 | <0.00001 | |||||
| Segment 4 (HA) | WSN | 0.70 | <0.00001 | 0.17 | <0.00001 | 0.10 | 2.00 × 10−5 |
| H1N1pdm | 0.16 | <0.00001 | 0.30 | <0.00001 | |||
| H3N2 | 0.11 | <0.00001 | |||||
| Segment 5 (NP) | WSN | 0.41 | <0.00001 | 0.25 | <0.00001 | 0.22 | <0.00001 |
| H1N1pdm | 0.30 | <0.00001 | 0.19 | <0.00001 | |||
| H3N2 | 0.01 | 0.67598 | |||||
| Segment 6 (NA) | WSN | 0.64 | <0.00001 | 0.20 | <0.00001 | −0.06 | 0.0283 |
| H1N1pdm | 0.35 | <0.00001 | 0.01 | 0.81643 | |||
| H3N2 | −0.08 | 0.00274 | |||||
| Segment 7 (M) | WSN | 0.34 | <0.00001 | 0.70 | <0.00001 | −0.04 | 0.20718 |
| H1N1pdm | 0.19 | <0.00001 | −0.03 | 0.30731 | |||
| H3N2 | −0.07 | 0.01504 | |||||
| Segment 8 (NS) | WSN | −0.06 | 0.03796 | 0.36 | <0.00001 | 0.29 | <0.00001 |
| H1N1pdm | 0.45 | <0.00001 | −0.10 | 0.00067 | |||
| H3N2 | 0.13 | 2.00 × 10−5 | |||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Sage, V.; Nanni, A.V.; Bhagwat, A.R.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S.; Lee, N. Non-Uniform and Non-Random Binding of Nucleoprotein to Influenza A and B Viral RNA. Viruses 2018, 10, 522. https://doi.org/10.3390/v10100522
Le Sage V, Nanni AV, Bhagwat AR, Snyder DJ, Cooper VS, Lakdawala SS, Lee N. Non-Uniform and Non-Random Binding of Nucleoprotein to Influenza A and B Viral RNA. Viruses. 2018; 10(10):522. https://doi.org/10.3390/v10100522
Chicago/Turabian StyleLe Sage, Valerie, Adalena V. Nanni, Amar R. Bhagwat, Dan J. Snyder, Vaughn S. Cooper, Seema S. Lakdawala, and Nara Lee. 2018. "Non-Uniform and Non-Random Binding of Nucleoprotein to Influenza A and B Viral RNA" Viruses 10, no. 10: 522. https://doi.org/10.3390/v10100522
APA StyleLe Sage, V., Nanni, A. V., Bhagwat, A. R., Snyder, D. J., Cooper, V. S., Lakdawala, S. S., & Lee, N. (2018). Non-Uniform and Non-Random Binding of Nucleoprotein to Influenza A and B Viral RNA. Viruses, 10(10), 522. https://doi.org/10.3390/v10100522