Sustained IFN-I Expression during Established Persistent Viral Infection: A “Bad Seed” for Protective Immunity

 , ,
, ,
Abstract
1. IFN-I and Its Beneficial Role
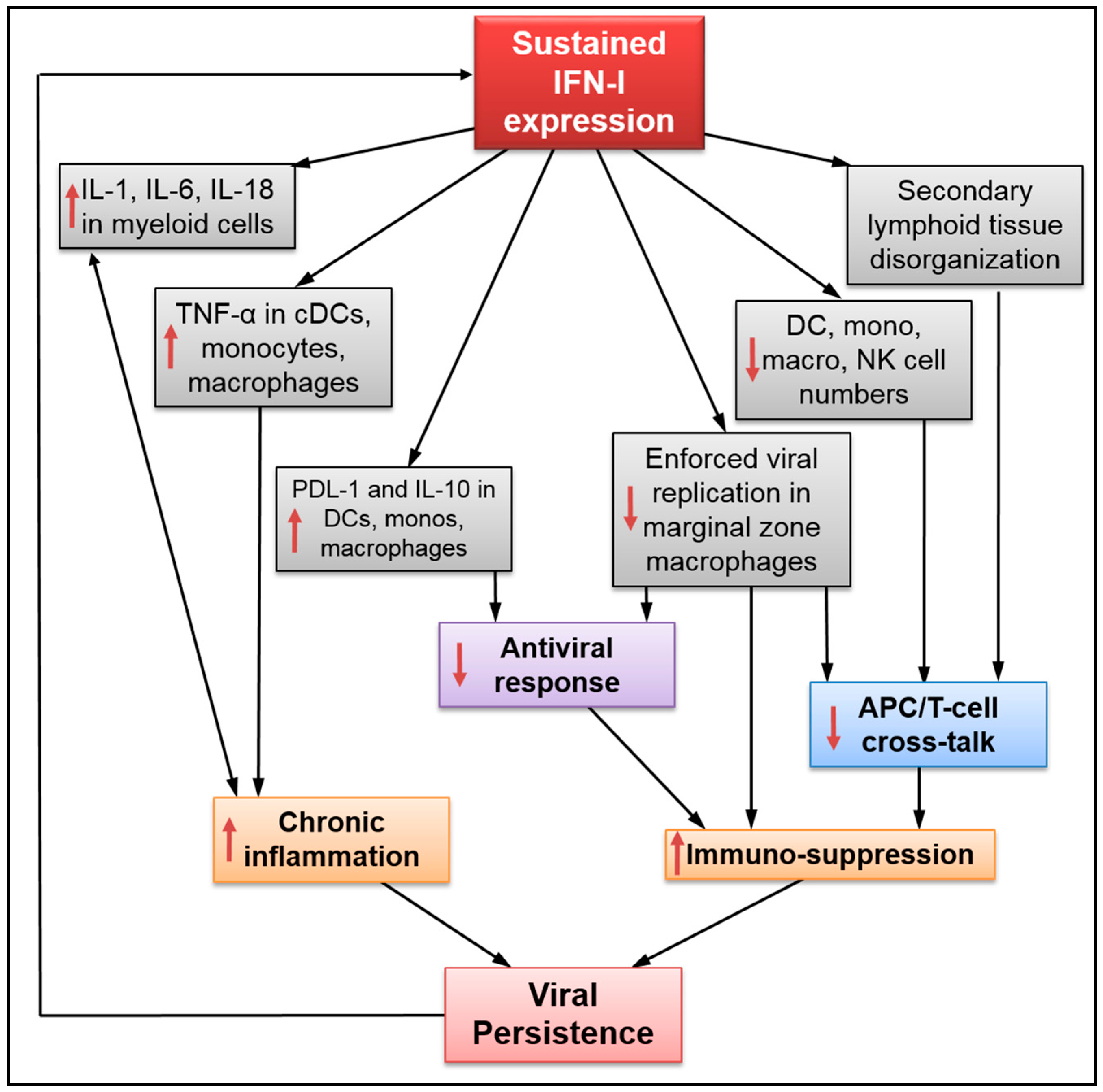
2. Sustained IFN-I Expression Drives Disease Progression during Persistent Infection
3. Impact of Sustained IFN-I Expression on the Innate Immune Response
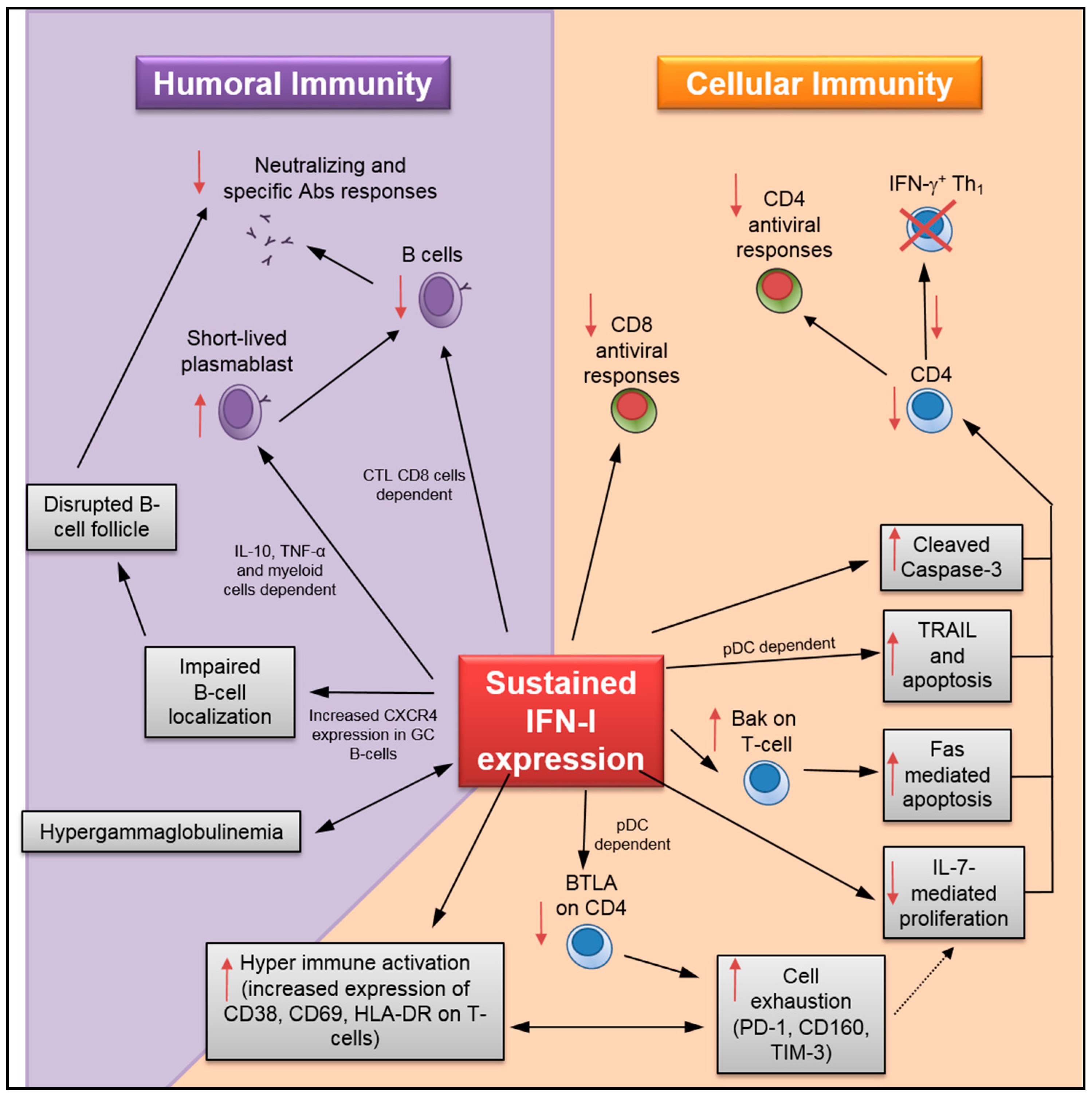
4. Detrimental Role of Sustained IFN-I Expression on the Humoral Immune Response
5. Impact of Sustained IFN-I on T-Cell Maintenance and Antiviral Response
- (i)
- T-cell loss: Although IFN-α administration initially prevents systemic SIV infection in rhesus macaques, prolonged treatment accelerates CD4 T-cell loss [6]. A recent study by Chen and colleagues has shown that sustained IFN-I expression increased HIV-1-induced apoptosis and caspase-3 activity in CD4 T-cells [19] (Figure 2). Importantly, INFR blockade rescued HIV-specific T-cell and total T-cell numbers during persistent infection, as well as reduced the apoptosis in CD4 T-cells (Table 1). Herbeuval and colleagues have shown that IFNR blockade results in decreased TRAIL/DR5-mediated apoptosis and caspase-3 activity in CD4 T-cells using an in vitro HIV-1 infection model [53]. In a separate study, they also reported a lower frequency of TRAIL+ and apoptotic CD4 T-cells in HIV-1-infected human samples after treatment with anti-IFNα/β neutralizing Abs [59]. In addition, it has also been shown that IFNR blockade causes downregulation of cell-death signal cascades by the reduction in Bak expression and Fas-mediated apoptosis in CD4 T-cells using an in vitro HIV-1 infection model [66]. IFNR blockade also increases total splenic T-lymphocyte and antiviral specific CD4 T-cell numbers during LCMVCL13 infection [26,29,63]. Finally, data collected by Cha and colleagues indicates that sustained IFN-I expression in HIV-1-infected patients undergoing ART may promote T-cell loss by accelerating cell turnover and activation-induced cell death while decreasing T-cell homeostasis mediated by IL-7 [7,60]. Similarly, prolonged exposure to IFN-I in mice under lymphopenic conditions has been found to alter CD4 T-cell homeostasis [67].
- (ii)
- T-cell hyperactivation: Elevated expression of T-cell activation/proliferation markers such as CD38, HLA-DR, and Ki67, which correlates with sustained IFN-I expression during persistent HIV-1 infection [8], is significantly reduced by IFNR blockade [18,38]. Similarly, INFR blockade reduces HIV-induced CD80 expression in CCR5+ T-cells, and CD69 and CD38 in T-cells during in vitro infection [46,68]. HIV-1-induced BTLA downregulation in T-cells, which may also contribute to hyperactivation, can be prevented by IFNR blockade [69].
- (iii)
- T-cell exhaustion: During chronic HIV-1 infection, the IFN-I pathway is associated with CD4 T-cell exhaustion [70]. Recent results in humanized mice infected with HIV-1 have shown that IFNR blockade resulted in reduced expression of several exhaustion markers in CD8 T-cells—including PD-1, CD160, and TIM-3—along with enhanced IFN-γ and IL-2 production in virus-specific T-cells [18,19,38]. Relatedly, IFNR blockade in LCMVCL13-infected mice enhances virus-specific CD4 T-cell response [26,29]. Finally, sustained IFN-I expression during LCMVCL13 infection also suppresses de novo Th1 differentiation in late primed virus-specific CD4 [71]. In this study, the authors have shown that, although reduced Th1 differentiation was not mediated through direct IFN-I sensing by CD4 T-cells, it could be rescued by IFNR blockade.
- (iv)
- Impact on immunosuppressive Treg: Although the effect of IFN-I signaling on hyperactivation and cell exhaustion is evident, its impact on regulatory T cells (Treg) during viral infections remains unclear [3]. In the case of acute LCMV infection, studies provide contradicting information showing either no effect of IFN-I on Treg [72], or a direct effect in reducing their numbers resulting in lower viral load [73]. Moreover, the effect of Treg depletion during chronic LCMV infection failed to increase viral clearance due to PD-L1 expression on infected cells despite an increase in virus-specific CD8 T-cell activity [74].
6. Concluding Remarks
Conflicts of Interest
References
- Acchioni, C.; Marsili, G.; Perrotti, E.; Remoli, A.L.; Sgarbanti, M.; Battistini, A. Type I IFN—A blunt spear in fighting HIV-1 infection. Cytokine Growth Factor Rev. 2015, 26, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I interferon in chronic virus infection and cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R. Type I interferons in viral control and immune regulation. Curr. Opin. Virol. 2016, 16, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.A.; Keeffe, E.B. Chronic viral hepatitis: Epidemiology, molecular biology, and antiviral therapy. Front. Biosci. 2011, 16, 225–250. [Google Scholar] [CrossRef]
- Sandler, N.G.; Bosinger, S.E.; Estes, J.D.; Zhu, R.T.; Tharp, G.K.; Boritz, E.; Levin, D.; Wijeyesinghe, S.; Makamdop, K.N.; del Prete, G.Q.; et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 2014, 511, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Cha, L.; Berry, C.M.; Nolan, D.; Castley, A.; Fernandez, S.; French, M.A. Interferon-α, immune activation and immune dysfunction in treated HIV infection. Clin. Transl. Immunol. 2014, 3, e10. [Google Scholar] [CrossRef] [PubMed]
- Hardy, G.A.; Sieg, S.; Rodriguez, B.; Anthony, D.; Asaad, R.; Jiang, W.; Mudd, J.; Schacker, T.; Funderburg, N.T.; Pilch-Cooper, H.A.; et al. Interferon-α is the primary plasma type-I IFN in HIV-1 infection and correlates with immune activation and disease markers. PLoS ONE 2013, 8, e56527. [Google Scholar] [CrossRef] [PubMed]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Up regulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Said, E.A.; Halwani, R.; Janbazian, L.; Chomont, N.; El-Far, M.; Breton, G.; Haddad, E.K.; Sekaly, R.P. Programmed death 1: A critical regulator of T-cell function and a strong target for immunotherapies for chronic viral infections. Curr. Opin. HIV AIDS 2007, 2, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Van Grevenynghe, J.; Cubas, R.A.; DaFonseca, S.; Metcalf, T.; Tremblay, C.L.; Trautmann, L.; Sekaly, R.P.; Schatzle, J.; Haddad, E.K. Foxo3a: An integrator of immune dysfunction during HIV infection. Cytokine Growth Factor Rev. 2012, 23, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Van Grevenynghe, J.; Cubas, R.A.; Noto, A.; DaFonseca, S.; He, Z.; Peretz, Y.; Filali-Mouhim, A.; Dupuy, F.P.; Procopio, F.A.; Chomont, N.; et al. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J. Clin. Investig. 2011, 121, 3877–3888. [Google Scholar] [CrossRef] [PubMed]
- Van Grevenynghe, J.; Procopio, F.A.; He, Z.; Chomont, N.; Riou, C.; Zhang, Y.; Gimmig, S.; Boucher, G.; Wilkinson, P.; Shi, Y.; et al. Transcription factor Foxo3a controls the persistence of memory CD4(+) T cells during hiv infection. Nat. Med. 2008, 14, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Beltra, J.C.; Decaluwe, H. Cytokines and persistent viral infections. Cytokine 2016, 82, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Bolen, C.R.; Robek, M.D.; Brodsky, L.; Schulz, V.; Lim, J.K.; Taylor, M.W.; Kleinstein, S.H. The blood transcriptional signature of chronic hepatitis C virus is consistent with an ongoing interferon-mediated antiviral response. J. Interferon Cytokine Res. 2013, 33, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Bosinger, S.E.; Utay, N.S. Type I interferon: Understanding its role in HIV pathogenesis and therapy. Curr. HIV/AIDS Rep. 2015, 12, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Ma, J.; Li, J.; Li, D.; Li, G.; Li, F.; Zhang, Q.; Yu, H.; Yasui, F.; Ye, C.; et al. Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J. Clin. Investig. 2017, 127, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Yu, H.; Li, G.; Li, F.; Ma, J.; Li, J.; Chi, L.; Zhang, L.; Su, L. Type I interferons suppress viral replication but contribute to T cell depletion and dysfunction during chronic HIV-1 infection. JCI Insight 2017, 2, e94366. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.; Tanaskovic, S.; Helbig, K.; Rajasuriar, R.; Kramski, M.; Murray, J.M.; Beard, M.; Purcell, D.; Lewin, S.R.; Price, P.; et al. CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J. Infect. Dis. 2011, 204, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Hardy, G.A.; Sieg, S.F.; Rodriguez, B.; Jiang, W.; Asaad, R.; Lederman, M.M.; Harding, C.V. Desensitization to type I interferon in HIV-1 infection correlates with markers of immune activation and disease progression. Blood 2009, 113, 5497–5505. [Google Scholar] [CrossRef] [PubMed]
- Honke, N.; Shaabani, N.; Merches, K.; Gassa, A.; Kraft, A.; Ehrhardt, K.; Haussinger, D.; Lohning, M.; Dittmer, U.; Hengel, H.; et al. Immunoactivation induced by chronic viral infection inhibits viral replication and drives immunosuppression through sustained IFN-I responses. Eur. J. Immunol. 2016, 46, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Marsili, G.; Remoli, A.L.; Sgarbanti, M.; Perrotti, E.; Fragale, A.; Battistini, A. HIV-1, interferon and the interferon regulatory factor system: An interplay between induction, antiviral responses and viral evasion. Cytokine Growth Factor Rev. 2012, 23, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; et al. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kang, W.; Zuo, J.; Kang, W.; Sun, Y. The significance of type-I interferons in the pathogenesis and therapy of human immunodeficiency virus 1 infection. Front. Immunol. 2017, 8, 1431. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Brooks, D.G. Decoding the complexity of type I interferon to treat persistent viral infections. Trends Microbiol. 2013, 21, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.D.; Tabb, B.; Sodora, D.L.; Paiardini, M.; Klatt, N.R.; Douek, D.C.; Silvestri, G.; Muller-Trutwin, M.; Vasile-Pandrea, I.; Apetrei, C.; et al. Down regulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J. Virol. 2010, 84, 7886–7891. [Google Scholar] [CrossRef] [PubMed]
- Jacquelin, B.; Mayau, V.; Targat, B.; Liovat, A.S.; Kunkel, D.; Petitjean, G.; Dillies, M.A.; Roques, P.; Butor, C.; Silvestri, G.; et al. Nonpathogenic SIV infection of african green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Investig. 2009, 119, 3544–3555. [Google Scholar] [CrossRef] [PubMed]
- Mandl, J.N.; Barry, A.P.; Vanderford, T.H.; Kozyr, N.; Chavan, R.; Klucking, S.; Barrat, F.J.; Coffman, R.L.; Staprans, S.I.; Feinberg, M.B. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat. Med. 2008, 14, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Rotger, M.; Dalmau, J.; Rauch, A.; McLaren, P.; Bosinger, S.E.; Martinez, R.; Sandler, N.G.; Roque, A.; Liebner, J.; Battegay, M.; et al. Comparative transcriptomics of extreme phenotypes of human HIV-1 infection and SIV infection in sooty mangabey and rhesus macaque. J. Clin. Investig. 2011, 121, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Shearer, G.M. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin. Immunol. 2008, 126, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, A.R.; German, J.; Teslovich, T.M.; Cofrancesco, J., Jr.; Jie, C.C.; Talbot, C.C., Jr.; Siliciano, R.F. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: Type I interferon-mediated disruption of T-cell dynamics. J. Virol. 2008, 82, 1870–1883. [Google Scholar] [CrossRef] [PubMed]
- Stylianou, E.; Aukrust, P.; Bendtzen, K.; Muller, F.; Froland, S.S. Interferons and interferon (IFN)-inducible protein 10 during highly active anti-retroviral therapy (HAART)-possible immunosuppressive role of IFN-alpha in HIV infection. Clin. Exp. Immunol. 2000, 119, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Van Grevenynghe, J.; Halwani, R.; Chomont, N.; Ancuta, P.; Peretz, Y.; Tanel, A.; Procopio, F.A.; shi, Y.; Said, E.A.; Haddad, E.K.; et al. Lymph node architecture collapse and consequent modulation of FOXO3a pathway on memory T- and B-cells during HIV infection. Semin. Immunol. 2008, 20, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.; Rezek, V.; Youn, C.; Lam, B.; Chang, N.; Rick, J.; Carrillo, M.; Martin, H.; Kasparian, S.; Syed, P.; et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J. Clin. Investig. 2017, 127, 260–268. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.M.; Jiang, J.; Abbas, A.R.; Morimoto, A.; Li, J.; Maciuca, R.; Townsend, M.; Wallace, D.J.; Kennedy, W.P.; Drappa, J. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: Results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012, 64, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Higgs, B.W.; Chang, L.; Vainshtein, I.; Liu, Z.; Streicher, K.; Liang, M.; White, W.I.; Yoo, S.; Richman, L.; et al. Pharmacogenomics and translational simulations to bridge indications for an anti-interferon-α receptor antibody. Clin. Pharmacol. Ther. 2013, 93, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Brooks, D.G. Interfering with type I interferon: A novel approach to purge persistent viral infection. Cell Cycle 2013, 12, 2919–2920. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Nilsson, J.; Boasso, A.; Hardy, A.W.; Kruhlak, M.J.; Anderson, S.A.; Dolan, M.J.; Dy, M.; Andersson, J.; Shearer, G.M. Differential expression of IFN-α and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl. Acad. Sci. USA 2006, 103, 7000–7005. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Harper, J.M.; Taubert, D.; Hartmann, P.; Fatkenheuer, G.; Jung, N.; van Lunzen, J.; Stellbrink, H.J.; Gallo, R.C.; Romerio, F. Increased interferon α expression in circulating plasmacytoid dendritic cells of HIV-1-infected patients. J. Acquir. Immune Defic. Syndr. 2008, 48, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, R.T.; Freeman, Z.T.; Korleski, J.; Cohen, L.K.; Massaccesi, G.; Tomasi, A.; Boesch, A.W.; Ackerman, M.E.; Margolick, J.B.; Blankson, J.N.; et al. HIV-antibody complexes enhance production of type I interferon by plasmacytoid dendritic cells. J. Clin. Investig. 2017, 127, 4352–4364. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Wang, Y.; Vermi, W.; Gilfillan, S.; Schreiber, R.D.; Colonna, M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 2011, 208, 2367–2374. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Hardy, A.W.; Landay, A.L.; Martinson, J.L.; Anderson, S.A.; Dolan, M.J.; Clerici, M.; Shearer, G.M. PDL-1 upregulation on monocytes and T cells by HIV via type I interferon: Restricted expression of type I interferon receptor by CCR5-expressing leukocytes. Clin. Immunol. 2008, 129, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, M.; Zeremski, M.; Talal, A.H.; Ginwala, R.; Elrod, E.; Grakoui, A.; Li, Q.G.; Philip, R.; Khan, Z.K.; Jain, P. IFN-alpha-induced downregulation of miR-221 in dendritic cells: Implications for HCV pathogenesis and treatment. J. Interferon Cytokine Res. 2015, 35, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.R.; Champhekar, A.; Tullius, M.V.; Dillon, B.J.; Zhen, A.; de la Fuente, J.R.; Herskovitz, J.; Elsaesser, H.; Snell, L.M.; Wilson, E.B.; et al. Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog. 2016, 12, e1005356. [Google Scholar] [CrossRef] [PubMed]
- Honke, N.; Shaabani, N.; Cadeddu, G.; Sorg, U.R.; Zhang, D.E.; Trilling, M.; Klingel, K.; Sauter, M.; Kandolf, R.; Gailus, N.; et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat. Immunol. 2011, 13, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Norris, B.A.; Uebelhoer, L.S.; Nakaya, H.I.; Price, A.A.; Grakoui, A.; Pulendran, B. Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity. Immunity 2013, 38, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Taleb, K.; Auffray, C.; Villefroy, P.; Pereira, A.; Hosmalin, A.; Gaudry, M.; Le Bon, A. Chronic type I IFN is sufficient to promote immunosuppression through accumulation of myeloid-derived suppressor cells. J. Immunol. 2017, 198, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Rempel, H.; Sun, B.; Calosing, C.; Pillai, S.K.; Pulliam, L. Interferon-α drives monocyte gene expression in chronic unsuppressed HIV-1 infection. AIDS 2010, 24, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Herbeuval, J.P.; Grivel, J.C.; Boasso, A.; Hardy, A.W.; Chougnet, C.; Dolan, M.J.; Yagita, H.; Lifson, J.D.; Shearer, G.M. CD4+ T-cell death induced by infectious and noninfectious HIV-1: Role of type 1 interferon-dependent, TRAIL/DR5-mediated apoptosis. Blood 2005, 106, 3524–3531. [Google Scholar] [CrossRef] [PubMed]
- Kardava, L.; Moir, S.; Shah, N.; Wang, W.; Wilson, R.; Buckner, C.M.; Santich, B.H.; Kim, L.J.; Spurlin, E.E.; Nelson, A.K.; et al. Abnormal B cell memory subsets dominate HIV-specific responses in infected individuals. J. Clin. Investig. 2014, 124, 3252–3262. [Google Scholar] [CrossRef] [PubMed]
- Fallet, B.; Narr, K.; Ertuna, Y.I.; Remy, M.; Sommerstein, R.; Cornille, K.; Kreutzfeldt, M.; Page, N.; Zimmer, G.; Geier, F.; et al. Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Moir, S.; Fauci, A.S. Insights into B cells and HIV-specific B-cell responses in HIV-infected individuals. Immunol. Rev. 2013, 254, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Moseman, E.A.; Wu, T.; de la Torre, J.C.; Schwartzberg, P.L.; McGavern, D.B. Type I interferon suppresses virus-specific B cell responses by modulating CD8+ T cell differentiation. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Sammicheli, S.; Kuka, M.; Di Lucia, P.; de Oya, N.J.; De Giovanni, M.; Fioravanti, J.; Cristofani, C.; Maganuco, C.G.; Fallet, B.; Ganzer, L.; et al. Inflammatory monocytes hinder antiviral B cell responses. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Herbeuval, J.P.; Hardy, A.W.; Boasso, A.; Anderson, S.A.; Dolan, M.J.; Dy, M.; Shearer, G.M. Regulation of tnf-related apoptosis-inducing ligand on primary CD4+ T cells by HIV-1: Role of type I IFN-producing plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2005, 102, 13974–13979. [Google Scholar] [CrossRef] [PubMed]
- Cha, L.; de Jong, E.; French, M.A.; Fernandez, S. IFN-alpha exerts opposing effects on activation-induced and IL-7-induced proliferation of T cells that may impair homeostatic maintenance of CD4+ T cell numbers in treated HIV infection. J. Immunol. 2014, 193, 2178–2186. [Google Scholar] [CrossRef] [PubMed]
- Moir, S.; Fauci, A.S. B-cell responses to HIV infection. Immunol. Rev. 2017, 275, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; Pantaleo, G. B-cell abnormalities and impact on antibody response in HIV infection. Curr. Opin. HIV AIDS 2017, 12, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Daugan, M.; Murira, A.; Mindt, B.C.; Germain, A.; Tarrab, E.; Lapierre, P.; Fritz, J.H.; Lamarre, A. Type I interferon impairs specific antibody responses early during establishment of LCMV infection. Front. Immunol. 2016, 7, 564. [Google Scholar] [CrossRef] [PubMed]
- Aounallah, M.; Dagenais-Lussier, X.; El-Far, M.; Mehraj, V.; Jenabian, M.A.; Routy, J.P.; van Grevenynghe, J. Current topics in HIV pathogenesis, part 2: Inflammation drives a warburg-like effect on the metabolism of HIV-infected subjects. Cytokine Growth Factor Rev. 2016, 28, 1–10. [Google Scholar] [CrossRef] [PubMed]
- El-Far, M.; Halwani, R.; Said, E.; Trautmann, L.; Doroudchi, M.; Janbazian, L.; Fonseca, S.; van Grevenynghe, J.; Yassine-Diab, B.; Sekaly, R.P.; et al. T-cell exhaustion in HIV infection. Curr. HIV/AIDS Rep. 2008, 5, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Mueller, Y.M.; Yang, G.; Boesteanu, A.C.; Gracias, D.T.; Do, D.H.; Hope, J.L.; Kathuria, N.; McGettigan, S.E.; Lewis, M.G.; et al. Type I interferon upregulates bak and contributes to T cell loss during human immunodeficiency virus (HIV) infection. PLoS Pathog. 2013, 9, e1003658. [Google Scholar] [CrossRef] [PubMed]
- Le Saout, C.; Hasley, R.B.; Imamichi, H.; Tcheung, L.; Hu, Z.; Luckey, M.A.; Park, J.H.; Durum, S.K.; Smith, M.; Rupert, A.W.; et al. Chronic exposure to type-I IFN under lymphopenic conditions alters CD4 T cell homeostasis. PLoS Pathog. 2014, 10, e1003976. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Hardy, A.W.; Anderson, S.A.; Dolan, M.J.; Shearer, G.M. HIV-induced type I interferon and tryptophan catabolism drive T cell dysfunction despite phenotypic activation. PLoS ONE 2008, 3, e2961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, X.; Lu, J.; Zhang, S.; Gu, L.; Fu, J.; Jin, L.; Li, H.; Zhao, M.; Zhang, J.; et al. B and T lymphocyte attenuator down-regulation by HIV-1 depends on type I interferon and contributes to T-cell hyperactivation. J. Infect. Dis. 2011, 203, 1668–1678. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crawford, A.; Angelosanto, J.M.; Kao, C.; Doering, T.A.; Odorizzi, P.M.; Barnett, B.E.; Wherry, E.J. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 2014, 40, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Osokine, I.; Snell, L.M.; Cunningham, C.R.; Yamada, D.H.; Wilson, E.B.; Elsaesser, H.J.; de la Torre, J.C.; Brooks, D. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc. Natl. Acad. Sci. USA 2014, 111, 7409–7414. [Google Scholar] [CrossRef] [PubMed]
- Che, J.W.; Kraft, A.R.; Selin, L.K.; Welsh, R.M. Regulatory T cells resist virus infection-induced apoptosis. J. Virol. 2015, 89, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Koch, M.A.; Pepper, M.; Campbell, D.J. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J. Exp. Med. 2014, 211, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Penaloza-MacMaster, P.; Kamphorst, A.O.; Wieland, A.; Araki, K.; Iyer, S.S.; West, E.E.; O’Mara, L.; Yang, S.; Konieczny, B.T.; Sharpe, A.H.; et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J. Exp. Med. 2014, 211, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Swiecki, M.; Cella, M.; Alber, G.; Schreiber, R.D.; Gilfillan, S.; Colonna, M. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 2012, 11, 631–642. [Google Scholar] [CrossRef] [PubMed]

 : impact on both sides.
: impact on both sides.
: impact on both sides.
: impact on both sides.

{kind=link}
{kind=link}
| Phenotype Improvement(s) | Model | Virus(es) | Reference(s) |
|---|---|---|---|
| Increased cytokine producing virus-specific CD4/CD8 (IFN-γ, TNF-α, IL-2); improved antiviral responses |  | HIV, LCMV | [12,13,23,26,34] |
| Reduced expression of PD-1, TIM-3, TIGIT, BATF, CD160 on CD8 (decreased cell exhaustion) |  | HIV | [12,34] |
| Reduced KI67+ population in CD4/CD8; decrease of HIV-mediated T-cell hyperactivation |  | HIV | [12] |
| Reduced HLA-DR, CD38, CD69, CD80 expression in CD4/CD8; decrease of HIV-mediated T-cell hyperactivation |  | HIV | [12,34,41*,53*] |
| Decrease of caspase-3-dependent apoptosis in total and specific CD4 T-cells |  | HIV, LCMV | [13,54,55*] |
| Accelerating neutralizing Abs production |  | LCMV | [18] |
| Reduced PD-L1 and IL-10 expression in DCs, mono and macro. Decreased IL-10 levels in plasma/sera |  | HIV, LCMV | [23,26,41*] |
| Reduced IL-1 and IL-18 levels, and inflammasome activation in DCs and monos; decreased CD80 expression in monos |  | HIV, LCMV | [23,26,41*] |
| Increase of splenocyte cell numbers (DCs, macrophages, CD4, CD8, B and matural killers) |  | LCMV | [23,26,52] |
| Proper splenic architecture organization |  | LCMV | [23,26] |
| Decreased CXCR4 expression on GC B Increased levels of specific Ab production and specific ASCs |  | LCMV | [52] |
| Decreased caspase-3+ apoptotic virus-specific GC B (by counteracting plasmablast differentiation); B expansion |  | LCMV | [54] |
| Increased virus-specific B number; Decreased CTL CD8-mediated kill of specific B |  | LCMV | [56] |
| Reduced TRAIL/DR5-mediated apoptosis in CD4 |  | HIV | [55*] |
| Decreased % of TRAIL+ and apoptotic CD4 |  | HIV | [57] |
| Increased Fas-mediated apoptosis in CD4 and CD8 T-cells |  | HIV | [58*] |
| Increased expression of BTLA on CD4 T-cells; reduced hyper-immune activation |  | HIV | [59*] |
| Increased Th1 differentiation in late primed virus-specific CD4 |  | LCMV | [60] |
 : Humans;
: Humans;  : Humanized mice;
: Humanized mice;  ; Mice; *: in vitro viral infection; ASC: antibody-secreting cells; CD: cluster of Differentiation; TNF: tumor Necrosis Factor; IL: interleukin; PD: programmed Death Protein; TIM: T-cell Immunoglobulin and Mucin; TIGIT: T-cell Ig and ITIM domain; BATF: basic leucine zipper transcription factor ATF-like; LCMV: Lymphocytic choriomeningitis virus; KI: proliferation Marker, Ki-67 is a prototype monoclonal Ab and was first produced in Kiel, Germany; HLA-DR: human leukocyte antigen - antigen D related; DCs: dendritic cells; CXCR: C-X-C motif chemokine receptor; GC: germinal center; ASCs: antibody secreting cells; CTL: cytotoxic T lymphocytes; TRAIL: TNF-related apoptosis inducing ligand; Th1: Type I T helper cells; BTLA: B and T lymphocyte attenuator.
; Mice; *: in vitro viral infection; ASC: antibody-secreting cells; CD: cluster of Differentiation; TNF: tumor Necrosis Factor; IL: interleukin; PD: programmed Death Protein; TIM: T-cell Immunoglobulin and Mucin; TIGIT: T-cell Ig and ITIM domain; BATF: basic leucine zipper transcription factor ATF-like; LCMV: Lymphocytic choriomeningitis virus; KI: proliferation Marker, Ki-67 is a prototype monoclonal Ab and was first produced in Kiel, Germany; HLA-DR: human leukocyte antigen - antigen D related; DCs: dendritic cells; CXCR: C-X-C motif chemokine receptor; GC: germinal center; ASCs: antibody secreting cells; CTL: cytotoxic T lymphocytes; TRAIL: TNF-related apoptosis inducing ligand; Th1: Type I T helper cells; BTLA: B and T lymphocyte attenuator.© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagenais-Lussier, X.; Loucif, H.; Murira, A.; Laulhé, X.; Stäger, S.; Lamarre, A.; Van Grevenynghe, J. Sustained IFN-I Expression during Established Persistent Viral Infection: A “Bad Seed” for Protective Immunity. Viruses 2018, 10, 12. https://doi.org/10.3390/v10010012
Dagenais-Lussier X, Loucif H, Murira A, Laulhé X, Stäger S, Lamarre A, Van Grevenynghe J. Sustained IFN-I Expression during Established Persistent Viral Infection: A “Bad Seed” for Protective Immunity. Viruses. 2018; 10(1):12. https://doi.org/10.3390/v10010012
Chicago/Turabian StyleDagenais-Lussier, Xavier, Hamza Loucif, Armstrong Murira, Xavier Laulhé, Simona Stäger, Alain Lamarre, and Julien Van Grevenynghe. 2018. "Sustained IFN-I Expression during Established Persistent Viral Infection: A “Bad Seed” for Protective Immunity" Viruses 10, no. 1: 12. https://doi.org/10.3390/v10010012
APA StyleDagenais-Lussier, X., Loucif, H., Murira, A., Laulhé, X., Stäger, S., Lamarre, A., & Van Grevenynghe, J. (2018). Sustained IFN-I Expression during Established Persistent Viral Infection: A “Bad Seed” for Protective Immunity. Viruses, 10(1), 12. https://doi.org/10.3390/v10010012