Interplay between Cellular Metabolism and Cytokine Responses during Viral Infection


Abstract
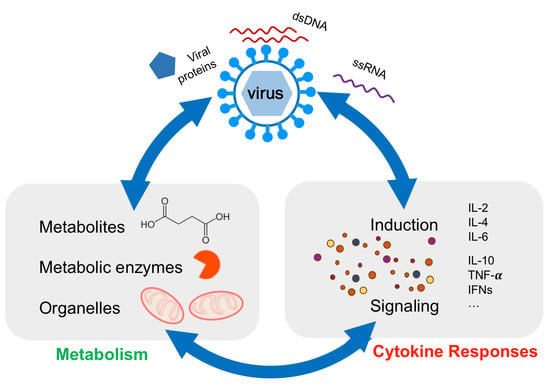
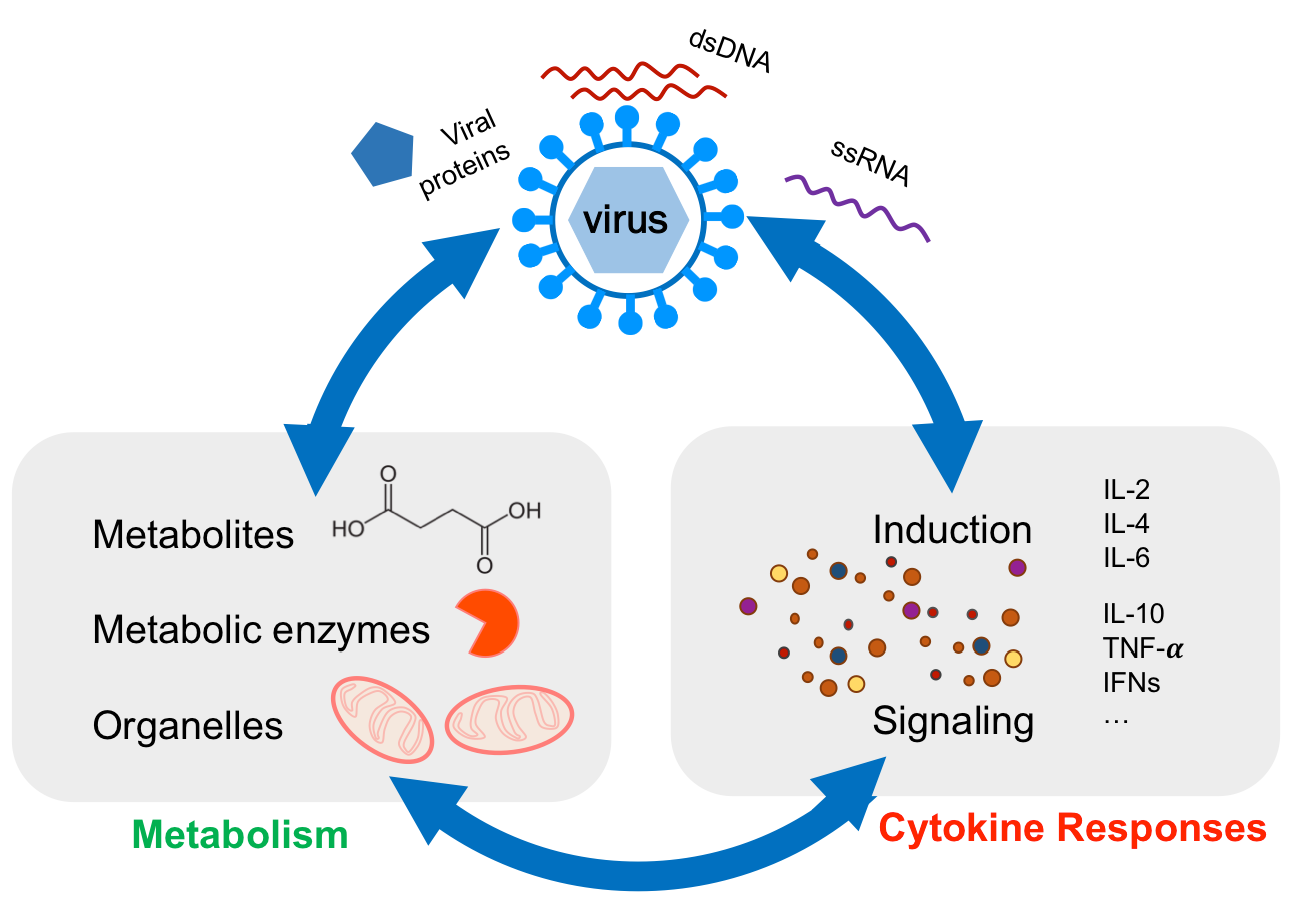
1. Introduction
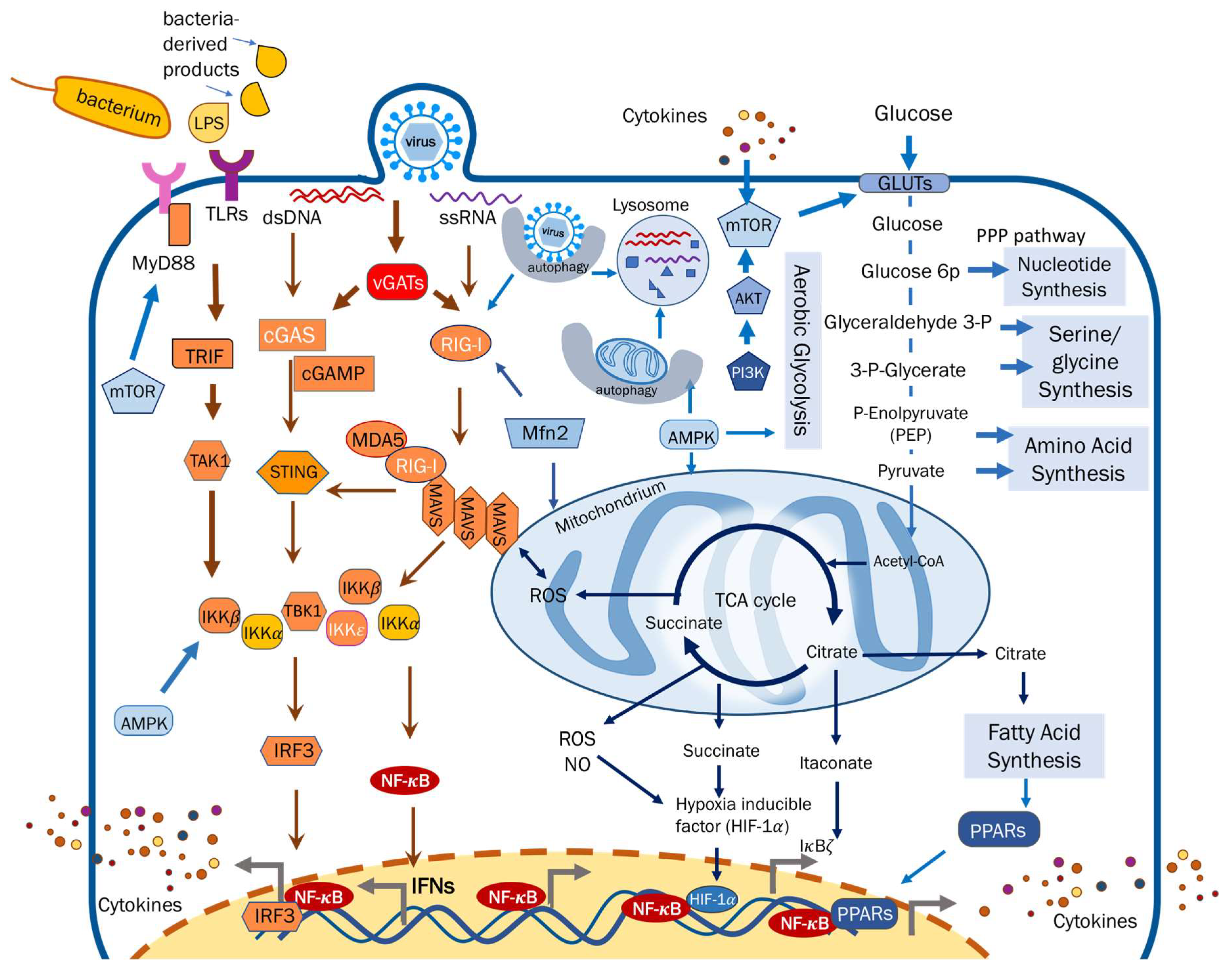
2. Metabolic Reprogramming Regulates Cytokine Responses against Viral Infection
2.1. Metabolites
2.1.1. Carbohydrates
2.1.2. Lipids and Fatty Acids
2.1.3. Amino Acids
2.1.4. Nucleotides
2.2. Metabolic Enzymes
Glutamine Amidotransferases and Deamidases
2.3. Eukaryotic Organelles
2.3.1. Mitochondria
2.3.2. Lysosome
3. Cytokine Responses Guiding Host Metabolic Activity
4. Perspectives
4.1. Molecular Mechanism of Virus-Induced Metabolic Reprogramming
4.2. Metabolic Reprogramming by Anti- and Pro-Inflammatory Cytokines
4.3. Immunometabolism in the Context of Inter-Kingdom Microbial Infections
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Cheng, S.C.; Joosten, L.A.B.; Netea, M.G. The interplay between central metabolism and innate immune responses. Cytokine Growth Factor Rev. 2014, 25, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.H.; Liu, Z.; et al. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.H.; et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J. How low cholesterol is good for anti-viral immunity. Cell 2015, 163, 1572–1574. [Google Scholar]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Lomvardas, S.; Parekh, B.; Yie, J.; Maniatis, T.; Thanos, D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell 2000, 103, 667–678. [Google Scholar] [CrossRef]
- Zhao, J.; He, S.; Minassian, A.; Li, J.; Feng, P. Recent advances on viral manipulation of NF-κB signaling pathway. Curr. Opin. Virol. 2015, 15, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, A.; Bowie, A.G. Innate immune recognition of DNA: A recent history. Virology 2015, 479–480, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and Rig-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Gale, M. Interference with virus infection. J. Immunol. 2015, 195, 1909–1910. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H.; Paludan, S.R. Reading the viral signature by Toll-like receptors and other pattern recognition receptors. J. Mol. Med. 2005, 83, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Rowe, D.C.; Barnes, B.J.; Caffrey, D.R.; Visintin, A.; Latz, E.; Monks, B.; Pitha, P.M.; Golenbock, D.T. Lps-TLR4 signaling to IRF-3/7 and NF-κB involves the Toll adapters TRAM and TRIF. J. Exp. Med. 2003, 198, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. Tak1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E. Nuclear factor—κB and its role in sepsis-associated organ failure. J. Infect. Dis. 2003, 187, S364–S369. [Google Scholar] [CrossRef] [PubMed]
- Tenoever, B.R.; Ng, S.L.; Chua, M.A.; McWhirter, S.M.; García-Sastre, A.; Maniatis, T. Multiple functions of the IKK-related kinase IKK epsilon in interferon-mediated antiviral immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.M.; Xu, S.; Munger, J. Stealing the keys to the kitchen: Viral manipulation of the host cell metabolic network. Trends Microbiol. 2015, 23, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Clippinger, A.J.; Alwine, J.C. Viral effects on metabolism: Changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.C.; Griss, T.; et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Pathirana, R.D.; Kaparakis-Liaskos, M. Bacterial membrane vesicles: Biogenesis, immune regulation and pathogenesis. Cell. Microbiol. 2016, 18, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, R.G.; Sintsova, A.; Buckwalter, C.M.; Leung, N.; Cochrane, A.; Li, J.; Cox, A.D.; Moffat, J.; Gray-Owen, S.D. Cytosolic detection of the bacterial metabolite HBP activates TIFA-dependent innate immunity. Science 2015, 348, 1251–1255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; She, Y.; Dong, N.; Li, P.; He, H.; Borio, A.; Wu, Q.; Lu, S.; Ding, X.; Cao, Y.; et al. Alpha-kinase 1 is a cytosolic innate immune receptor for bacterial ADP-heptose. Nature 2018, 561, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Aounallah, M.; Dagenais-Lussier, X.; El-Far, M.; Mehraj, V.; Jenabian, M.A.; Routy, J.P.; van Grevenynghe, J. Current topics in HIV pathogenesis, part 2: Inflammation drives a warburg-like effect on the metabolism of HIV-infected subjects. Cytokine Growth Factor Rev. 2016, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalili, L.; Bouzakri, K.; Glund, S.; Lönnqvist, F.; Koistinen, H.A.; Krook, A. Signaling specificity of interleukin-6 action on glucose and lipid metabolism in skeletal muscle. Mol. Endocrinol. 2006, 20, 3364–3375. [Google Scholar] [CrossRef] [PubMed]
- Pathmaperuma, A.N.; Maña, P.; Cheung, S.N.; Kugathas, K.; Josiah, A.; Koina, M.E.; Broomfield, A.; Delghingaro-Augusto, V.; Ellwood, D.A.; Dahlstrom, J.E.; et al. Fatty acids alter glycerolipid metabolism and induce lipid droplet formation, syncytialisation and cytokine production in human trophoblasts with minimal glucose effect or interaction. Placenta 2010, 31, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shi, Y.; Wang, G.; Wang, X.; Zeng, S.; Dunn, S.E.; Fairn, G.D.; Li, Y.J.; Spaner, D.E. PPAR-delta modulates membrane cholesterol and cytokine signaling in malignant B cells. Leukemia 2017, 32, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Kim, J.S.; Kang, S.J.; Park, C.G. Lipid metabolism regulator, Acetyl-coA carboxylase: A possible therapeutic target for dendritic cell-based immunotherapy (IRC10P.414). J. Immunol. 2015, 194, 4039–4048. [Google Scholar]
- Panther, E.; Idzko, M.; Corinti, S.; Ferrari, D.; Herouy, Y.; Mockenhaupt, M.; Dichmann, S.; Gebicke-Haerter, P.; Di Virgilio, F.; Girolomoni, G.; et al. The influence of lysophosphatidic acid on the functions of human dendritic cells. J. Immunol. 2002, 169, 4129–4135. [Google Scholar] [CrossRef] [PubMed]
- Horio, Y.; Osawa, S.; Takagaki, K.; Hishida, A.; Furuta, T.; Ikuma, M. Glutamine supplementation increases Th1-cytokine responses in murine intestinal intraepithelial lymphocytes. Cytokine 2008, 44, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Maguire, T.G.; Alwine, J.C. Glutamine metabolism is essential for human cytomegalovirus infection. J. Virol. 2010, 84, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Te, H.S.; Randall, G.; Jensen, D.M. Mechanism of action of ribavirin in the treatment of chronic hepatitis C. Gastroenterol Hepatol (N Y) 2007, 3, 218–225. [Google Scholar]
- Calder, P.C.; Yaqoob, P. Glutamine and the immune system. Amino Acids 1999, 17, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Sartori, T.; Galvão dos Santos, G.; Nogueira-Pedro, A.; Makiyama, E.; Rogero, M.M.; Borelli, P.; Fock, R.A. Effects of glutamine, taurine and their association on inflammatory pathway markers in macrophages. Inflammopharmacology 2018, 26, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hoshino, Y.; Dowdell, K.; Bosch-Marce, M.; Myers, T.G.; Sarmiento, M.; Pesnicak, L.; Krause, P.R.; Cohen, J.I. Glutamine supplementation suppresses herpes simplex virus reactivation. J. Clin. Invest. 2017, 127, 2626–2630. [Google Scholar] [CrossRef] [PubMed]
- Coëffier, M.; Marion, R.; Ducrotté, P.; Déchelotte, P. Modulating effect of glutamine on IL-1β-induced cytokine production by human gut. Clin. Nutr. 2003, 22, 407–413. [Google Scholar] [CrossRef]
- Hubert-Buron, A.l.; Leblond, J.; Jacquot, A.; Ducrotté, P.; Déchelotte, P.; Coëffier, M.S. Glutamine pretreatment reduces IL-8 production in human intestinal epithelial cells by limiting IkappaBalpha ubiquitination. J. Nutr. 2006, 136, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, M.; Toy, T.; Shin, A.; Wagner, M.; Cebon, J.; Maraskovsky, E. Extracellular nucleotide signaling by P2 receptors inhibits IL-12 and enhances IL-23 expression in human dendritic cells: A novel role for the camp pathway. Blood 2005, 105, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.P.; Buch-Pedersen, M.J.; Preben Morth, J.; Palmgren, M.G.; Nissen, P. Crystal structure of the plasma membrane proton pump. Nature 2007, 450, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.Y.; Constantinides, C.; Kebschull, M.; Papapanou, P.N. Micrornas regulate cytokine responses in gingival epithelial cells. Infect. Immun. 2016, 84, 3282–3289. [Google Scholar] [CrossRef] [PubMed]
- Asirvatham, A.J.; Magner, W.J.; Tomasi, T.B. Mirna regulation of cytokine genes. Cytokine 2009, 45, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Danilchanka, O.; Mekalanos, J.J. Cyclic dinucleotides and the innate immune response. Cell 2013, 154, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. Sting is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H. Viral encounters with 2′,5′-oligoadenylate synthetase and rnase l during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [PubMed]
- Rebouillat, D.; Hovanessian, A.G. The human 2′,5′-oligoadenylate synthetase family: Interferon-induced proteins with unique enzymatic properties. J. Interferon Cytokine Res. 1999, 19, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase l amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Kern, E.R.; Kushner, N.L.; Hartline, C.B.; Williams-Aziz, S.L.; Harden, E.A.; Zhou, S.; Zemlicka, J.; Prichard, M.N. In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob. Agents Chemother. 2005, 49, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Isorce, N.; Testoni, B.; Locatelli, M.; Fresquet, J.; Rivoire, M.; Luangsay, S.; Zoulim, F.; Durantel, D. Antiviral activity of various interferons and pro-inflammatory cytokines in non-transformed cultured hepatocytes infected with hepatitis B virus. Antiviral Res. 2016, 130, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mtor inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.; Richard, D.; Laplante, M. The roles of mtor complexes in lipid metabolism. Annu. Rev. Nutr. 2015, 35, 321–348. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.; Heit, A.; Dreher, S.; Eisenächer, K.; Mages, J.; Haas, T.; Krug, A.; Janssen, K.P.; Kirschning, C.J.; Wagner, H. Mammalian target of rapamycin (mtor) orchestrates the defense program of innate immune cells. Eur. J. Immunol. 2008, 38, 2981–2992. [Google Scholar] [CrossRef] [PubMed]
- Loftus, R.M.; Finlay, D.K. Immunometabolism: Cellular metabolism turns immune regulator. J. Biol. Chem. 2016, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into rna biology from an atlas of mammalian mrna-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Richmond, A.L.; Kabi, A.; Homer, C.R.; Marina-García, N.; Nickerson, K.P.; Nesvizhskii, A.I.; Sreekumar, A.; Chinnaiyan, A.M.; Nuñez, G.; McDonald, C. The nucleotide synthesis enzyme cad inhibits nod2 antibacterial function in human intestinal epithelial cells. Gastroenterology 2012, 142, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Gilst, M.R.V.; Hadjivassiliou, H.; Jolly, A.; Yamamoto, K.R. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. Elegans. PLOS Biol. 2005, 3, e53. [Google Scholar] [CrossRef] [PubMed]
- Ratnappan, R.; Ward, J.D.; Yamamoto, K.R.; Ghazi, A. Nuclear hormone receptors as mediators of metabolic adaptability following reproductive perturbations. Worm 2016, 5, e1151609. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.C. Regulation of cytokine receptor signaling by nuclear hormone receptors: A new paradigm for receptor interaction. DNA Cell Biol. 2004, 23, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Viollet, B. Beyond energy homeostasis: The expanding role of amp-activated protein kinase in regulating metabolism. Cell Metab. 2015, 21, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Théret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Bess, E.; Fisslthaler, B.; Frömel, T.; Fleming, I. Nitric oxide-induced activation of the AMP-activated protein kinase α2 subunit attenuates IκB kinase activity and inflammatory responses in endothelial cells. PLoS ONE 2011, 6, e20848. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, C.; Speirs, C.; Williams, J.J.L.; Ewart, M.A.; Mancini, S.J.; Hawley, S.A.; Delles, C.; Viollet, B.; Costa-Pereira, A.P.; Baillie, G.S.; et al. Phosphorylation of janus kinase 1 (JAK1) by amp-activated protein kinase (AMPK) links energy sensing to anti-inflammatory signaling. Sci. Signal. 2016, 9, ra109. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Hankinson, O. The role of ahr-inducible cytochrome P450s in metabolism of polyunsaturated fatty acids. Drug Metab. Rev. 2016, 48, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.B.; Winans, B.; Martin, K.C.; Lawrence, B.P. New insights into the role of the aryl hydrocarbon receptor in the function of CD11C+ cells during respiratory viral infection. Eur. J. Immunol. 2014, 44, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links Th17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Amaro, R.E.; Sethi, A.; Myers, R.S.; Davisson, V.J.; Luthey-Schulten, Z.A. A network of conserved interactions regulates the allosteric signal in a glutamine amidotransferase. Biochemistry 2007, 46, 2156–2173. [Google Scholar] [CrossRef] [PubMed]
- Dho, S.H.; Deverman, B.E.; Lapid, C.; Manson, S.R.; Gan, L.; Riehm, J.J.; Aurora, R.; Kwon, K.S.; Weintraub, S.J. Control of cellular Bcl-xL levels by deamidation-regulated degradation. PLOS Biol. 2013, 11, e1001588. [Google Scholar] [CrossRef] [PubMed]
- Flatau, G.; Lemichez, E.; Gauthier, M.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the g protein p21 Rho by deamidation of glutamine. Nature 1997, 387, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Sugai, M.; Hatazaki, K.; Mogami, A.; Ohta, H.; Pérès, S.Y.; Hérault, F.; Horiguchi, Y.; Masuda, M.; Ueno, Y.; Komatsuzawa, H.; et al. Cytotoxic Necrotizing Factor Type 2 Produced by Pathogenic Escherichia coli Deamidates a Gln Residue in the Conserved G-3 Domain of the Rho Family and Preferentially Inhibits the GTPase Activity of RhoA and Rac1. Infect. Immun. 1999, 67, 6550–6557. [Google Scholar] [PubMed]
- Zhao, J.; Li, J.; Xu, S.; Feng, P. Emerging roles of protein deamidation in innate immune signaling. J. Virol. 2016, 90, 4262–4268. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhao, J.; Song, S.; He, X.; Minassian, A.; Zhou, Y.; Zhang, J.; Brulois, K.; Wang, Y.; Cabo, J.; et al. Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 2015, 58, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zeng, Y.; Xu, S.; Chen, J.; Shen, G.; Yu, C.; Knipe, D.; Yuan, W.; Peng, J.; Xu, W.; et al. A viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 2016, 20, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, J.; Xu, S.; Li, J.; He, S.; Zeng, Y.; Xie, L.; Xie, N.; Liu, T.; Lee, K.; et al. Species-specific deamidation of cGAS facilitates herpes simplex virus lytic replication. Cell Host Microbe 2018. [Google Scholar] [CrossRef] [PubMed]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Stürzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposi's sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLOS Pathog. 2014, 10, e1003863. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Wies, E.; Thurau, M.; Marquardt, G.; Naschberger, E.; Hentschel, S.; Jochmann, R.; Schulz, T.F.; Erfle, H.; Brors, B.; et al. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-κB activation. J. Virol. 2009, 83, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Van Skike, N.D.; Minkah, N.K.; Hogan, C.H.; Wu, G.; Benziger, P.T.; Oldenburg, D.G.; Kara, M.; Kim-Holzapfel, D.M.; White, D.W.; Tibbetts, S.A.; et al. Viral fgarat ORF75A promotes early events in lytic infection and gammaherpesvirus pathogenesis in mice. PLOS Pathog. 2018, 14, e1006843. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Imamura, H.; Taku, T.; Kuroki, T.; Kawaguchi, A.; Ishikawa, K.; Nakada, K.; Koshiba, T. RLR-mediated antiviral innate immunity requires oxidative phosphorylation activity. Sci. Rep. 2017, 7, 5379. [Google Scholar] [CrossRef] [PubMed]
- Calfee, C.S.; Matthay, M.A. Culprits with evolutionary ties. Nature 2010, 464, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, X.; Nie, X.; Sun, L.; Tang, T.S.; Chen, D.; Sun, Q. Cox5b regulates mavs-mediated antiviral signaling through interaction with ATG5 and repressing ROS production. PLOS Pathog. 2012, 8, e1003086. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Hur, S. How RIG-I like receptors activate MAVS. Curr. Opin. Virol. 2015, 12, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the rig-i antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. Visa is an adapter protein required for virus-triggered IFN- beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. Ips-1, an adaptor triggering RIG-I and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ahn, D.G.; Syed, G.H.; Siddiqui, A. The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion 2018, 41, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S. Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza Virus Protein PB1-F2 Inhibits the Induction of Type I Interferon by Binding to MAVS and Decreasing Mitochondrial Membrane Potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, K.; Oshiumi, H.; Takeda, M.; Ishihara, N.; Yanagi, Y.; Seya, T.; Kawabata, S.I.; Koshiba, T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009, 2, ra47. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis c virus protease NS3/4a cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Ge, W.; Li, D.; Gao, Y.; Cao, X. The roles of lysosomes in inflammation and autoimmune diseases. Int. Rev. Immunol. 2015, 34, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Iyer, J.K.; Khurana, T.; Langer, M.; West, C.M.; Ballard, J.D.; Metcalf, J.P.; Merkel, T.J.; Coggeshall, K.M. Inflammatory cytokine response to bacillus anthracis peptidoglycan requires phagocytosis and lysosomal trafficking. Infect. Immun. 2010, 78, 2418–2428. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.L.; Denton, P.W.; O’Neill, E.; Luo, T.; Foster, J.L.; Garcia, J.V. Inhibition of lysosome and proteasome function enhances human immunodeficiency virus type 1 infection. J. Virol. 2005, 79, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The ATG5–ATG12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [PubMed]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of autophagy results in reactive oxygen species-dependent amplification of rlr signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef] [PubMed]
- Bartee, E.; McFadden, G. Cytokine synergy: An underappreciated contributor to innate anti-viral immunity. Cytokine 2013, 63, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Escoll, P.; Buchrieser, C. Metabolic reprogramming of host cells upon bacterial infection: Why shift to a warburg-like metabolism? FEBS J. 2018, 285, 2146–2160. [Google Scholar] [CrossRef] [PubMed]
- Yogev, O.; Lagos, D.; Enver, T.; Boshoff, C. Kaposi’s sarcoma herpesvirus micrornas induce metabolic transformation of infected cells. PLOS Pathog. 2014, 10, e1004400. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.A.; Garcia-Smith, R.; Trujillo, K.A.; Bisoffi, M. Tumor necrosis factor alpha increases aerobic glycolysis and reduces oxidative metabolism in prostate epithelial cells. Prostate 2013, 73, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, R.; Kanety, H.; Papa, M.Z.; Lunenfeld, B.; Karasik, A. Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J. Biol. Chem. 1993, 268, 26055–26058. [Google Scholar] [PubMed]
- Hummel, D.M.; Fetahu, I.S.; Gröschel, C.; Manhardt, T.; Kállay, E. Role of proinflammatory cytokines on expression of vitamin d metabolism and target genes in colon cancer cells. J. Steroid Biochem. Mol. Biol. 2014, 144, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Ray, J.P.; Staron, M.M.; Shyer, J.A.; Ho, P.C.; Marshall, H.D.; Gray, S.M.; Laidlaw, B.J.; Araki, K.; Ahmed, R.; Kaech, S.M.; et al. The interleukin-2-mtorc1 kinase axis defines the signaling, differentiation, and metabolism of t helper 1 and follicular b helper T cells. Immunity 2015, 43, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Szczepankiewicz, D.; Skrzypski, M.; Pruszyńska-Oszmałek, E.; Kołodziejski, P.A.; Sassek, M.; Stefańska, B.; Nowak, K.W.; Szczepankiewicz, A. Interleukin 4 affects lipid metabolism and the expression of pro-inflammatory factors in mature rat adipocytes. Immunobiology 2018, 223, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Tsao, C.H.; Shiau, M.Y.; Chuang, P.H.; Chang, Y.H.; Hwang, J. Interleukin-4 regulates lipid metabolism by inhibiting adipogenesis and promoting lipolysis. J. Lipid Res. 2014, 55, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Steensberg, A. The role of IL-6 in exercise-induced immune changes and metabolism. Exerc. Immunol. Rev. 2003, 9, 40–47. [Google Scholar] [PubMed]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Dagdeviren, S.; Jung, D.Y.; Friedline, R.H.; Noh, H.L.; Kim, J.H.; Patel, P.R.; Tsitsilianos, N.; Inashima, K.; Tran, D.A.; Hu, X.; et al. IL-10 prevents aging-associated inflammation and insulin resistance in skeletal muscle. FASEB J. 2017, 31, 701–710. [Google Scholar] [CrossRef] [PubMed]
- York, A.G.; Williams, K.J.; Argus, J.P.; Zhou, Q.D.; Brar, G.; Vergnes, L.; Gray, E.E.; Zhen, A.; Wu, N.C.; Yamada, D.H.; et al. Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 2015, 163, 1716–1729. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Brönneke, H.S.; et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Vazquez, I.; Fernández-Veledo, S.; de Alvaro, C.; Lorenzo, M. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes 2008, 57, 3211–3221. [Google Scholar] [CrossRef] [PubMed]
- Modrow, S.; Falke, D.; Truyen, U.; Schätzl, H. Cytokines, chemokines and interferons. In Molecular Virology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 95–114. [Google Scholar]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Yu, Y.; Zhong, Y.; Giannopoulou, E.G.; Hu, X.; Liu, H.; Cross, J.R.; Rätsch, G.; Rice, C.M.; Ivashkiv, L.B. Interferon-γ regulates cellular metabolism and mrna translation to potentiate macrophage activation. Nat. Immunol. 2015, 16, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.K.; Castrillo, A.; Shahangian, A.; Pei, L.; O’Connell, R.M.; Modlin, R.L.; Tontonoz, P.; Cheng, G. A role for IRF3-dependent RXRα repression in hepatotoxicity associated with viral infections. J. Exp. Med. 2006, 203, 2589–2602. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Naka, T.; Kubo, M. Socs proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Piñeros Alvarez, A.R.; Glosson-Byers, N.; Brandt, S.; Wang, S.; Wong, H.; Sturgeon, S.; McCarthy, B.P.; Territo, P.R.; Alves-Filho, J.C.; Serezani, C.H. SOCS1 is a negative regulator of metabolic reprogramming during sepsis. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Lagunoff, M. Activation of cellular metabolism during latent Kaposi’s Sarcoma herpesvirus infection. Curr. Opin. Virol. 2016, 19, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Breen, E.C. Pro- and anti-inflammatory cytokines in human immunodeficiency virus infection and acquired immunodeficiency syndrome. Pharmacol. Ther. 2002, 95, 295–304. [Google Scholar] [CrossRef]
- Nakamura, S.; Davis, K.M.; Weiser, J.N. Synergistic stimulation of type i interferons during influenza virus coinfection promotes streptococcus pneumoniae colonization in mice. J. Clin. Invest. 2011, 121, 3657–3665. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Merino, N.; Okamoto, A.; Gedalanga, P. Interkingdom microbial consortia mechanisms to guide biotechnological applications. Microb. Biotechnol. 2018, 11, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, S.H.; Kim, M.; Moon, J.S.; Kim, G.W.; Jung, H.G.; Kim, I.H.; Oh, J.E.; Jung, H.E.; Lee, H.K.; et al. Vibrio vulnificus quorum-sensing molecule cyclo(Phe-Pro) inhibits RIG-I-mediated antiviral innate immunity. Nat. Commun. 2018, 9, 1606. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cytokine | Effects on Metabolism | References |
|---|---|---|
| TNF-α | Induces Insulin resistance; increase glycolysis, adenosine triphosphate (ATP) production, and lactate export; reduce vitamin metabolism | [125,126,127] |
| IL-2 | Increases glucose metabolism via Akt-mTOR signaling to promote T cell differentiation | [128] |
| IL-4 | Up-regulates the expression of glucose transporter 4 (GLUT4); enhance glucose and lipid metabolism | [129,130] |
| IL-6 | Reduces vitamin metabolism; enhance lipolysis | [127,131] |
| IL-10 | Promotes insulin sensitivity; inhibits aerobic glycolysis and promotes oxidative phosphorylation. | [132,133] |
| IFNs | Induce fatty acid oxidation; reduce lipid biosynthesis | [2,134] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Carriere, J.; Lin, X.; Xie, N.; Feng, P. Interplay between Cellular Metabolism and Cytokine Responses during Viral Infection. Viruses 2018, 10, 521. https://doi.org/10.3390/v10100521
Zhang S, Carriere J, Lin X, Xie N, Feng P. Interplay between Cellular Metabolism and Cytokine Responses during Viral Infection. Viruses. 2018; 10(10):521. https://doi.org/10.3390/v10100521
Chicago/Turabian StyleZhang, Shu, Jessica Carriere, Xiaoxi Lin, Na Xie, and Pinghui Feng. 2018. "Interplay between Cellular Metabolism and Cytokine Responses during Viral Infection" Viruses 10, no. 10: 521. https://doi.org/10.3390/v10100521
APA StyleZhang, S., Carriere, J., Lin, X., Xie, N., & Feng, P. (2018). Interplay between Cellular Metabolism and Cytokine Responses during Viral Infection. Viruses, 10(10), 521. https://doi.org/10.3390/v10100521