Risk of Human Papillomavirus Infection in Cancer-Prone Individuals: What We Know


{kind=link}
Abstract
1. HPV Infection, a Significant Threat to Public Health
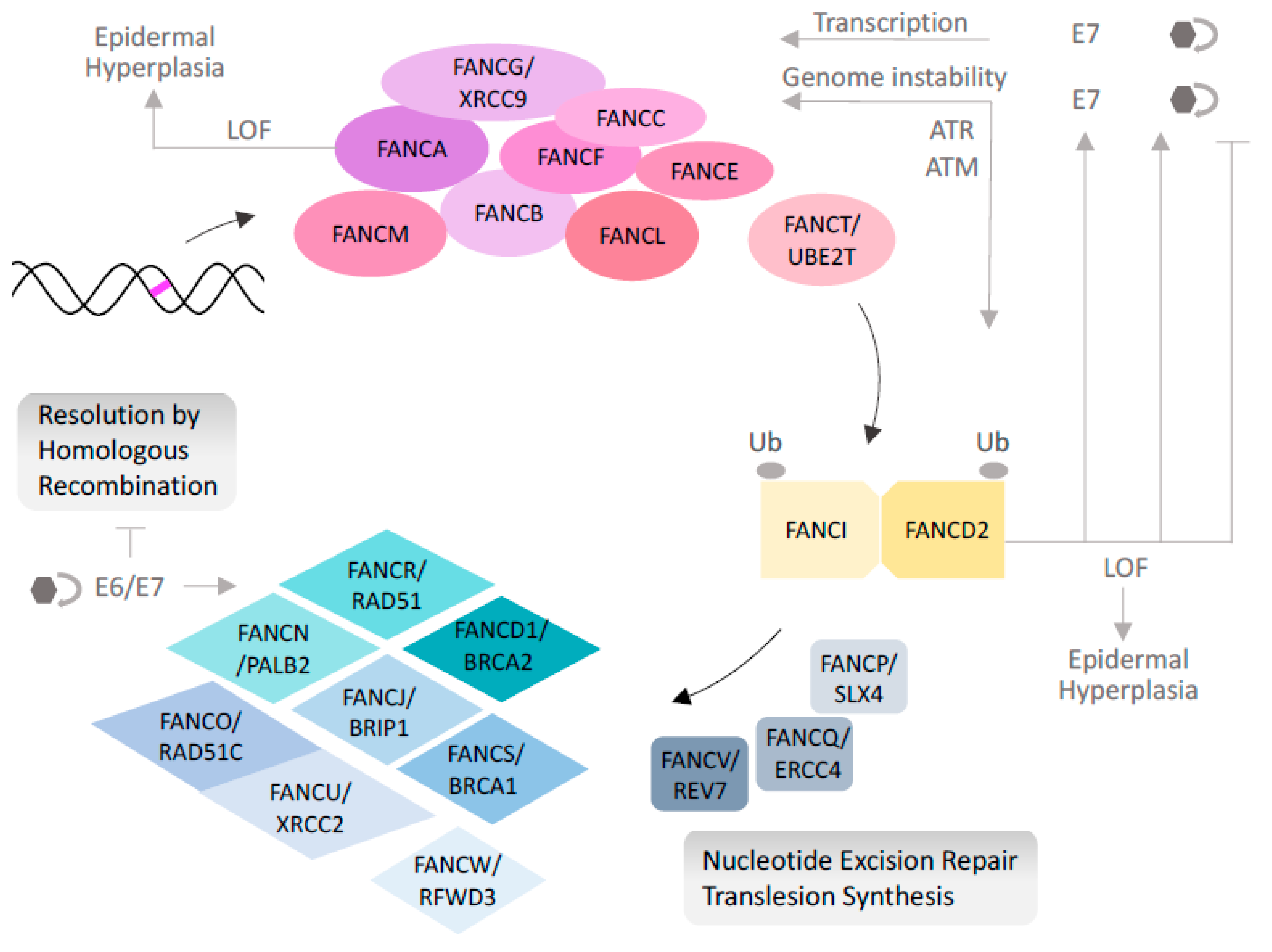
2. Fanconi Anemia, an Inherited DNA Repair Syndrome
3. The Many Phenotypes of Fanconi Anemia
4. FA Loss Stimulates HPV Genome Amplification, Integration, and Oncogenicity
5. Immune dysfunction in FA and HPV susceptibility
6. Epidemiological Studies in Fanconi Anemia Demonstrate Increased Oral HPV Prevalence
7. Response to HPV Vaccine in Individuals with FA
8. Epidermal abnormalities in Fanconi Anemia
9. Summary
Acknowledgments
Conflicts of Interest
References
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global burden of human papillomavirus and related diseases. Vaccine 2012, 30, F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. The search for infectious causes of human cancers: Where and why (Nobel lecture). Angew. Chem. Int. Ed. 2009, 48, 5798–5808. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E. Human papillomaviruses and non-melanoma skin cancer. Semin. Oncol. 2015, 42, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Prigge, E.S.; von Knebel Doeberitz, M.; Reuschenbach, M. Clinical relevance and implications of HPV-induced neoplasia in different anatomical locations. Mutat. Res. Rev. Mutat. Res. 2017, 772, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Frazer, I.H. Prevention of cervical cancer through papillomavirus vaccination. Nat. Rev. Immunol. 2004, 4, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Muller, M. Next generation prophylactic human papillomavirus vaccines. Lancet Oncol. 2015, 16, e217–e225. [Google Scholar] [CrossRef]
- Hofstetter, A.M.; Rosenthal, S.L. Factors impacting HPV vaccination: Lessons for health care professionals. Expert Rev. Vaccines 2014, 13, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Bodily, J.; Laimins, L.A. Persistence of human papillomavirus infection: Keys to malignant progression. Trends Microbiol. 2011, 19, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T. Model systems to study the life cycle of human papillomaviruses and HPV-associated cancers. Virol. Sin. 2015, 30, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Watt, F.M. Stem cell heterogeneity and plasticity in epithelia. Cell Stem Cell 2015, 16, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Model systems of human papillomavirus-associated disease. J. Pathol. 2016, 238, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.M.; Herrera, R.; Chin-Hong, P.; Veluppillai, P.; Greenspan, D.; Michael Berry, J.; Pilcher, C.D.; Shiboski, C.H.; Jay, N.; Rubin, M.; et al. HIV-associated disruption of mucosal epithelium facilitates paracellular penetration by human papillomavirus. Virology 2013, 446, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Brooke, M.A.; Nitoiu, D.; Kelsell, D.P. Cell-cell connectivity: Desmosomes and disease. J. Pathol. 2012, 226, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Broussard, J.A.; Getsios, S.; Green, K.J. Desmosome regulation and signaling in disease. Cell Tissue Res. 2015, 360, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Ishida-Yamamoto, A.; Igawa, S. Genetic skin diseases related to desmosomes and corneodesmosomes. J. Dermatol. Sci. 2014, 74, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Brandner, J.M.; Zorn-Kruppa, M.; Yoshida, T.; Moll, I.; Beck, L.A.; de Benedetto, A. Epidermal tight junctions in health and disease. Tissue Barriers 2015, 3, e974451. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, M.; Zheng, Z.M. Oncogenes and RNA splicing of human tumor viruses. Emerg. Microbes Infect. 2014, 3, e63. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mo.l Cell. Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 2009, 11, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.P.; Cho, Y.J.; Huang, H.; Kim, H.Y.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Minko, I.G.; Lloyd, R.S.; Harris, T.M.; et al. Interstrand DNA cross-links induced by α, β-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 2008, 41, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Ghittoni, R.; Accardi, R.; Hasan, U.; Gheit, T.; Sylla, B.; Tommasino, M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes 2010, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Munger, K. Mechanisms of genomic instability in human cancer: Insights from studies with human papillomavirus oncoproteins. Int. J. Cancer 2004, 109, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.A.; Tolar, J. Fanconi Anemia. In Genereviews(r); Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.S.; Huang, Y.; Alter, B.P. Individualized risks of first adverse events in patients with Fanconi anemia. Blood 2004, 104, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.A.; Davies, S.M.; Leemhuis, T.; Myers, K.; Kernan, N.A.; Prockop, S.E.; Scaradavou, A.; O’Reilly, R.J.; Williams, D.A.; Lehmann, L.; et al. Radiation-free, alternative-donor HCT for Fanconi anemia patients: Results from a prospective multi-institutional study. Blood 2017, 129, 2308–2315. [Google Scholar] [CrossRef] [PubMed]
- Boulad, F.; Gillio, A.; Small, T.N.; George, D.; Prasad, V.; Torok-Castanza, J.; Regan, A.D.; Collins, N.; Auerbach, A.D.; Kernan, N.A.; et al. Stem cell transplantation for the treatment of Fanconi anaemia using a fludarabine-based cytoreductive regimen and T-cell-depleted related HLA-mismatched peripheral blood stem cell grafts. Br. J. Haematol. 2000, 111, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Zecca, M.; Pession, A.; Morreale, G.; Longoni, D.; Di Bartolomeo, P.; Porta, F.; Fagioli, F.; Nobili, B.; Bernardo, M.E.; et al. The outcome of children with Fanconi anemia given hematopoietic stem cell transplantation and the influence of fludarabine in the conditioning regimen: A report from the Italian pediatric group. Haematologica 2007, 92, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Patel, K.R.; Auerbach, A.D.; Kennedy, J.; Lach, F.P.; Sanborn, E.; Cohen, M.A.; Kuhel, W.I.; Smogorzewska, A. Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. Laryngoscope 2016, 126, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Singh, B.; Satagopan, J.; Batish, S.D.; Berwick, M.; Giampietro, P.F.; Hanenberg, H.; Auerbach, A.D. A 20-year perspective on the international Fanconi anemia registry (IFAR). Blood 2003, 101, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, H.; Beech, T.; Nicholson, T.; El-Hariry, I.; McConkey, C.; Paleri, V.; Roberts, S. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer—Systematic review and meta-analysis of trends by time and region. Head Neck 2013, 35, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Van Zeeburg, H.J.; Snijders, P.J.; Wu, T.; Gluckman, E.; Soulier, J.; Surralles, J.; Castella, M.; van der Wal, J.E.; Wennerberg, J.; Califano, J.; et al. Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J. Natl. Cancer Inst. 2008, 100, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N.; Savage, S.A.; Quint, W.G.; de Koning, M.N.; Schiffman, M. Squamous cell carcinomas in patients with Fanconi anemia and dyskeratosis congenita: A search for human papillomavirus. Int. J. Cancer 2013, 133, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, M.R.; Rubira-Bullen, I.R.; Santos, C.F.; Dionisio, T.J.; Bonfim, C.M.; De Marco, L.; Gillio-Tos, A.; Merletti, F. High prevalence of oral human papillomavirus infection in Fanconi’s anemia patients. Oral Dis. 2011, 17, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Sauter, S.L.; Wells, S.I.; Zhang, X.; Hoskins, E.E.; Davies, S.M.; Myers, K.C.; Mueller, R.; Panicker, G.; Unger, E.R.; Sivaprasad, U.; et al. Oral human papillomavirus is common in individuals with Fanconi anemia. Cancer Epidemiol. Prev. Biomark. 2015, 24, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Gulbahce, N.; Yan, H.; Dricot, A.; Padi, M.; Byrdsong, D.; Franchi, R.; Lee, D.S.; Rozenblatt-Rosen, O.; Mar, J.C.; Calderwood, M.A.; et al. Viral perturbations of host networks reflect disease etiology. PLoS Comput. Biol. 2012, 8, e1002531. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, L.A.; van Baars, R.; Terlou, A.; Heijmans-Antonissen, C.; Swagemakers, S.M.; van der Spek, P.J.; Ewing, P.C.; van Beurden, M.; van der Meijden, W.I.; Helmerhorst, T.J.; et al. Different DNA damage and cell cycle checkpoint control in low- and high-risk human papillomavirus infections of the vulva. Int. J. Cancer 2012, 130, 2874–2885. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Gunawardena, R.W.; Habash, K.B.; Wise-Draper, T.M.; Jansen, M.; Knudsen, E.S.; Wells, S.I. Coordinate regulation of Fanconi anemia gene expression occurs through the RB/E2F pathway. Oncogene 2008, 27, 4798–4808. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Morreale, R.J.; Werner, S.P.; Higginbotham, J.M.; Laimins, L.A.; Lambert, P.F.; Brown, D.R.; Gillison, M.L.; Nuovo, G.J.; Witte, D.P.; et al. The Fanconi anemia pathway limits human papillomavirus replication. J. Virol. 2012, 86, 8131–8138. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, E.E.; Morris, T.A.; Higginbotham, J.M.; Spardy, N.; Cha, E.; Kelly, P.; Williams, D.A.; Wikenheiser-Brokamp, K.A.; Duensing, S.; Wells, S.I. Fanconi anemia deficiency stimulates HPV-associated hyperplastic growth in organotypic epithelial raft culture. Oncogene 2009, 28, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Laimins, L.A. FANCD2 binds human papillomavirus genomes and associates with a distinct set of DNA repair proteins to regulate viral replication. mBio 2017, 8, e02340-16. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High-risk alphapapillomavirus oncogenes impair the homologous recombination pathway. J. Virol. 2017, 91, e01084-17. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Pitot, H.C.; Strati, K.; Spardy, N.; Duensing, S.; Grompe, M.; Lambert, P.F. Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer. Cancer Res. 2010, 70, 9959–9968. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Shin, M.K.; Lambert, P.F. High incidence of female reproductive tract cancers in FA-deficient HPV16-transgenic mice correlates with E7’s induction of DNA damage response, an activity mediated by E7’s inactivation of pocket proteins. Oncogene 2014, 33, 3383–3391. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Shin, M.K.; Pitot, H.C.; Lambert, P.F. High incidence of HPV-associated head and neck cancers in FA deficient mice is associated with E7’s induction of DNA damage through its inactivation of pocket proteins. PLoS ONE 2013, 8, e75056. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Lui, V.W.; Grandis, J.R.; Wells, S.I. The Fanconi anemia pathway: Repairing the link between DNA damage and squamous cell carcinoma. Mutat. Res. 2013, 743, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Hoskins, E.E.; Vinnedge, L.M.P.; Foglesong, G.D.; Brusadelli, M.G.; Potter, S.S.; Komurov, K.; Brugmann, S.A.; Lambert, P.F.; Kimple, R.J.; et al. Defects in the Fanconi anemia pathway in head and neck cancer cells stimulate tumor cell invasion through DNA-PK and RAC1 signaling. Clin. Cancer Res. 2016, 22, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Park, J.W.; Pitot, H.C.; Lambert, P.F. Loss of dependence on continued expression of the human papillomavirus 16 E7 oncogene in cervical cancers and precancerous lesions arising in Fanconi anemia pathway-deficient mice. mBio 2016, 7, e00628-16. [Google Scholar] [CrossRef] [PubMed]
- Froom, P.; Aghai, E.; Dobinsky, J.B.; Quitt, M.; Lahat, N. Reduced natural killer activity in patients with Fanconi’s anemia and in family members. Leuk. Res. 1987, 11, 197–199. [Google Scholar] [CrossRef]
- Hersey, P.; Edwards, A.M.; Lewis, R.; Kemp, A.H.; McInnes, J. Deficient natural killer cell activity in a patient with Fanconi’s anaemia and squamous cell carcinoma. Clin. Exp. Immunol. 1982, 48, 205–212. [Google Scholar] [PubMed]
- Petridou, M.; Barrett, A. Physical and laboratory characteristics of heterozygote carriers of the Fanconi aplasia gene. Acta Paediatr. 1990, 79, 1069–1074. [Google Scholar] [CrossRef]
- Lebbe, C.; Pinquier, L.; Rybojad, M.; Chomienne, C.; Ochonisky, S.; Miclea, J.M.; Gluckman, E.; Morel, P. Fanconi’s anaemia associated with multicentric bowen’s disease and decreased NK cytotoxicity. Br. J. Dermatol. 1993, 129, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Shahidi, N. Tumor necrosis factor-alpha overproduction in Fanconi’s anemia. Am. J. Hematol. 1993, 42, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Rosselli, F.; Sanceau, J.; Gluckman, E.; Wietzerbin, J.; Moustacchi, E. Abnormal lymphokine production: A novel feature of the genetic disease Fanconi anemia. II. In vitro and in vivo spontaneous overproduction of tumor necrosis factor alpha. Blood 1994, 83, 1216–1225. [Google Scholar] [PubMed]
- Roxo, P., Jr.; Arruda, L.K.; Nagao, A.T.; Carneiro-Sampaio, M.M.; Ferriani, V.P. Allergic and immunologic parameters in patients with Fanconi’s anemia. Int. Arch. Allergy Immunol. 2001, 125, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Suzergoz, F.; Gurol, A.; Evcimik, F.; Yalman, N. Lymphoproliferative response of Fanconi anemia patients to mitogens, bacterial and viral antigens in vitro. Harran Üniv. Tıp Fak. Derg. 2008, 5, 19–23. [Google Scholar]
- Castello, G.; Gallo, C.; Napolitano, M.; Ascierto, P.A. Immunological phenotype analysis of patients with Fanconi’s anaemia and their family members. Acta Haematol. 1998, 100, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Justo, G.A.; Bitencourt, M.A.; Pasquini, R.; Castelo-Branco, M.T.; Almeida-Oliveira, A.; Diamond, H.R.; Rumjanek, V.M. Immune status of Fanconi anemia patients: Decrease in T CD8 and CD56dim CD16+ NK lymphocytes. Ann. Hematol. 2014, 93, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.C.; Bleesing, J.J.; Davies, S.M.; Zhang, X.; Martin, L.J.; Mueller, R.; Harris, R.E.; Filipovich, A.H.; Kovacic, M.B.; Wells, S.I.; et al. Impaired immune function in children with Fanconi anaemia. Br. J. Haematol. 2011, 154, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Giri, N.; Alter, B.P.; Penrose, K.; Falk, R.T.; Pan, Y.; Savage, S.A.; Williams, M.; Kemp, T.J.; Pinto, L.A. Immune status of patients with inherited bone marrow failure syndromes. Am. J. Hematol. 2015, 90, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.C.; Sauter, S.; Zhang, X.; Bleesing, J.J.; Davies, S.M.; Wells, S.I.; Mehta, P.A.; Kumar, A.; Marmer, D.; Marsh, R.; et al. Impaired immune function in children and adults with Fanconi anemia. Pediatr. Blood Cancer 2017, 64, e26599. [Google Scholar] [CrossRef] [PubMed]
- Winer, R.L.; Huang, C.E.; Cherne, S.; Stern, J.E.; Butsch Kovacic, M.S.; Mehta, P.A.; Sauter, S.L.; Galloway, D.A.; Katzenellenbogen, R.A. Detection of human papillomavirus in the oral cavities of persons with Fanconi anemia. Oral Dis. 2015, 21, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Ryndock, E.J.; Meyers, C. A risk for non-sexual transmission of human papillomavirus? Expert Rev. Anti-Infect. Ther. 2014, 12, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Dinshaw, J.E.; Frazer, I.H.; Garcia, P.J.; Kahn, J.; Markowitz, L.E.; Munoz, N.; Ndumbe, P.M.; Pitisuttithum, P.; Beutels, P.; Chirenje, M.; et al. Human Papillomavirus (HPV) Vaccine Background Paper; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Katzenellenbogen, R.A.; Carter, J.J.; Stern, J.E.; Butsch Kovacic, M.S.; Mehta, P.A.; Sauter, S.L.; Galloway, D.A.; Winer, R.L. Skin and mucosal human papillomavirus seroprevalence in persons with Fanconi anemia. Clin. Vaccine Immunol. 2015, 22, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.A.; Sauter, S.; Zhang, X.; Davies, S.M.; Wells, S.I.; Myers, K.C.; Panicker, G.; Unger, E.R.; Butsch Kovacic, M. Antibody response to human papillomavirus vaccination and natural exposure in individuals with Fanconi anemia. Vaccine 2017, 35, 6712–6719. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Giri, N.; Pan, Y.; Savage, S.A.; Pinto, L.A. Antibody response to human papillomavirus vaccine in subjects with inherited bone marrow failure syndromes. Vaccine 2014, 32, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- De Araujo, M.R.; de Oliveira Ribas, M.; Koubik, A.C.; Mattioli, T.; de Lima, A.A.; Franca, B.H. Fanconi’s anemia: Clinical and radiographic oral manifestations. Oral Dis. 2007, 13, 291–295. [Google Scholar] [CrossRef] [PubMed]
- De Kerviler, E.; Guermazi, A.; Zagdanski, A.M.; Gluckman, E.; Frija, J. The clinical and radiological features of Fanconi’s anaemia. Clin. Radiol. 2000, 55, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Karalis, A.; Tischkowitz, M.; Millington, G.W. Dermatological manifestations of inherited cancer syndromes in children. Br. J. Dermatol. 2011, 164, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Tischkowitz, M.; Dokal, I. Fanconi anaemia and leukaemia—Clinical and molecular aspects. Br. J. Haematol. 2004, 126, 176–191. [Google Scholar] [CrossRef] [PubMed]
- Han, T.J.; Lee, C.H.; Yoo, C.W.; Shin, H.J.; Park, H.J.; Cho, K.H.; Park, J.Y.; Choi, S.W.; Kim, J.Y. Synchronous multifocal HPV-related neoplasm involving both the genital tract and the head-and-neck area: A case report of Fanconi anemia. Radiother. Oncol. 2009, 92, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Kutler, D.I.; Wreesmann, V.B.; Goberdhan, A.; Ben-Porat, L.; Satagopan, J.; Ngai, I.; Huvos, A.G.; Giampietro, P.; Levran, O.; Pujara, K.; et al. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J. Natl. Cancer Inst. 2003, 95, 1718–1721. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P. Fanconi’s anemia and malignancies. Am. J. Hematol. 1996, 53, 99–110. [Google Scholar] [CrossRef]
- Rosenberg, P.S.; Greene, M.H.; Alter, B.P. Cancer incidence in persons with Fanconi anemia. Blood 2003, 101, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.S.; Alter, B.P.; Ebell, W. Cancer risks in Fanconi anemia: Findings from the German Fanconi anemia registry. Haematologica 2008, 93, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Gorell, E.S.; Nguyen, N.; Siprashvili, Z.; Marinkovich, M.P.; Lane, A.T. Characterization of patients with dystrophic epidermolysis bullosa for collagen VII therapy. Br. J. Dermatol. 2015, 173, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Pourreyron, C.; Cox, G.; Mao, X.; Volz, A.; Baksh, N.; Wong, T.; Fassihi, H.; Arita, K.; O’Toole, E.A.; Ocampo-Candiani, J.; et al. Patients with recessive dystrophic epidermolysis bullosa develop squamous-cell carcinoma regardless of type VII collagen expression. J. Investig. Dermatol. 2007, 127, 2438–2444. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E. Cincinnati Children’s Hospital Medical Center, USA, Unpublished work. 2016.
- Li, S.L.; Duo, L.N.; Wang, H.J.; Dai, W.; Zhou, E.H.; Xu, Y.N.; Zhao, T.; Xiao, Y.Y.; Xia, L.; Yang, Z.H.; et al. Identification of LCK mutation in a family with atypical epidermodysplasia verruciformis with T-cell defects and virus-induced squamous cell carcinoma. Br. J. Dermatol. 2016, 175, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Vuillier, F.; Gaud, G.; Guillemot, D.; Commere, P.H.; Pons, C.; Favre, M. Loss of the HPV-infection resistance EVER2 protein impairs NF-kappaB signaling pathways in keratinocytes. PLoS ONE 2014, 9, e89479. [Google Scholar] [CrossRef] [PubMed]
- Amanuma, Y.; Ohashi, S.; Itatani, Y.; Tsurumaki, M.; Matsuda, S.; Kikuchi, O.; Nakai, Y.; Miyamoto, S.; Oyama, T.; Kawamoto, T.; et al. Protective role of ALDH2 against acetaldehyde-derived DNA damage in oesophageal squamous epithelium. Sci. Rep. 2015, 5, 14142. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, A.; Ohashi, S.; Hirohashi, K.; Amanuma, Y.; Matsuda, T.; Muto, M. Molecular mechanisms of acetaldehyde-mediated carcinogenesis in squamous epithelium. Int. J. Mol. Sci. 2017, 18, 1943. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, M.; Yoshimura, K.; Suzuki, Y.; Uchida, G.; Kitano, Y.; Harii, K.; Imokawa, G. The mechanism of epidermal hyperpigmentation in cafe-au-lait macules of neurofibromatosis type 1 (von Recklinghausen’s disease) may be associated with dermal fibroblast-derived stem cell factor and hepatocyte growth factor. Br. J. Dermatol. 2003, 148, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, M.; Yoshimura, K.; Uchida, G.; Suzuki, Y.; Kitano, Y.; Harii, K. Epidermal hyperpigmentation in non-syndromic solitary cafe-au-lait macules may be associated with increased secretion of endothelin-1 by lesional keratinocytes. Scand. J. Plast. Reconstr. Surg. Hand Surg. 2005, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoury, R.; Sauter, S.; Butsch Kovacic, M.; Nelson, A.S.; Myers, K.C.; Mehta, P.A.; Davies, S.M.; Wells, S.I. Risk of Human Papillomavirus Infection in Cancer-Prone Individuals: What We Know. Viruses 2018, 10, 47. https://doi.org/10.3390/v10010047
Khoury R, Sauter S, Butsch Kovacic M, Nelson AS, Myers KC, Mehta PA, Davies SM, Wells SI. Risk of Human Papillomavirus Infection in Cancer-Prone Individuals: What We Know. Viruses. 2018; 10(1):47. https://doi.org/10.3390/v10010047
Chicago/Turabian StyleKhoury, Ruby, Sharon Sauter, Melinda Butsch Kovacic, Adam S. Nelson, Kasiani C. Myers, Parinda A. Mehta, Stella M. Davies, and Susanne I. Wells. 2018. "Risk of Human Papillomavirus Infection in Cancer-Prone Individuals: What We Know" Viruses 10, no. 1: 47. https://doi.org/10.3390/v10010047
APA StyleKhoury, R., Sauter, S., Butsch Kovacic, M., Nelson, A. S., Myers, K. C., Mehta, P. A., Davies, S. M., & Wells, S. I. (2018). Risk of Human Papillomavirus Infection in Cancer-Prone Individuals: What We Know. Viruses, 10(1), 47. https://doi.org/10.3390/v10010047