Biomarkers of Progression after HIV Acute/Early Infection: Nothing Compares to CD4+ T-cell Count?

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Ethical Considerations
2.3. Samples
2.4. HIV-1 Viral Load, CD4+ T-cell Count and Immune Activation
2.5. Human Leukocyte Antigen (HLA) and CCR5 Genotyping
2.6. HIV-Specific Cellular Immune Responses
2.7. Quantitation of Soluble Plasma Factors
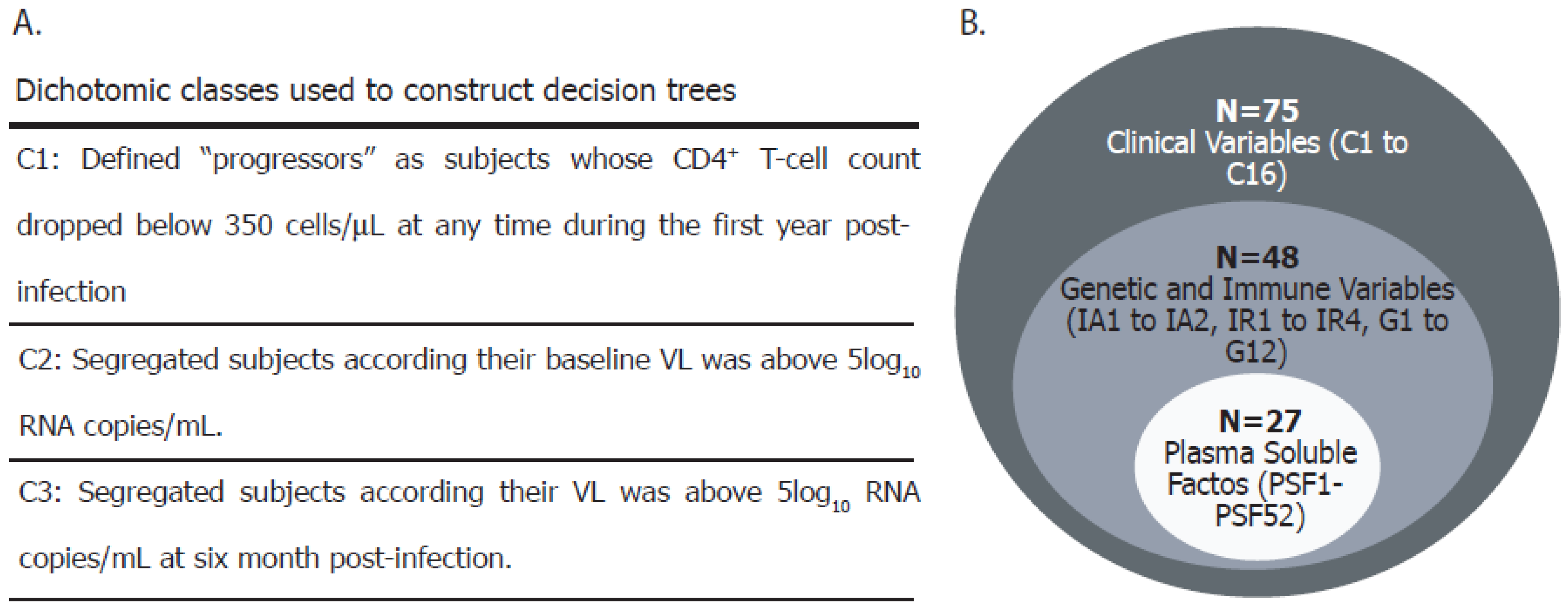
2.8. Definitions and Data Analysis
3. Results
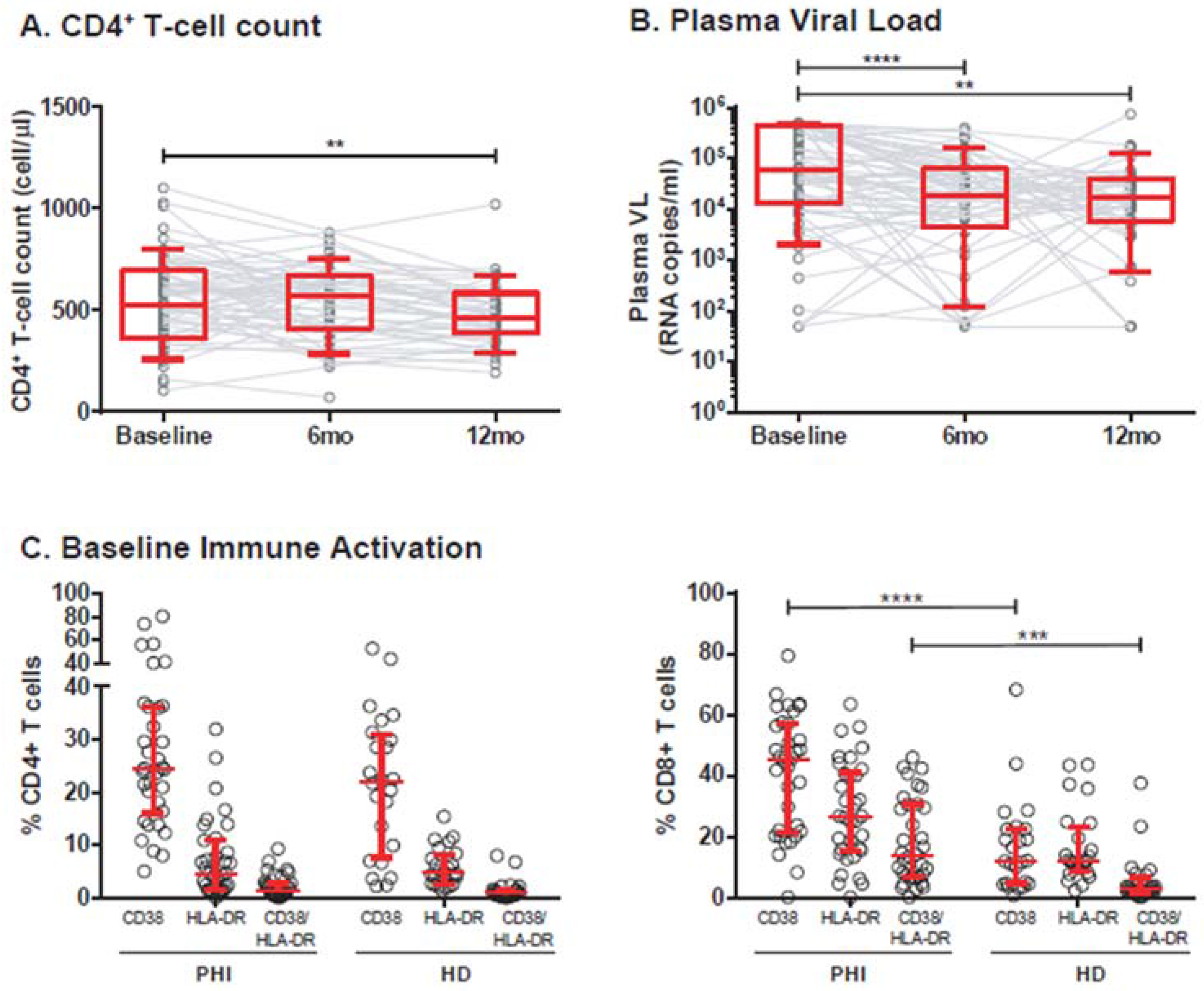
3.1. Cohort Description
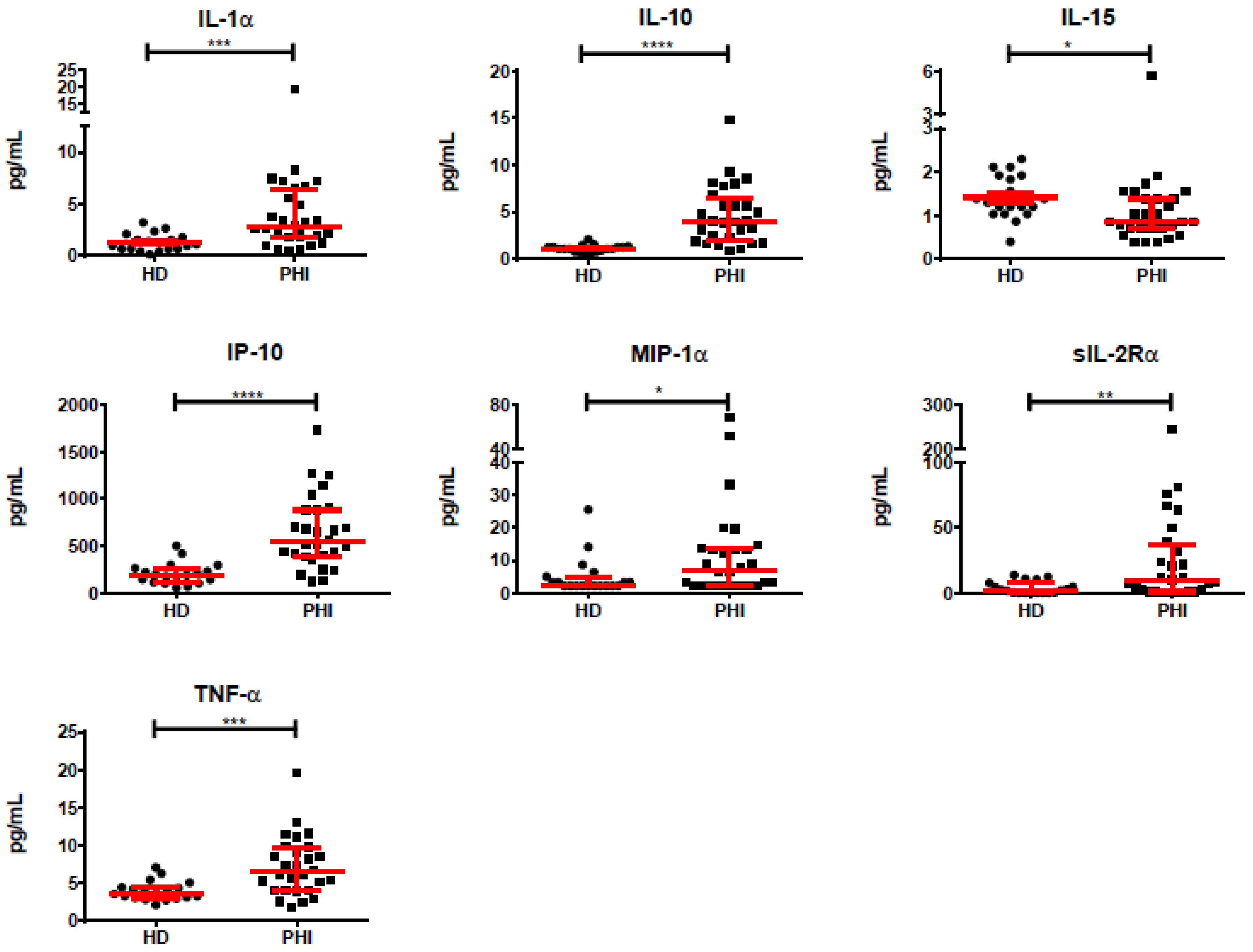
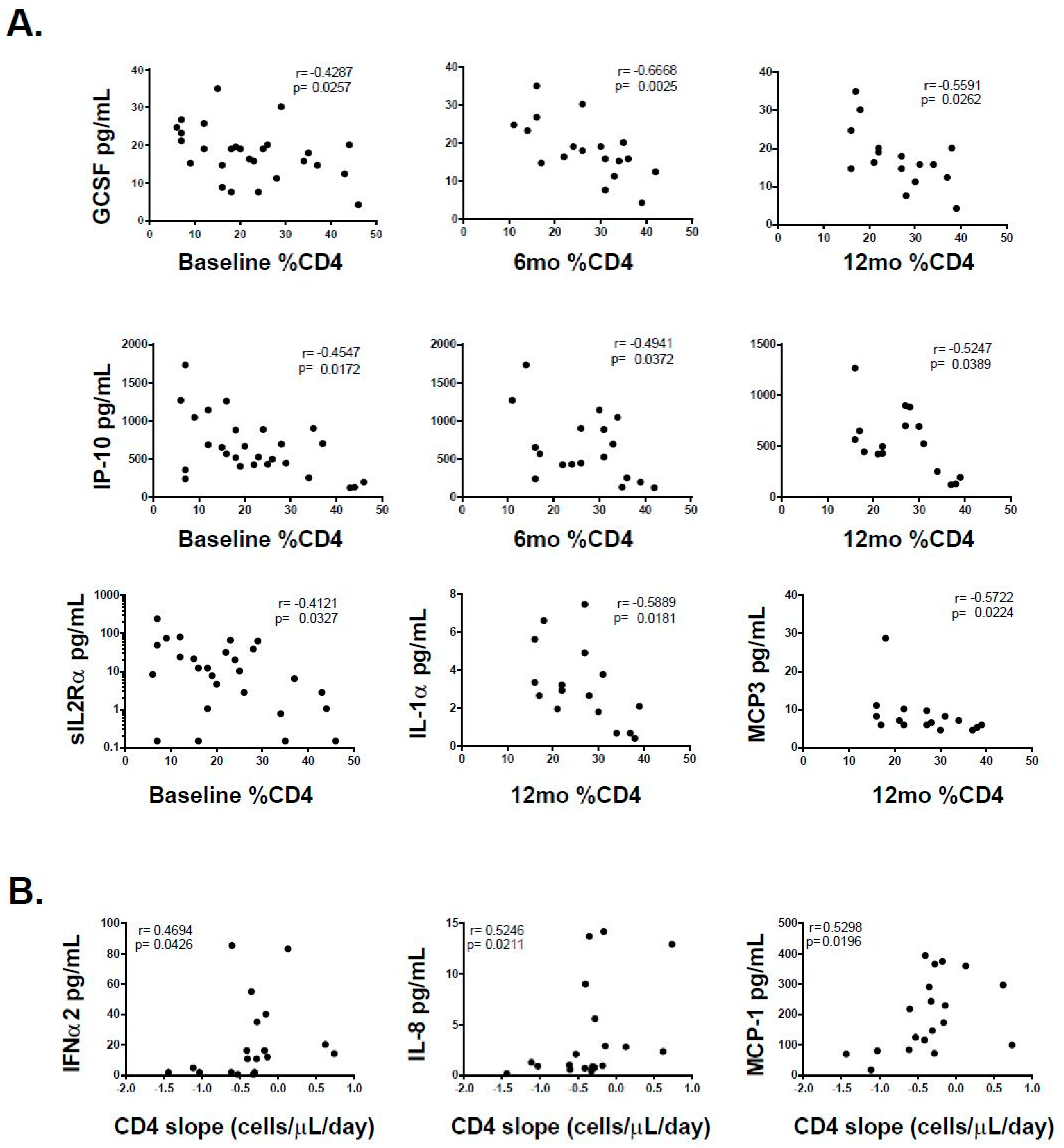
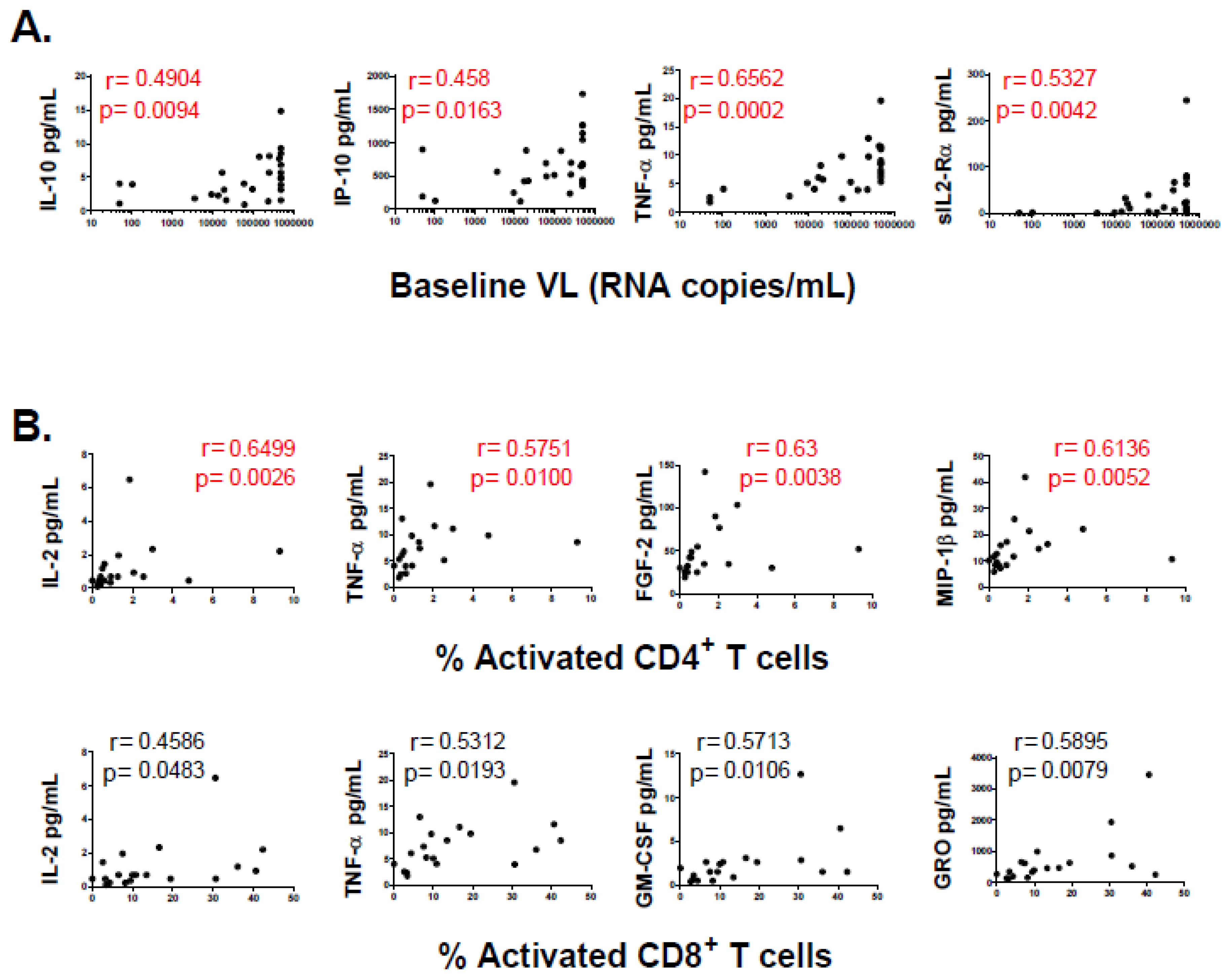
3.2. Association of Individual Parameters with Disease Progression
3.3. Baseline CD4+ T-cell Count Was the Most Potent Variable to Distinguish “Progressors” from “Non-Progressors”
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable ID | Variable Name | Variable Description |
|---|---|---|
| C1 | Baseline CD4 | Absolute CD4+ T-cell count evaluated at enrollment |
| C2 | Baseline %CD4 | Percentage of CD4+ T-cells evaluated at enrollment |
| C3 | Baseline CD4/CD8 | CD4/CD8 ratio evaluated at enrollment |
| C4 | 6mo CD4 | Absolute CD4+ T-cell count evaluated at 6 months postinfection |
| C5 | 6mo %CD4 | Percentage of CD4+ T-cells evaluated at 6 months postinfection |
| C6 | 6mo CD4/CD8 | CD4/CD8 ratio evaluated at 6 months postinfection |
| C7 | 12mo CD4 | Absolute CD4+ T-cell count evaluated at 12 months postinfection |
| C8 | 12mo %CD4 | Percentage of CD4+ T-cells evaluated at 12 months postinfection |
| C9 | 12mo CD4/CD8 | CD4/CD8 ratio evaluated at 12 months postinfection |
| C10 | CD4 Slope | Rate of CD4+ T-cell decay over the first year postinfection (cells/µL/day) |
| C11 | Baseline VL | Plasma viral load evaluated at enrollment (RNA copies/mL) |
| C12 | Baseline log10VL | Log10 plasma viral load evaluated at enrollment |
| C13 | 6mo VL | Plasma viral load evaluated at 6 months postinfection (RNA copies/mL) |
| C14 | 6mo log10VL | Log10 plasma viral load evaluated at 6 months postinfection |
| C15 | 12mo VL | Plasma viral load evaluated at 12 months postinfection (RNA copies/mL) |
| C16 | 12mo log10VL | Log10 plasma viral load evaluated at 12 months postinfection |
| IA1 | CD4CD38 | Percentage of CD4+ CD38+ T-cells evaluated at enrollment |
| IA2 | CD4HLADR | Percentage of CD4+ HLA-DR+ T-cells evaluated at enrollment |
| IA3 | CD4double | Percentage of CD4+ CD38+ HLA-DR+ T-cells evaluated at enrollment |
| IA4 | CD8CD38 | Percentage of CD8+ CD38+ T-cells evaluated at enrollment |
| IA5 | CD8HLADR | Percentage of CD8+ HLA-DR+ T-cells evaluated at enrollment |
| IA6 | CD8double | Percentage of CD8+ CD38+ HLA-DR+ T-cells evaluated at enrollment |
| IR1 | %Nef response | Percentage of T-cell response directed to Nef (over total HIV-specific response) evaluated at enrollment by ELISPOT |
| IR2 | Absolute Nef response | Magnitude of Nef-specific response evaluated at enrollment by ELISPOT (SFU/million PBMC) |
| IR3 | %Gag response | Percentage of T-cell response directed to Gag (over total HIV-specific response) evaluated at enrollment by ELISPOT |
| IR4 | Absolute Gag response | Magnitude of Gag-specific response evaluated at enrollment by ELISPOT (SFU/million PBMC) |
| G1 | GS1 | Additive genetic score constructed based on the presence or absence of certain HLA alleles, as described previously [30]. Alleles with a previous reported protective effect were added (+1), and risk alleles were subtracted (-1). Based on relevant bibliography, HLA-A*02, HLA-A*32, HLA-A*68, HLA-B*15, HLA-B*13, HLA-B*27, HLA-B*32, HLA-B*39, HLA-B*44, HLA-B*51 and HLA-B*57 were considered as protective. HLA-A*11, HLA-A*23, HLA-A*24, HLA-B*08, HLA-B*35, HLA-B*53, HLA-C*04 and HLA-C*07 were considered as deleterious. Other HLA alleles were considered as neutral (0). Heterozygosis for HLA was considered as protective (1) and homozygosis as deleterious (–1). |
| G2 | GS3 | Additive genetic score constructed based on the presence or absence of certain HLA alleles and CCR5 genotypes, as described previously [30]. HLA alleles associated with protection or risk were considered as in GS1. For CCR5 polymorphisms, Δ32 and CCR2-64I alleles were considered as protective. CCR5 genotypes HHC/HHF*2 and HHC/HHG*2 were considered as protective (+1), HHC/HHE, HHE/HHE and HHE/HHG*2 were considered as deleterious (–1), and the others as neutral (0). |
| G3 | GS5 | Additive genetic score constructed based on adding +1 when a protective allele was present and subtracting −1 when a risk allele was present. Protective and risk alleles were determined based on the odd ratio obtained for each allele in our cohort with a p < 0.05. HLA alleles with p > 0.05 were considered as neutral (0). |
| G4 | GS6 | Idem to GS5 but the cut-off level for significance was established at p < 0.03. |
| G5 | GS7 | Constructed by multiplying the odds ratio corresponding to the 6-month CD4+ T-cell count for each of the 6 HLA alleles (A, B and C) of each individual. |
| G6 | GS8 | Constructed by multiplying the odds ratio corresponding to the 6-month VL for each of the 6 HLA alleles (A, B and C) of each individual. |
| G7 | GS9 | Constructed by multiplying the odds ratio corresponding to the 6-month CD4+ T-cell count for each of the 2 CCR5 haplotypes of each individual. |
| G8 | GS10 | Constructed by multiplying the odds ratio corresponding to the 6-month VL for each of the 2 CCR5 haplotypes of each individual. |
| G9 | GS11 | Constructed by multiplying the odds ratio corresponding to the 6-month CD4+ T-cell count for each of the 6 HLA alleles (A, B and C) and the 2 CCR5 haplotypes of each individual. |
| G10 | GS12 | Constructed by multiplying the odds ratio corresponding to the 6-month VL for each of the 6 HLA alleles (A, B and C) and the 2 CCR5 haplotypes of each individual. |
| PSF1 | EGF | Plasma level of EGF (Endothelial growth factor) evaluated at enrollment by Luminex (pg/mL) |
| PSF2 | Eotaxin | Plasma level of Eotaxin evaluated at enrollment by Luminex (pg/mL) |
| PSF3 | FGF2 | Plasma level of FGF-2 (fibroblast growth factor 2) evaluated at enrollment by Luminex (pg/mL) |
| PSF4 | Flt3Ligand | Plasma level of Flt-3 (Fms-like tyrosine kinase 3) ligand evaluated at enrollment by Luminex (pg/mL) |
| PSF5 | Fractalkine | Plasma level of Fractalkine evaluated at enrollment by Luminex (pg/mL) |
| PSF6 | GCSF | Plasma level of G-CSF (granulocyte colony-stimulating factor) evaluated at enrollment by Luminex (pg/mL) |
| PSF7 | GMCSF | Plasma level of GM-CSF (granulocyte monocyte colony-stimulating factor) evaluated at enrollment by Luminex (pg/mL) |
| PSF8 | GRO | Plasma level of GRO evaluated at enrollment by Luminex (pg/mL) |
| PSF9 | IFN-α2 | Plasma level of IFN-α2 (interferon alpha 2) evaluated at enrollment by Luminex (pg/mL) |
| PSF10 | IFN-γ | Plasma level of IFN-γ (interferon gamma) evaluated at enrollment by Luminex (pg/mL) |
| PSF11 | IL1α | Plasma level of IL-1α (interleukin 1 alpha) evaluated at enrollment by Luminex (pg/mL) |
| PSF12 | IL1β | Plasma level of IL-1β (interleukin 1 beta) evaluated at enrollment by Luminex (pg/mL) |
| PSF13 | IL1ra | Plasma level of IL-1ra (interleukin 1 receptor antagonist) evaluated at enrollment by Luminex (pg/mL) |
| PSF14 | IL2 | Plasma level of IL-2 (interleukin 2) evaluated at enrollment by Luminex (pg/mL) |
| PSF15 | IL3 | Plasma level of IL-3 (interleukin 3) evaluated at enrollment by Luminex (pg/mL) |
| PSF16 | IL4 | Plasma level of IL-4 (interleukin 4) evaluated at enrollment by Luminex (pg/mL) |
| PSF17 | IL5 | Plasma level of IL-5 (interleukin 5) evaluated at enrollment by Luminex (pg/mL) |
| PSF18 | IL6 | Plasma level of IL-6 (interleukin 6) evaluated at enrollment by Luminex (pg/mL) |
| PSF19 | IL7 | Plasma level of IL-7 (interleukin 7) evaluated at enrollment by Luminex (pg/mL) |
| PSF20 | IL8 | Plasma level of IL-8 (interleukin 8) evaluated at enrollment by Luminex (pg/mL) |
| PSF21 | IL9 | Plasma level of IL-9 (interleukin 9) evaluated at enrollment by Luminex (pg/mL) |
| PSF22 | IL10 | Plasma level of IL-10 (interleukin 10) evaluated at enrollment by Luminex (pg/mL) |
| PSF23 | IL12p40 | Plasma level of IL-12p40 (interleukin 12 subunit p40) evaluated at enrollment by Luminex (pg/mL) |
| PSF24 | IL12p70 | Plasma level of IL-12p70 (interleukin 12) evaluated at enrollment by Luminex (pg/mL) |
| PSF25 | IL13 | Plasma level of IL-13 (interleukin 13) evaluated at enrollment by Luminex (pg/mL) |
| PSF26 | IL15 | Plasma level of IL-15 (interleukin 15) evaluated at enrollment by Luminex (pg/mL) |
| PSF27 | IL17 | Plasma level of IL-17 (interleukin 17) evaluated at enrollment by Luminex (pg/mL) |
| PSF28 | IP10 | Plasma level of IP10 (interferon gamma-induced protein 10, CXCL10) evaluated at enrollment by Luminex (pg/mL) |
| PSF29 | MCP1 | Plasma level of MCP1 (monocyte chemoattractant protein 1) evaluated at enrollment by Luminex (pg/mL) |
| PSF30 | MCP3 | Plasma level of MCP3 (monocyte chemoattractant protein 3) evaluated at enrollment by Luminex (pg/mL) |
| PSF31 | MDC | Plasma level of MDC (macrophage derived chemokine, CCL22) evaluated at enrollment by Luminex (pg/mL) |
| PSF32 | MIP1α | Plasma level of MIP-1α (macrophage inflammatory protein 1 alpha) evaluated at enrollment by Luminex (pg/mL) |
| PSF33 | MIP1β | Plasma level of MIP-1β (macrophage inflammatory protein 1 beta) evaluated at enrollment by Luminex (pg/mL) |
| PSF34 | sCD40L | Plasma level of sCD40L (soluble CD40 ligand) evaluated at enrollment by Luminex (pg/mL) |
| PSF35 | sIL2Rα | Plasma level of sIL-2Rα (soluble interleukin 2 receptor alpha) evaluated at enrollment by Luminex (pg/mL) |
| PSF36 | TGFα | Plasma level of TGF-α (tumor growth factor alpha) evaluated at enrollment by Luminex (pg/mL) |
| PSF37 | TNFα | Plasma level of TNF-α (tumor necrosis factor alpha) evaluated at enrollment by Luminex (pg/mL) |
| PSF38 | TNFβ | Plasma level of TNF-β (tumor necrosis factor beta) evaluated at enrollment by Luminex (pg/mL) |
| PSF39 | VEGF | Plasma level of VEGF (vascular endothelial growth factor) evaluated at enrollment by Luminex (pg/mL) |
| PSF40 | LPS | Plasma level of LPS (lipopolysaccharide) evaluated at enrollment by Lal assay (EU/mL) |
| PSF41 | CSVL1 | Cytokines that significantly correlated with baseline VL were considered (sIL-2Rα, TNF-α, IP-10 and IL-10) to construct an additive score based on 1s and −1s. If the cytokine value of the subject was above 75% IQR corresponding to the group of healthy donors (HD), then this cytokine was assigned a value of 1. If the value was below 25% IQR corresponding to HD, it was assigned a value of −1. If the value was within the range IQR25–75% corresponding to HD, it was assigned a value of 0. |
| PSF42 | CSCD41 | Cytokines that significantly correlated with baseline CD4+ T-cell counts were considered (sIL-2Rα, IP-10 and G-CSF) to construct an additive score based on 1s and −1s. If the cytokine value of the subject was above 75% IQR corresponding to the group of healthy donors (HD), then this cytokine was assigned a value of −1. If the value was below 25% IQR corresponding to HD, it was assigned a value of 1. If the value was within the range IQR25–75% corresponding to HD, it was assigned a value of 0. |
| PSF43 | CST1 | Score defined as the arithmetic sum of CSVL1 + CSCD41. |
| PSF44 | CSVL2 | Cytokines that significantly correlated with baseline VL were considered (sIL-2Rα, TNF-α, IP-10 and IL-10) to construct an additive score based on adding the corresponding Spearman’s R values. If the cytokine value of the subject was above 75% IQR corresponding to the group of healthy donors (HD), then the Spearman´s R value corresponding to that cytokine was added to the score. If the value was below 25% IQR corresponding to HD, the Spearman´s R value corresponding to that cytokine was subtracted from the score. If the value was within the range IQR25–75% corresponding to HD, it was assigned a value of 0. |
| PSF45 | CSCD42 | Cytokines that significantly correlated with baseline CD4+ T-cell counts were considered (sIL-2Rα, IP-10 and G-CSF) to construct an additive score based on adding the corresponding Spearman’s R values. If the cytokine value of the subject was above 75% IQR corresponding to the group of healthy donors (HD), then the Spearman´s R value corresponding to that cytokine was subtracted from the score. If the value was below 25% IQR corresponding to HD, the Spearman´s R value corresponding to that cytokine was added to the score. If the value was within the range IQR25–75% corresponding to HD, it was assigned a value of 0. |
| PSF46 | CST2 | Score defined as the arithmetic sum of CSVL2 + CSCD42 |
| PSF47 | CSVL3 | Cytokines that significantly correlated with baseline VL were considered (sIL-2Rα, TNF-α, IP-10 and IL-10) to construct an additive score based on normalizing the value of each of these cytokines over the cytokine median of the PHI group and multiplying this adjusted value by the corresponding Spearman´s R. |
| PSF48 | CSCD43 | Cytokines that significantly correlated with baseline CD4+ T-cell counts were considered (sIL-2Rα, IP-10 and G-CSF) to construct an additive score based on normalizing the value of each of these cytokines over the cytokine median of the PHI group and multiplying this adjusted value by the corresponding Spearman´s R. |
| PSF49 | CST3 | Score defined as the arithmetic sum of CSVL3 + CSCD43. |
| PSF50 | CSVL4 | Cytokines that significantly correlated with baseline VL were considered (sIL-2Rα, TNF-α, IP-10 and IL-10) to construct an additive score based on multiplying the log10 value of each of these cytokines by the corresponding Spearman´s R. |
| PSF51 | CSCD44 | Cytokines that significantly correlated with baseline CD4+ T-cell counts were considered (sIL-2Rα, IP-10 and G-CSF) to construct an additive score based on normalizing the log10 value of each of these cytokines by the corresponding Spearman´s R |
| PSF52 | CST4 | It was defined as the arithmetic sum of CSVL4 + CSCD44 |
References
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- Rutstein, S.E.; Ananworanich, J.; Fidler, S.; Johnson, C.; Sanders, E.J.; Sued, O.; Saez-Cirion, A.; Pilcher, C.D.; Fraser, C.; Cohen, M.S.; et al. Clinical and public health implications of acute and early HIV detection and treatment: A scoping review. J. Int. AIDS Soc. 2017, 20, 21579. [Google Scholar] [CrossRef] [PubMed]
- McDermott, A.B.; Koup, R.A. CD8(+) T cells in preventing HIV infection and disease. Aids 2012, 26, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Burger, S.; Poles, M.A. Natural history and pathogenesis of human immunodeficiency virus infection. Semin. Liver Dis. 2003, 23, 115–124. [Google Scholar] [PubMed]
- Mayeux, R. Biomarkers: Potential uses and limitations. NeuroRx J. Am. Soc. Exp. NeuroTher. 2004, 1, 182–188. [Google Scholar] [CrossRef] [PubMed]
- De Wit, S.; Battegay, M.; D’Arminio Monforte, A.; Lundgren, J.D.; Oprea, C.; Antinori, A.; Bhagani, S.; Fätkenheuer, G.; Friis-Moller, N.; Furrer, H.; et al. European AIDS Clinical Society; EACS: Brussels, Belgium, 2017. [Google Scholar]
- Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents; Department of Health and Human Services: Washington, DC, USA, 2015.
- IAS-USA. Antiretroviral Treatment of Adult HIV Infection: 2014 Recommendations of the International Antiviral Society-USA Panel; IAS-USA: San Francisco, CA, USA, 2014. [Google Scholar]
- WHO. Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection: Recommendations for a Public Health Approach, 2nd ed.; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Younesi, E.; Toldo, L.; Muller, B.; Friedrich, C.M.; Novac, N.; Scheer, A.; Hofmann-Apitius, M.; Fluck, J. Mining biomarker information in biomedical literature. BMC Med. Inform. Decis. Mak. 2012, 12, 148. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.P.; Hurst, J.; Stohr, W.; Robinson, N.; Brown, H.; Fisher, M.; Kinloch, S.; Cooper, D.; Schechter, M.; Tambussi, G.; et al. HIV-1 DNA predicts disease progression and post-treatment virological control. eLife 2014, 3, e03821. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Song, K.; Sauer, M.M.; Nason, M.C.; Giret, M.T.; Carvalho, K.I.; Costa, P.R.; Roederer, M.; Kallas, E.G. Early immunologic and virologic predictors of clinical HIV-1 disease progression. Aids 2013, 27, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Kitchen, C.M.; Liu, L.; Guo, H.; Gascon, R.; Narvaez, A.B.; Hunt, P.; Martin, J.N.; Kahn, J.O.; Levy, J.; et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 2004, 104, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.; Passmore, J.A.; Williamson, C.; Little, F.; Bebell, L.M.; Mlisana, K.; Burgers, W.A.; van Loggerenberg, F.; Walzl, G.; Djoba Siawaya, J.F.; et al. Plasma cytokine levels during acute HIV-1 infection predict HIV disease progression. Aids 2010, 24, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Liovat, A.S.; Rey-Cuille, M.A.; Lecuroux, C.; Jacquelin, B.; Girault, I.; Petitjean, G.; Zitoun, Y.; Venet, A.; Barre-Sinoussi, F.; Lebon, P.; et al. Acute plasma biomarkers of T cell activation set-point levels and of disease progression in HIV-1 infection. PLoS ONE 2012, 7, e46143. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.M.; Golub, E.T.; Nowicki, M.; Young, M.; Anastos, K.; Crystal, H.; Cohen, M.H.; Zhang, J.; Greenblatt, R.M.; Desai, S.; et al. The effect of HIV infection and HAART on inflammatory biomarkers in a population-based cohort of women. Aids 2011, 25, 1823–1832. [Google Scholar] [CrossRef] [PubMed]
- Katsikis, P.D.; Mueller, Y.M.; Villinger, F. The cytokine network of acute HIV infection: A promising target for vaccines and therapy to reduce viral set-point? PLoS Pathog. 2011, 7, e1002055. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, R.; Lucchetti, A.; Galvan, P.; Sanchez, S.; Sanchez, C.; Hernandez, A.; Sanchez, H.; Frahm, N.; Linde, C.H.; Hewitt, H.S.; et al. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 2006, 80, 3122–3125. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Currier, J.R.; Herrmann, E.; Haule, A.; Kuta, E.; McCutchan, F.; Njovu, L.; Geis, S.; Hoffmann, O.; Maboko, L.; et al. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 2007, 81, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Kiepiela, P.; Ngumbela, K.; Thobakgale, C.; Ramduth, D.; Honeyborne, I.; Moodley, E.; Reddy, S.; de Pierres, C.; Mncube, Z.; Mkhwanazi, N.; et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 2007, 13, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Streeck, H.; Jolin, J.S.; Qi, Y.; Yassine-Diab, B.; Johnson, R.C.; Kwon, D.S.; Addo, M.M.; Brumme, C.; Routy, J.P.; Little, S.; et al. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. J. Virol. 2009, 83, 7641–7648. [Google Scholar] [CrossRef] [PubMed]
- Masemola, A.; Mashishi, T.; Khoury, G.; Mohube, P.; Mokgotho, P.; Vardas, E.; Colvin, M.; Zijenah, L.; Katzenstein, D.; Musonda, R.; et al. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: Correlation with viral load. J. Virol. 2004, 78, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
- Radebe, M.; Nair, K.; Chonco, F.; Bishop, K.; Wright, J.K.; van der Stok, M.; Bassett, I.V.; Mncube, Z.; Altfeld, M.; Walker, B.D.; et al. Limited immunogenicity of HIV CD8+ T-cell epitopes in acute Clade C virus infection. J. Infect. Dis. 2011, 204, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Ferrando-Martinez, S.; Casazza, J.P.; Leal, M.; Machmach, K.; Munoz-Fernandez, M.A.; Viciana, P.; Koup, R.A.; Ruiz-Mateos, E. Differential Gag-specific polyfunctional T cell maturation patterns in HIV-1 elite controllers. J. Virol. 2012, 86, 3667–3674. [Google Scholar] [CrossRef] [PubMed]
- Naranbhai, V.; Carrington, M. Host genetic variation and HIV disease: From mapping to mechanism. Immunogenetics 2017, 69, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Socias, M.E.; Sued, O.; Laufer, N.; Lazaro, M.E.; Mingrone, H.; Pryluka, D.; Remondegui, C.; Figueroa, M.I.; Cesar, C.; Gun, A.; et al. Acute retroviral syndrome and high baseline viral load are predictors of rapid HIV progression among untreated Argentinean seroconverters. J. Int. AIDS Soc. 2011, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Turk, G.; Ghiglione, Y.; Falivene, J.; Socias, M.E.; Laufer, N.; Coloccini, R.S.; Rodriguez, A.M.; Ruiz, M.J.; Pando, M.A.; Giavedoni, L.D.; et al. Early Gag immunodominance of the HIV-specific T-cell response during acute/early infection is associated with higher CD8+ T-cell antiviral activity and correlates with preservation of the CD4+ T-cell compartment. J. Virol. 2013, 87, 7445–7462. [Google Scholar] [CrossRef] [PubMed]
- Ghiglione, Y.; Falivene, J.; Ruiz, M.J.; Laufer, N.; Socias, M.E.; Cahn, P.; Giavedoni, L.; Sued, O.; Gherardi, M.M.; Salomon, H.; et al. Early skewed distribution of total and HIV-specific CD8+ T-cell memory phenotypes during primary HIV infection is related to reduced antiviral activity and faster disease progression. PLoS ONE 2014, 9, e104235. [Google Scholar] [CrossRef] [PubMed]
- Coloccini, R.S.; Dilernia, D.; Ghiglione, Y.; Turk, G.; Laufer, N.; Rubio, A.; Socias, M.E.; Figueroa, M.I.; Sued, O.; Cahn, P.; et al. Host genetic factors associated with symptomatic primary HIV infection and disease progression among Argentinean seroconverters. PLoS ONE 2014, 9, e113146. [Google Scholar] [CrossRef] [PubMed]
- Falivene, J.; Ghiglione, Y.; Laufer, N.; Socias, M.E.; Holgado, M.P.; Ruiz, M.J.; Maeto, C.; Figueroa, M.I.; Giavedoni, L.D.; Cahn, P.; et al. Th17 and Th17/Treg ratio at early HIV infection associate with protective HIV-specific CD8(+) T-cell responses and disease progression. Sci. Rep. 2015, 5, 11511. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.A.; Aberg, J.A.; Hoy, J.F.; Telenti, A.; Benson, C.; Cahn, P.; Eron, J.J.; Gunthard, H.F.; Hammer, S.M.; Reiss, P.; et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-USA panel. JAMA 2012, 308, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Sociedad Argentina de Infectología (SADI). V Consenso Argentino de Terapia Antirretroviral; SADI: Buenos Aires, Argentina, 2014; p. 200. [Google Scholar]
- Turk, G.; Gherardi, M.M.; Laufer, N.; Saracco, M.; Luzzi, R.; Cox, J.H.; Cahn, P.; Salomon, H. Magnitude, breadth, and functional profile of T-cell responses during human immunodeficiency virus primary infection with B and BF viral variants. J. Virol. 2008, 82, 2853–2866. [Google Scholar] [CrossRef] [PubMed]
- Giavedoni, L.D. Simultaneous detection of multiple cytokines and chemokines from nonhuman primates using luminex technology. J. Immunol. Methods 2005, 301, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.J.; Ghiglione, Y.; Falivene, J.; Laufer, N.; Holgado, M.P.; Socias, M.E.; Cahn, P.; Sued, O.; Giavedoni, L.; Salomon, H.; et al. Env-Specific IgA from Viremic HIV-Infected Subjects Compromises Antibody-Dependent Cellular Cytotoxicity. J. Virol. 2016, 90, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench. In Online Appendix for “Data Mining: Practical Machine Learning Tools and Techniques”, 4th ed.; Morgan Kauffmann Publishers Inc.: San Francisco, CA, USA, 2016. [Google Scholar]
- Cohen, J. A Coefficient of Agreement for Nominal Scales. Educ. Psychol. Meas. 1960, 20, 37–46. [Google Scholar] [CrossRef]
- Huang, X.; Liu, X.; Meyers, K.; Liu, L.; Su, B.; Wang, P.; Li, Z.; Li, L.; Zhang, T.; Li, N.; et al. Cytokine cascade and networks among MSM HIV seroconverters: Implications for early immunotherapy. Sci. Rep. 2016, 6, 36234. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The immune response during acute HIV-1 infection: Clues for vaccine development. Nat. Rev. Immunol. 2010, 10, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.; Dibben, O.; Anderson, J.A.; Stacey, A.; Mayo, A.J.; Norris, P.J.; Kuruc, J.D.; Salazar-Gonzalez, J.F.; Li, H.; Keele, B.F.; et al. Cross-sectional detection of acute HIV infection: Timing of transmission, inflammation and antiretroviral therapy. PLoS ONE 2011, 6, e19617. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Catalfamo, M.; Le Saout, C.; Lane, H.C. The role of cytokines in the pathogenesis and treatment of HIV infection. Cytokine Growth Factor Rev. 2012, 23, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Mueller, Y.M.; Katsikis, P.D. IL-15 in HIV infection: Pathogenic or therapeutic potential? Eur. Cytokine Netw. 2010, 21, 219–221. [Google Scholar] [PubMed]
- Noel, N.; Boufassa, F.; Lecuroux, C.; Saez-Cirion, A.; Bourgeois, C.; Dunyach-Remy, C.; Goujard, C.; Rouzioux, C.; Meyer, L.; Pancino, G.; et al. Elevated IP10 levels are associated with immune activation and low CD4(+) T-cell counts in HIV controller patients. Aids 2014, 28, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.M.; Hong, H.A.; Young, K.; Lewis, D.A.; Fallows, D.; Manca, C.; Kaplan, G. Plasma interferon-gamma-inducible protein 10 can be used to predict viral load in HIV-1-infected individuals. J. Acquir. Immune Defic. Syndr. 2013, 63, e115–e116. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.; Misra, V.; Cassol, E.; Ancuta, P.; Yan, Z.; Li, C.; Morgello, S.; Gabuzda, D. A plasma biomarker signature of immune activation in HIV patients on antiretroviral therapy. PLoS ONE 2012, 7, e30881. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, G.; Steel, H.C.; Cassol, S.; de Oliveira, T.; Seebregts, C.J.; Anderson, R.; Cassol, E.; Rossouw, T.M. Circulating biomarkers of immune activation distinguish viral suppression from nonsuppression in HAART-treated patients with advanced HIV-1 subtype C infection. Mediat. Inflamm. 2014, 2014, 198413. [Google Scholar] [CrossRef] [PubMed]
- Norris, P.J.; Zhang, J.; Worlock, A.; Nair, S.V.; Anastos, K.; Minkoff, H.L.; Villacres, M.C.; Young, M.; Greenblatt, R.M.; Desai, S.; et al. Systemic Cytokine Levels Do Not Predict CD4(+) T-Cell Recovery After Suppressive Combination Antiretroviral Therapy in Chronic Human Immunodeficiency Virus Infection. Open Forum Infect. Dis. 2016, 3, ofw025. [Google Scholar] [CrossRef] [PubMed]
- Stiksrud, B.; Lorvik, K.B.; Kvale, D.; Mollnes, T.E.; Ueland, P.M.; Troseid, M.; Tasken, K.; Dyrhol-Riise, A.M. Plasma IP-10 Is Increased in Immunological NonResponders and Associated With Activated Regulatory T Cells and Persisting Low CD4 Counts. J. Acquir. Immune Defic. Syndr. 2016, 73, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhang, T.; Wang, R.; Zhang, H.; Huang, X.; Yin, J.; Zhang, L.; Xu, X.; Wu, H. Plasma IP-10 is associated with rapid disease progression in early HIV-1 infection. Viral Immunol. 2012, 25, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Boyle, D.S.; Hawkins, K.R.; Steele, M.S.; Singhal, M.; Cheng, X. Emerging technologies for point-of-care CD4 T-lymphocyte counting. Trends Biotechnol. 2012, 30, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Duro, R.; Rocha-Pereira, N.; Figueiredo, C.; Pineiro, C.; Caldas, C.; Serrao, R.; Sarmento, A. Routine CD4 monitoring in HIV patients with viral suppression: Is it really necessary? A Portuguese cohort. J. Microbiol. Immunol. Infect. 2017. [Google Scholar] [CrossRef] [PubMed]
- Caniglia, E.C.; Cain, L.E.; Sabin, C.A.; Robins, J.M.; Logan, R.; Abgrall, S.; Mugavero, M.J.; Hernandez-Diaz, S.; Meyer, L.; Seng, R.; et al. Comparison of dynamic monitoring strategies based on CD4 cell counts in virally suppressed, HIV-positive individuals on combination antiretroviral therapy in high-income countries: A prospective, observational study. Lancet HIV 2017, 4, e251–e259. [Google Scholar] [CrossRef]
- Cori, A.; Pickles, M.; van Sighem, A.; Gras, L.; Bezemer, D.; Reiss, P.; Fraser, C. CD4+ cell dynamics in untreated HIV-1 infection: Overall rates, and effects of age, viral load, sex and calendar time. Aids 2015, 29, 2435–2446. [Google Scholar] [CrossRef] [PubMed]
- Ananworanich, J.; Chomont, N.; Eller, L.A.; Kroon, E.; Tovanabutra, S.; Bose, M.; Nau, M.; Fletcher, J.L.K.; Tipsuk, S.; Vandergeeten, C.; et al. HIV DNA Set Point is Rapidly Established in Acute HIV Infection and Dramatically Reduced by Early ART. EBioMedicine 2016, 11, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Vandergeeten, C.; Fromentin, R.; Chomont, N. The role of cytokines in the establishment, persistence and eradication of the HIV reservoir. Cytokine Growth Factor Rev. 2012, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]






| Total sample size (number of individuals) | 75 | ||
| Female:Male ratio | 1:3 | ||
| Age at enrollment (years, median and IQR25–75%) | 30 (24–38) | ||
| Estimated time of infection at enrollment (days, median and IQR25–75%) | 75 (54–113) | ||
| Follow-up of Virologic and Immune Characteristics: | |||
| Baseline (N = 75) | 6-month pi (N = 59) | 12-month pi (N = 46) | |
| VL (RNA copies/mL; median and IQR25–75%) a | 61,045 (12,736–455,417) | 18,951 (4298–62,739) | 16,988 (5695–40,105) |
| Log10VL (mean ± SD) a | 4.6 ± 1 | 4 ± 1 | 4 ±0.89 |
| CD4+ T-cell count (cells/μL, median and IQR25–75%) b | 525 (361–698) | 571 (406–673) | 464 (387–585) |
| CD4/CD8 Ratio (Median and IQR25–75%) | 0.6 (0.32–0.83) | 0.55 (0.34–0.8) | 0.61 (0.39–0.93) |
| CD4+ T-cell decay rate (cells/μL/day; median and IQR25–75%) | −0.62 (−0.31 – −0.03) | ||
| Baseline Immune Activation (%cells, median and IQR25–75%) c: | |||
| %CD4+CD38+ | 24.4 (16–36.2) | ||
| %CD4+HLA-DR+ | 4.6 (1.5–11.01) | ||
| %CD4+CD38+HLA-DR+ | 1.2 (0.47–2.8) | ||
| %CD8+CD38+ | 45.2 (21.3–57.1) | ||
| %CD8+HLA-DR+ | 26.5 (15.2–41) | ||
| %CD8+CD38+HLA-DR+ | 13.8 (6.7–30.6) | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turk, G.; Ghiglione, Y.; Hormanstorfer, M.; Laufer, N.; Coloccini, R.; Salido, J.; Trifone, C.; Ruiz, M.J.; Falivene, J.; Holgado, M.P.; et al. Biomarkers of Progression after HIV Acute/Early Infection: Nothing Compares to CD4+ T-cell Count? Viruses 2018, 10, 34. https://doi.org/10.3390/v10010034
Turk G, Ghiglione Y, Hormanstorfer M, Laufer N, Coloccini R, Salido J, Trifone C, Ruiz MJ, Falivene J, Holgado MP, et al. Biomarkers of Progression after HIV Acute/Early Infection: Nothing Compares to CD4+ T-cell Count? Viruses. 2018; 10(1):34. https://doi.org/10.3390/v10010034
Chicago/Turabian StyleTurk, Gabriela, Yanina Ghiglione, Macarena Hormanstorfer, Natalia Laufer, Romina Coloccini, Jimena Salido, César Trifone, María Julia Ruiz, Juliana Falivene, María Pía Holgado, and et al. 2018. "Biomarkers of Progression after HIV Acute/Early Infection: Nothing Compares to CD4+ T-cell Count?" Viruses 10, no. 1: 34. https://doi.org/10.3390/v10010034
APA StyleTurk, G., Ghiglione, Y., Hormanstorfer, M., Laufer, N., Coloccini, R., Salido, J., Trifone, C., Ruiz, M. J., Falivene, J., Holgado, M. P., Caruso, M. P., Figueroa, M. I., Salomón, H., Giavedoni, L. D., Pando, M. D. l. Á., Gherardi, M. M., Rabinovich, R. D., Pury, P. A., & Sued, O. (2018). Biomarkers of Progression after HIV Acute/Early Infection: Nothing Compares to CD4+ T-cell Count? Viruses, 10(1), 34. https://doi.org/10.3390/v10010034