Liver Cell Transformation in Chronic HBV Infection

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Animal Models of Hepadnavirus-Related Carcinogenesis
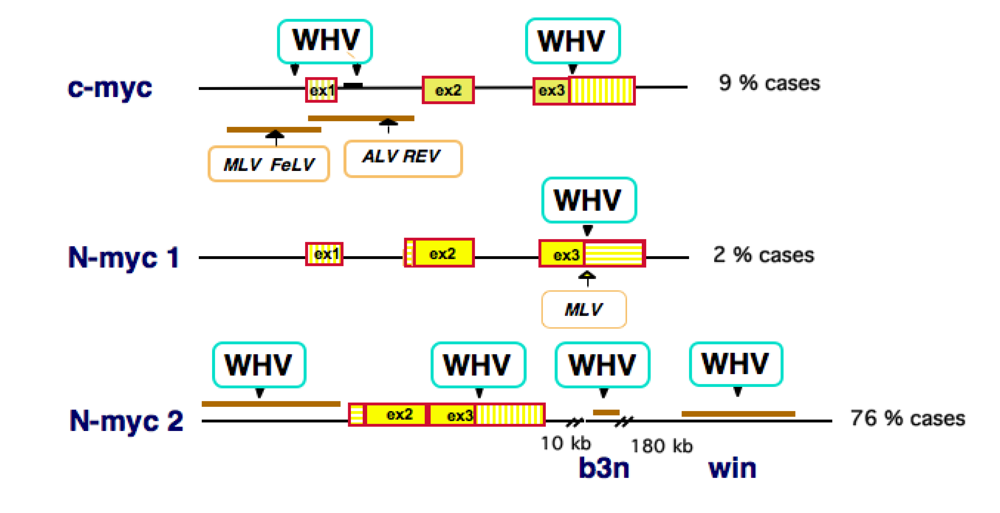
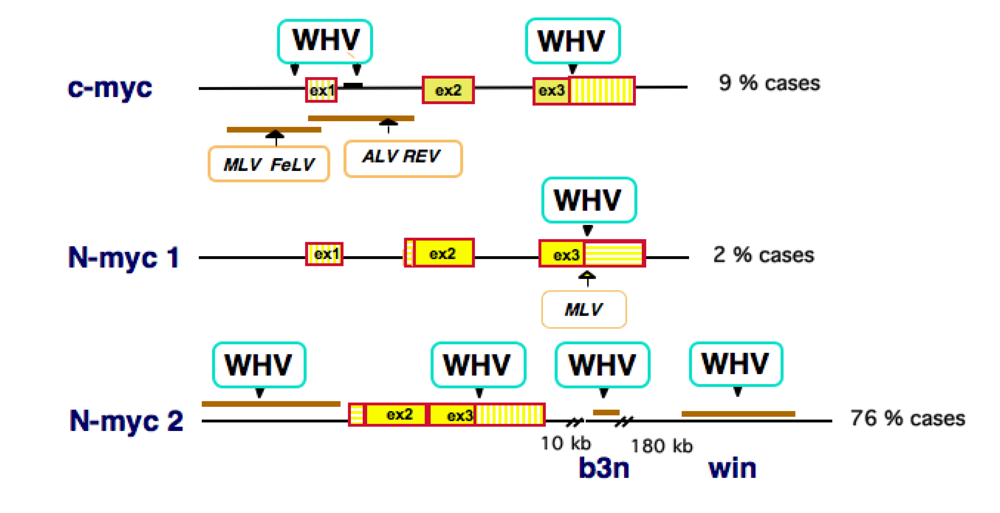
3. HBV DNA Integration into Human Chromosomes
4. Oncogenic Capabilities of the HBx Regulatory Protein
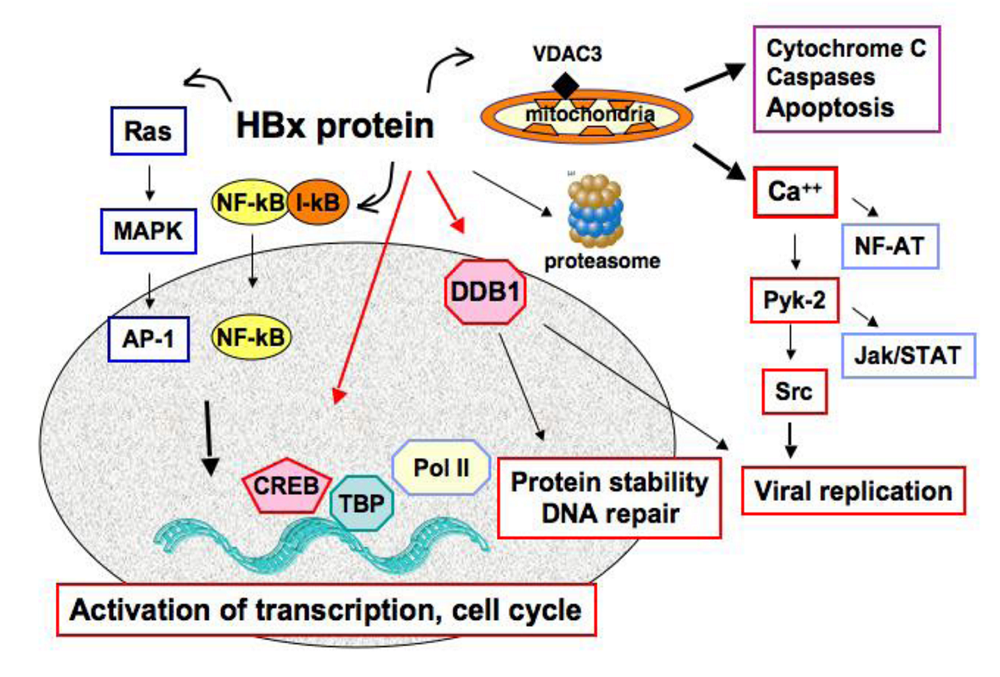
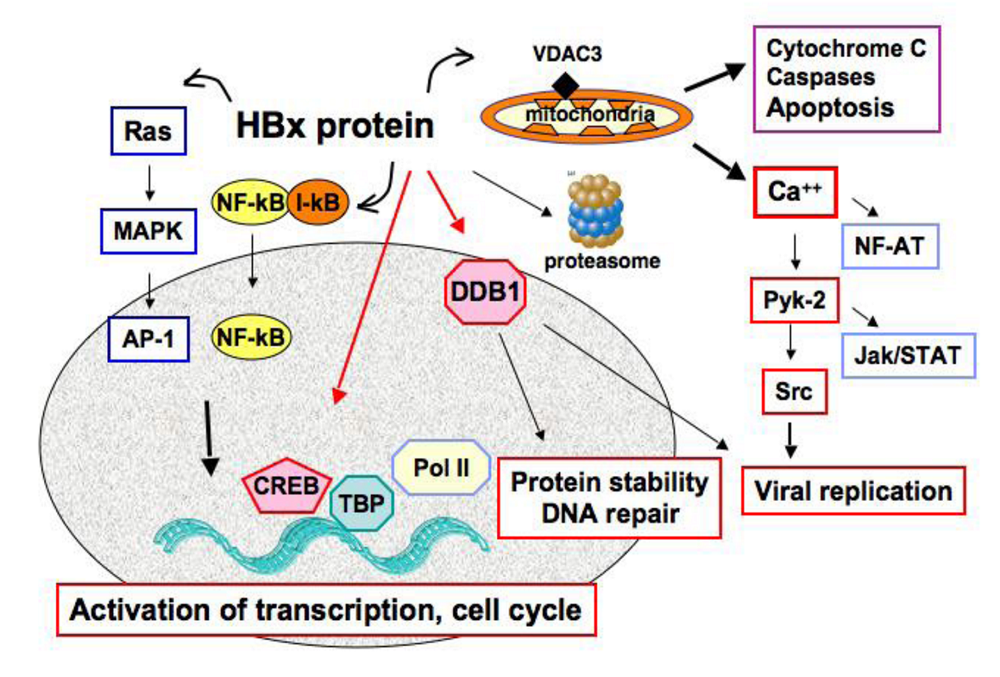
5. HBx, Replication Stress and Mitotic Defects
6. Conclusions
References and Notes
- Butel, J.S. Viral carcinogenesis: revelation of molecular mechanisms and etiology of human disease. Carcinogenesis 2000, 21, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Pagano, J.S.; Blaser, M.; Buendia, M.A.; Damania, B.; Khalili, K.; Raab-Traub, N.; Roizman, B. Infectious agents and cancer: criteria for a causal relation. Semin. Cancer Biol. 2004, 14, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Szmuness, W. Hepatocellular carcinoma and the hepatitis B virus : Evidence for a causal association. Prog. Med. Virol. 1978, 24, 40–69. [Google Scholar] [PubMed]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int J Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef] [PubMed]
- Paterlini, P.; Driss, F.; Pisi, E.; Franco, D.; Berthelot, P.; Bréchot, C. Persistence of hepatitis B and hepatitis C viral genomes in primary liver cancers from HBsAg negative patients: a study of a low endemic area. Hepatology 1993, 17, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Squadrito, G.; Cerenzia, G.; Cacciola, I.; Raffa, G.; Craxi, A.; Farinati, F.; Missale, G.; Smedile, A.; Tiribelli, C.; Villa, E.; Raimondo, G. Hepatitis B virus maintains its pro-oncogenic properties in the case of occult HBV infection. Gastroenterology 2004, 126, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Bréchot, C.; Pourcel, C.; Louise, A.; Rain, B.; Tiollais, P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature 1980, 286, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Saigo, K.; Takashima, H.; Minami, M.; Okanoue, T.; Brechot, C.; Paterlini-Brechot, P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 2005, 54, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, Y.; Guidotti, L.G.; Kuhlen, C.V.; Fowler, P.; Chisari, F.V. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 1998, 188, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Brechot, C.; Gozuacik, D.; Murakami, Y.; Paterlini-Brechot, P. Molecular bases for the development of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC). Semin. Cancer Biol. 2000, 10, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Kremsdorf, D.; Soussan, P.; Paterlini-Brechot, P.; Brechot, C. Hepatitis B virus-related hepatocellular carcinoma: paradigms for viral-related human carcinogenesis. Oncogene. 2006, 25, 3823–3833. [Google Scholar] [CrossRef] [PubMed]
- Cougot, D.; Buendia, M.A.; Neuveut, C. Carcinogenesis induced by Hepatitis B Virus . Chan, S.H.H., Ed.; Karger: Basel, 2008; pp. 108–136. [Google Scholar]
- Summers, J. Three recently described animal virus models for human hepatitis B virus. Hepatology 1981, 1, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Buendia, M.A. Hepatitis B viruses and hepatocellular carcinoma. Adv. Cancer Res. 1992, 59, 167–226. [Google Scholar] [PubMed]
- Kulkarni, K.; Jacobson, I.M.; Tennant, B.C. The role of the woodchuck model in the treatment of hepatitis B virus infection. Clin. Liver Dis. 2007, 11, 707–725. [Google Scholar] [CrossRef] [PubMed]
- Tennant, B.C.; Toshkov, I.A.; Peek, S.F.; Jacob, J.R.; Menne, S.; Hornbuckle, W.E.; Schinazi, R.D.; Korba, B.E.; Cote, P.J.; Gerin, J.L. Hepatocellular carcinoma in the woodchuck model of hepatitis B virus infection . Gastroenterology 2004, 127 , S283–293. [Google Scholar] [CrossRef] [PubMed]
- Korba, B.E.; Wells, F.V.; Baldwin, B.; Cote, P.J.; Tennant, B.C.; Popper, H.; Gerin, J.L. Hepatocellular carcinoma in woodchuck hepatitis virus-infected woodchucks: presence of viral DNA in tumor tissue from chronic carriers and animals serologically recovered from acute infections. Hepatology 1989, 9, 461–470. [Google Scholar] [CrossRef]
- Michalak, T.I.; Pardoe, I.U.; Coffin, C.S.; Churchill, N.D.; Freake, D.S.; Smith, P.; Trelegan, C.L. Occult lifelong persistence of infectious hepadnavirus and residual liver inflammation in woodchucks convalescent from acute viral hepatitis. Hepatology 1999, 29, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Paterlini, P.; Gerken, G.; Nakajima, E.; Terré, S.; D'Errico, A.; Grigioni, W.; Nalpas, B.; Franco, D.; Wands, J.; Kew, M.; Pisi, E.; Tiollais, P.; Bréchot, C. Polymerase chain reaction to detect hepatitis B virus DNA and RNA sequences in primary liver cancers from patients negative for hepatitis B surface antigen. New Engl. J. Med. 1990, 323, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Rogler, C.E.; Summers, J. Cloning and structural analysis of integrated woochuck hepatitis virus sequences from a chronically infected liver. J. Virol. 1984, 50, 832–837. [Google Scholar] [PubMed]
- Mason, W.S.; Jilbert, A.R.; Summers, J. Clonal expansion of hepatocytes during chronic woodchuck hepatitis virus infection. Proc. Natl. Acad. Sci. U S A 2005, 102, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Alt, E.; Rogler, C.E. Coordinate expression of N-myc 2 and insulin-like growth factor II in precancerous altered hepatic foci in woodchuck hepatitis virus carriers. Cancer Res. 1993, 53, 2020–2027. [Google Scholar] [PubMed]
- Hsu, T.; Moroy, T.; Etiemble, J.; Louise, A.; Trepo, C.; Tiollais, P.; Buendia, M.A. Activation of c-myc by woodchuck hepatitis virus insertion in hepatocellular carcinoma. Cell 1988, 55, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Fourel, G.; Trépo, C.; Bougueleret, L.; Henglein, B.; Ponzetto, A.; Tiollais, P.; Buendia, M.A. Frequent activation of N-myc genes by hepadnavirus insertion in woodchuck liver tumours. Nature 1990, 347, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Fourel, G.; Ponzetto, A.; Silvestro, M.; Tiollais, P.; Buendia, M.A. Hepadnavirus integration: mechanisms of activation of the N-myc2 retrotransposon in woodchuck liver tumors. J. Virol. 1992, 66, 5265–5276. [Google Scholar] [PubMed]
- Fourel, G.; Couturier, J.; Wei, Y.; Apiou, F.; Tiollais, P.; Buendia, M.A. Evidence for long-range oncogene activation by hepadnavirus insertion. EMBO J. 1994, 13, 2526–2534. [Google Scholar] [PubMed]
- Bruni, R.; D'Ugo, E.; Giuseppetti, R.; Argentini, C.; Rapicetta, M. Activation of the N-myc2 oncogene by woodchuck hepatitis virus integration in the linked downstream b3n locus in woodchuck hepatocellular carcinoma. Virology 1999, 257, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Bruni, R.; D'Ugo, E.; Villano, U.; Fourel, G.; Buendia, M.A.; Rapicetta, M. The win locus involved in activation of the distal N-myc2 gene upon WHV integration in woodchuck liver tumors harbors S/MAR elements. Virology 2004, 329, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.R.; Sterczer, A.; Toshkov, I.A.; Yeager, A.E.; Korba, B.E.; Cote, P.J.; Buendia, M.A.; Gerin, J.L.; Tennant, B.C. Integration of woodchuck hepatitis and N-myc rearrangement determine size and histologic grade of hepatic tumors. Hepatology 2004, 39, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Etiemble, J.; Degott, C.; Renard, C.A.; Fourel, G.; Shamoon, B.; Vitvitski-Trepo, L.; Hsu, T.Y.; Tiollais, P.; Babinet, C.; Buendia, M.A. Liver-specific expression and high oncogenic efficiency of a c-myc transgene activated by woodchuck hepatitis virus insertion. Oncogene 1994, 9, 727–737. [Google Scholar] [PubMed]
- Renard, C.A.; Fourel, G.; Bralet, M.P.; Degott, C.; De la Coste, A.; Perret, C.; Tiollais, P.; Buendia, M.A. Hepatocellular carcinoma in WHV/N-myc2 transgenic mice: oncogenic mutations of beta-catenin and synergistic effects of p53-null alleles. Oncogene 2000, 19, 2678–2686. [Google Scholar] [CrossRef] [PubMed]
- Kaposi-Novak, P.; Libbrecht, L.; Woo, H.G.; Lee, Y.H.; Sears, N.C.; Conner, E.A.; Factor, V.M.; Roskams, T.; Thorgeirsson, S.S. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009, 69, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D. Oncogenic viruses. Of marmots and men. Nature 1990, 347, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Lugassy, C.; Bernuau, J.; Thiers, V.; Krosgaard, K.; Degott, C.; Wantzin, P.; Schalm, S.W.; Rueff, B.; Benhamou, J.P.; Tiollais, P.; Bréchot, C. Sequences of hepatitis B virus DNA in the serum and liver of patients with acute benign and fulminant hepatitis. J. Infect. Dis. 1987, 155, 64–71. [Google Scholar] [PubMed]
- Yaginuma, K.; Kobayashi, H.; Kobayashi, M.; Morishima, T.; Matsuyama, K.; Koike, K. Multiple integration site of hepatitis B virus DNA in hepatocellular carcinoma and chronic active hepatitis tissues from children. J. Virol. 1987, 61, 1808–1813. [Google Scholar] [PubMed]
- Boender, P.J.; Schalm, S.W.; Heijtink, R.A. Detection of integration during active replication of hepatitis B virus in the liver. J. Med. Virol. 1985, 16, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Esumi, M.; Shikata, T. Frequent integration of hepatitis B virus DNA in noncancerous liver tissue from hepatocellular carcinoma patients. J. Med. Virol. 1988, 26, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; Chen, P.J.; Chen, J.Y.; Lai, M.Y.; Hsu, H.C.; Lian, D.C.; Liu, Y.G.; Chen, D.S. Hepatitis B virus integration in hepatitis B virus-related hepatocellular carcinoma in childhood. Hepatology 1991, 13, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Koshy, R.; Koch, S.; von Loringhoven, A.F.; Kahmann, R.; Murray, K.; Hofschneider, P.H. Integration of hepatitis B virus DNA: evidence for integration in the single-stranded gap. Cell 1983, 34, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.; Burke, K.; Chou, M.J.; Zeldis, J.B.; Yang, C.S.; Lee, C.S.; Isselbacher, K.J.; Wands, J.R.; Goodman, H. Tight clustering of human hepatitis B virus integration sites in hepatomas near a triple-stranded region. J. Virol. 1987, 61, 3491–3498. [Google Scholar] [PubMed]
- Nagaya, T.; Nakamura, T.; Tokino, T.; Tsurimoto, T.; Imai, M.; Mayumi, T.; Kamino, K.; Yamamura, K.; Matsubara, K. The mode of hepatitis B virus DNA integration in chromosomes of human hepatocellular carcinoma. Genes Dev. 1987, 1, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.P.; Rogler, C.E. Topoisomerase I-mediated integration of hepadnavirus DNA in vitro. J. Virol. 1991, 65, 2381–2392. [Google Scholar] [PubMed]
- Tokino, T.; Matsubara, K. Chromosomal sites for hepatitis B virus integration in human hepatocellular carcinoma. J. Virol. 1991, 65, 6761–6764. [Google Scholar] [PubMed]
- Dejean, A.; Bougueleret, L.; Grzeschik, K.H.; Tiollais, P. Hepatitis B virus DNA integration in a sequence homologous to v-erbA and steroid receptor genes in a hepatocellular carcinoma. Nature 1986, 322, 70–72. [Google Scholar] [CrossRef] [PubMed]
- De Thé, H.; Marchio, A.; Tiollais, P.; Dejean, A. A novel steroid/thyroid hormone receptor-related gene inappropriately expressed in human hepatocellular carcinoma. Nature 1987, 330, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; de The, H.; Tiollais, P.; Samarut, J.; Dejean, A. A hepatitis B virus pre-S-retinoic acid receptor beta chimera transforms erythrocytic progenitor cells in vitro. Proc. Natl. Acad. Sci. U S A 1993, 90, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chenivesse, X.; Henglein, B.; Bréchot, C. Hepatitis B virus integration in a cyclin A gene in a human hepatocellular carcinoma. Nature 1990, 343, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zindy, F.; Chenivesse, X.; Lamas, E.; Henglein, B.; Brechot, C. Modification of cyclin A expression by hepatitis B virus DNA integration in a hepatocellular carcinoma. Oncogene 1992, 7, 1653–1656. [Google Scholar] [PubMed]
- Berasain, C.; Patil, D.; Perara, E.; Huang, S.M.; Mouly, H.; Brechot, C. Oncogenic activation of a human cyclin A2 targeted to the endoplasmic reticulum upon hepatitis B virus genome insertion. Oncogene 1998, 16, 1277–1288. [Google Scholar] [PubMed]
- Paterlini-Brechot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Brechot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Mikkers, H.; Allen, J.; Knipscheer, P.; Romeijn, L.; Hart, A.; Vink, E.; Berns, A. High-throughput retroviral tagging to identify components of specific signaling pathways in cancer. Nat. Genet. 2002, 32, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, I.; Barrett, J.C. cis-Activation of the human telomerase gene (hTERT) by the hepatitis B virus genome. J. Natl. Cancer Inst. 2001, 93, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Ferber, M.J.; Montoya, D.P.; Yu, C.; Aderca, I.; McGee, A.; Thorland, E.C.; Nagorney, D.M.; Gostout, B.S.; Burgart, L.J.; Boix, L.; Bruix, J.; McMahon, B.J.; Cheung, T.H.; Chung, T.K.; Wong, Y.F.; Smith, D.I.; Roberts, L.R. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 2003, 22, 3813–3820. [Google Scholar] [CrossRef] [PubMed]
- Tamori, A.; Yamanishi, Y.; Kawashima, S.; Kanehisa, M.; Enomoto, M.; Tanaka, H.; Kubo, S.; Shiomi, S.; Nishiguchi, S. Alteration of gene expression in human hepatocellular carcinoma with integrated hepatitis B virus DNA. Clin. Cancer Res. 2005, 11, 5821–5826. [Google Scholar] [CrossRef]
- Yaginuma, K.; Kobayashi, M.; Yoshida, E.; Koike, K. Hepatitis B virus integration in hepatocellular carcinoma DNA: duplication of cellular flanking sequences at the integration site. Proc. Natl. Acad. Sci. U S A 1985, 82, 4458–4462. [Google Scholar] [CrossRef] [PubMed]
- Tokino, T.; Fukushige, S.; Nakamura, T.; Nagaya, T.; Murotsu, T.; Shiga, K.; Aoki, N.; Matsubara, K. Chromosomal translocation and inverted duplication associated with integrated hepatitis B virus in hepatocellular carcinomas. J. Virol. 1987, 61, 3848–3854. [Google Scholar] [PubMed]
- Hino, O.; Shows, T.B.; Rogler, C.E. Hepatitis B virus integration site in hepatocellular carcinoma at chromosome 17;18 translocation. Proc. Natl. Acad. Sci. U S A 1986, 83, 8338–8342. [Google Scholar] [CrossRef] [PubMed]
- Hino, O.; Tabata, S.; Hotta, Y. Evidence for increased in vitro recombination with insertion of human hepatitis B virus DNA. Proc. Natl. Acad. Sci. U S A 1991, 88, 9248–9252. [Google Scholar] [CrossRef] [PubMed]
- Tsuei, D.J.; Chang, M.H.; Chen, P.J.; Hsu, T.Y.; Ni, Y.H. Characterization of integration patterns and flanking cellular sequences of hepatitis B virus in childhood hepatocellular carcinomas. J. Med. Virol. 2002, 68, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Buendia, M.A. Genetics of Hepatocellular Carcinoma. Semin. Cancer Biol. 2000, 10, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Marchio, A.; Pineau, P.; Meddeb, M.; Terris, B.; Tiollais, P.; Bernheim, A.; Dejean, A. Distinct chromosomal abnormality pattern in primary liver cancer of non-B, non-C patients. Oncogene 2000, 19, 3733–3738. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Legoix, P.; Bluteau, O.; Belghiti, J.; Franco, D.; Binot, F.; Monges, G.; Thomas, G.; Bioulac-Sage, P.; Zucman-Rossi, J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001, 120, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Boyault, S.; Rickman, D.S.; de Reynies, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Herault, A.; Saric, J.; Belghiti, J.; Franco, D.; Bioulac-Sage, P.; Laurent-Puig, P.; Zucman-Rossi, J. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Heo, J.; Libbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; Nevens, F.; Roskams, T.; Thorgeirsson, S.S. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Kaneko, S.; Kawai, H.; Shirota, Y.; Kobayashi, K. Differential gene expression between chronic hepatitis B and C hepatic lesion. Gastroenterology 2001, 120, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Yamashita, T.; Ueda, T.; Takatori, H.; Nishino, R.; Kaneko, S. Different signaling pathways in the livers of patients with chronic hepatitis B or chronic hepatitis C. Hepatology 2006, 44, 1122–1138. [Google Scholar] [CrossRef] [PubMed]
- Andrisani, O.M.; Barnabas, S. The transcriptional function of the hepatitis B virus X protein and its role in hepatocarcinogenesis (Review). Int. J. Oncol. 1999, 15, 373–379. [Google Scholar] [PubMed]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular functions and biological roles of hepatitis B virus x protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Seifer, M.; Hohne, M.; Schaefer, S.; Gerlich, W.H. In vitro tumorigenicity of hepatitis B virus DNA and HBx protein . J. Hepatol. 1991, 13, S61–65. [Google Scholar] [CrossRef] [PubMed]
- Gottlob, K.; Pagano, S.; Levrero, M.; Graessmann, A. Hepatitis B virus X protein transcription activation domains are neither required nor sufficient for cell transformation. Cancer Res. 1998, 58, 3566–3570. [Google Scholar] [PubMed]
- Kim, Y.C.; Song, K.S.; Yoon, G.; Nam, M.J.; Ryu, W.S. Activated ras oncogene collaborates with HBx gene of hepatitis B virus to transform cells by suppressing HBx-mediated apoptosis. Oncogene 2001, 20, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Shilagardi, K.; Nakamoto, Y.; Honda, M.; Kaneko, S.; Murakami, S. Hepatitis B virus X protein overcomes oncogenic RAS-induced senescence in human immortalized cells. Cancer Sci. 2007, 98, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Oguey, D.; Dumenco, L.L.; Pierce, R.H.; Fausto, N. Analysis of the tumorigenicity of the X gene of hepatitis B virus in a nontransformed hepatocyte cell line and the effects of cotransfection with a murine p53 mutant equivalent to human codon 249. Hepatology 1996, 24, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Schuster, R.; Gerlich, W.H.; Schaefer, S. Induction of apoptosis by the transactivating domains of the hepatitis B virus X gene leads to suppression of oncogenic transformation of primary rat embryo fibroblasts. Oncogene 2000, 19, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Schrôder, C.H.; Hofman, W.J.; Otto, G.; Pichlmayr, R.; Bannasch, P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinoma. Hepatology 1998, 27, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Reifenberg, K.; Pudollek, H.P.; Schmitteckert, E.; Spindler, G.; Köck, J.; Schlicht, H.J. Long-term expression of hepatitis B virus core-e- and X-proteins does not cause pathologic changes in transgenic mice. J. Hepatol. 1997, 26, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Lee, T.H.; Medina, D.; Finegold, M.J.; Butel, J.S. Increased sensitivity to the hepatocarcinogen diethylnitrosamine in transgenic mice carrying the hepatitis B virus x gene. Mol. Carcinog. 1996, 15, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Terradillos, O.; Billet, O.; Renard, C.A.; Lévy, R.; Molina, T.; Briand, P.; Buendia, M.A. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene 1997, 14, 395–404. [Google Scholar] [PubMed]
- Kalra, N.; Kumar, V. The X protein of hepatitis B virus binds to the F box protein Skp2 and inhibits the ubiquitination and proteasomal degradation of c-Myc . FEBS Lett. 2006, 580 , 431–436. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, F.; Lv, Y.; Li, C.; Xu, X.; Deng, C.; Wang, D.; Sun, Y.; Hu, G.; Lang, Z.; Huang, C.; Yang, X. HBsAg and HBx knocked into the p21 locus causes hepatocellular carcinoma in mice. Hepatology 2004, 39, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Longato, L.; de la Monte, S.; Kuzushita, N.; Horimoto, M.; Rogers, A.B.; Slagle, B.L.; Wands, J.R. Overexpression of insulin receptor substrate-1 and hepatitis Bx genes causes premalignant alterations in the liver. Hepatology 2009, 49, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Madden, C.R.; Finegold, M.J.; Slagle, B.L. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J. Virol. 2001, 75, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Terradillos, O.; Pollicino, T.; Lecoeur, H.; Tripodi, M.; Gougeon, M.L.; Tiollais, P.; Buendia, M.A. p53-independent apoptotic effects of the hepatitis B virus HBx protein in vivo and in vitro. Oncogene 1998, 17, 2115–2123. [Google Scholar] [PubMed]
- Bouchard, M.; Giannakopoulos, S.; Wang, E.H.; Tanese, N.; Schneider, R.J. Hepatitis B virus HBx protein activation of cyclin A-cyclin-dependent kinase 2 complexes and G1 transit via a Src kinase pathway. J. Virol. 2001, 75, 4247–4257. [Google Scholar] [CrossRef] [PubMed]
- Chirillo, P.; Pagano, S.; Natoli, G.; Puri, P.L.; Burgio, V.L.; Balsano, C.; Levrero, M. The hepatitis B virus X gene induces p53-mediated programmed cell death. Proc. Natl. Acad. Sci. U S A 1997, 94, 8162–8167. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Leach, K.; McKinstry, R.; Gilfor, D.; Yacoub, A.; Park, J.S.; Grant, S.; Hylemon, P.B.; Fisher, P.B.; Dent, P. Hepatitis B virus X protein increases expression of p21(Cip-1/WAF1/MDA6) and p27(Kip-1) in primary mouse hepatocytes, leading to reduced cell cycle progression. Hepatology 2001, 34, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Park, U.S.; Park, S.K.; Lee, Y.I.; Park, J.G.; Lee, Y.I. Hepatitis B virus-X protein upregulates the expression of p21waf1/cip1 and prolongs G1-->S transition via a p53-independent pathway in human hepatoma cells. O. Oncogene 2000, 19, 3384–3394. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.K.; Li, C.C.; Chen, H.J.; Chang, J.L.; Jeng, K.S.; Chou, C.K.; Hsu, M.T.; Tsai, T.F. Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis B virus X protein transgenic mice. Biochem. Biophys. Res. Commun. 2006, 340, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Tralhao, J.G.; Roudier, J.; Morosan, S.; Giannini, C.; Tu, H.; Goulenok, C.; Carnot, F.; Zavala, F.; Joulin, V.; Kremsdorf, D.; Brechot, C. Paracrine in vivo inhibitory effects of hepatitis B virus X protein (HBx) on liver cell proliferation: an alternative mechanism of HBx-related pathogenesis. Proc. Natl. Acad. Sci. U S A 2002, 99, 6991–6996. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, A.J.; Keasler, V.V.; Slagle, B.L. Premature cell cycle entry induced by hepatitis B virus regulatory HBx protein during compensatory liver regeneration. Cancer Res. 2008, 68, 10341–10348. [Google Scholar] [CrossRef] [PubMed]
- Twu, J.S.; Robinson, W.S. Hepatitis B virus X gene can transactivate heterologous viral sequences. Proc. Natl. Acad. Sci. U S A 1989, 86, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Balsano, C.; Avantaggiati, M.L.; Natoli, G.; De Marzio, E.; Will, H.; Perricaudet, M.; Levrero, M. Full-length and truncated versions of the hepatitis B virus (HBV) X protein (pX) transactivate the cmyc protooncogene at the transcriptional level. Biochem. Biophys. Res. Commun. 1991, 176, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Benn, J.; Schneider, R.J. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc. Natl. Acad. Sci. U S A 1994, 91, 10350–10354. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Wang, L.H.; Schneider, R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef] [PubMed]
- Cougot, D.; Wu, Y.; Cairo, S.; Caramel, J.; Renard, C.A.; Levy, L.; Buendia, M.A.; Neuveut, C. The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J. Biol. Chem. 2007, 282, 4277–4287. [Google Scholar] [CrossRef] [PubMed]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lluesma, S.; Schaeffer, C.; Robert, E.I.; van Breugel, P.C.; Leupin, O.; Hantz, O.; Strubin, M. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 2008, 48, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Elledge, S.J.; Butel, J.S. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J. Virol. 1995, 69, 1107–1114. [Google Scholar] [PubMed]
- Sitterlin, D.; Bergametti, F.; Tiollais, P.; Tennant, B.C.; Transy, C. Correct binding of viral X protein to UVDDB-p127 cellular protein is critical for efficient infection by hepatitis B viruses. Oncogene 2000, 19, 4427–4431. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; McCall, C.M.; Ohta, T.; Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol 2004, 6, 1003–1009. [Google Scholar] [CrossRef]
- O'Connell, B.C.; Harper, J.W. Ubiquitin proteasome system (UPS): what can chromatin do for you? Curr. Opin. Cell Biol. 2007, 19, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Golubkov, V.S.; Strongin, A.Y.; Jiang, W.; Reed, J.C. Interaction of hepatitis B viral oncoprotein with cellular target HBXIP dysregulates centrosome dynamics and mitotic spindle formation. J. Biol. Chem. 2008, 283, 2793–2803. [Google Scholar] [CrossRef] [PubMed]
- Forgues, M.; Difilippantonio, M.J.; Linke, S.P.; Ried, T.; Nagashima, K.; Feden, J.; Valerie, K.; Fukasawa, K.; Wang, X.W. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol. Cell Biol. 2003, 23, 5282–5292. [Google Scholar] [CrossRef] [PubMed]
- Rakotomalala, L.; Studach, L.; Wang, W.H.; Gregori, G.; Hullinger, R.L.; Andrisani, O. Hepatitis B virus X protein increases the Cdt1-to-geminin ratio inducing DNA re-replication and polyploidy. J. Biol. Chem. 2008, 283, 28729–28740. [Google Scholar] [CrossRef] [PubMed]
- Studach, L.L.; Rakotomalala, L.; Wang, W.H.; Hullinger, R.L.; Cairo, S.; Buendia, M.A.; Andrisani, O.M. Polo-like kinase 1 inhibition suppresses hepatitis B virus X protein-induced transformation in an in vitro model of liver cancer progression . Hepatology 2009, . [Google Scholar]
- Kim, S.; Park, S.Y.; Yong, H.; Famulski, J.K.; Chae, S.; Lee, J.H.; Kang, C.M.; Saya, H.; Chan, G.K.; Cho, H. HBV X protein targets hBubR1, which induces dysregulation of the mitotic checkpoint. Oncogene 2008, 27, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Benhenda, S.; Cougot, D.; Neuveut, C.; Buendia, M.A. Liver Cell Transformation in Chronic HBV Infection. Viruses 2009, 1, 630-646. https://doi.org/10.3390/v1030630
Benhenda S, Cougot D, Neuveut C, Buendia MA. Liver Cell Transformation in Chronic HBV Infection. Viruses. 2009; 1(3):630-646. https://doi.org/10.3390/v1030630
Chicago/Turabian StyleBenhenda, Shirine, Delphine Cougot, Christine Neuveut, and Marie Annick Buendia. 2009. "Liver Cell Transformation in Chronic HBV Infection" Viruses 1, no. 3: 630-646. https://doi.org/10.3390/v1030630
APA StyleBenhenda, S., Cougot, D., Neuveut, C., & Buendia, M. A. (2009). Liver Cell Transformation in Chronic HBV Infection. Viruses, 1(3), 630-646. https://doi.org/10.3390/v1030630