The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
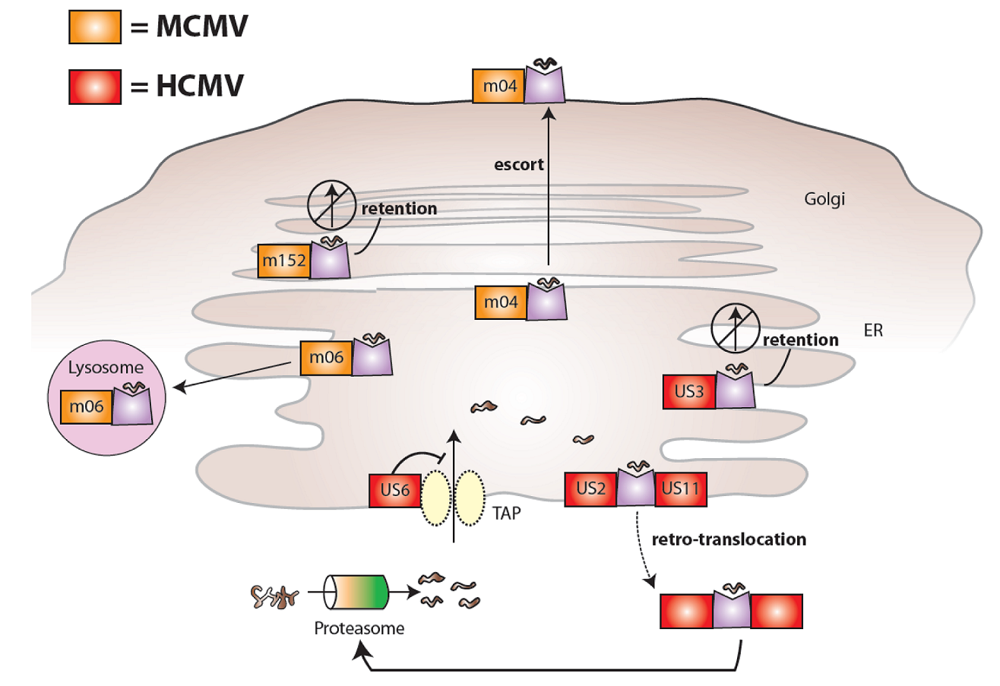
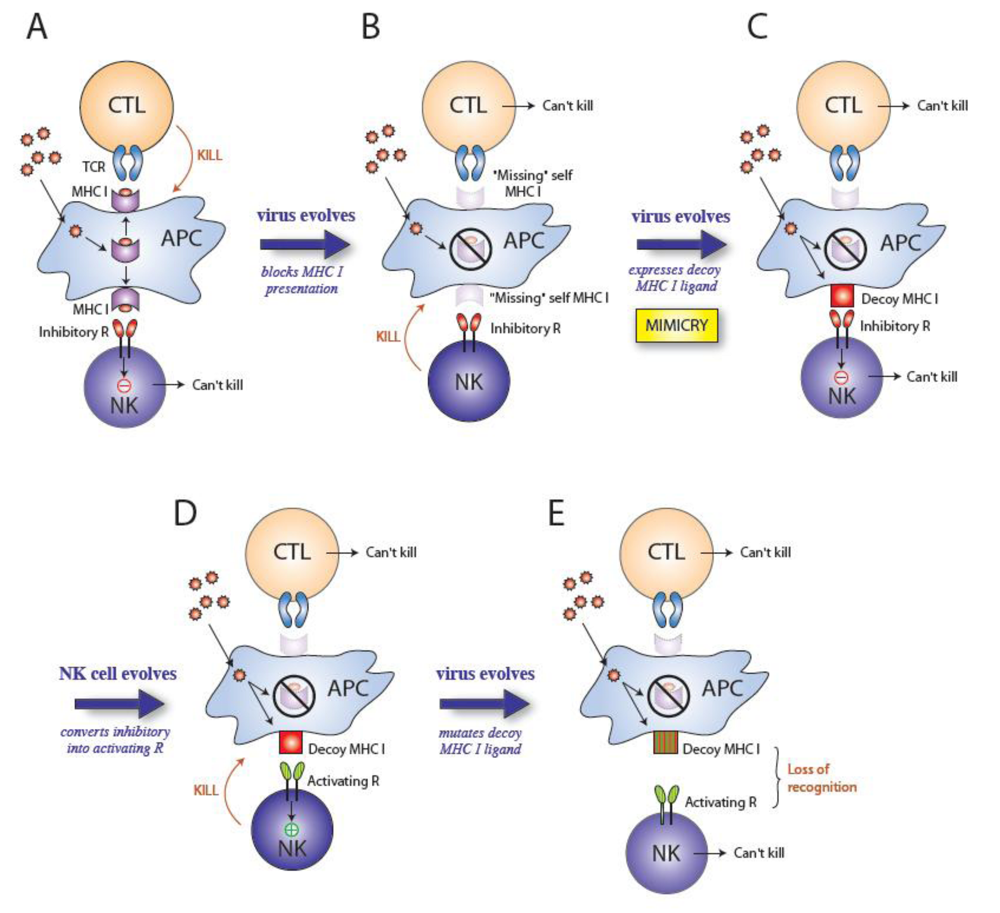
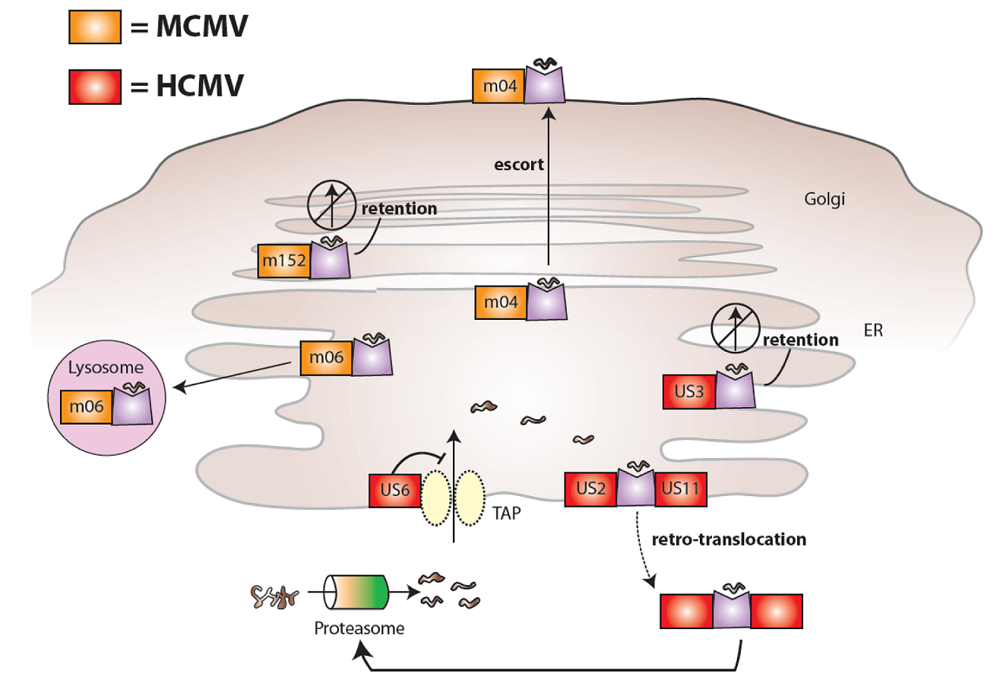
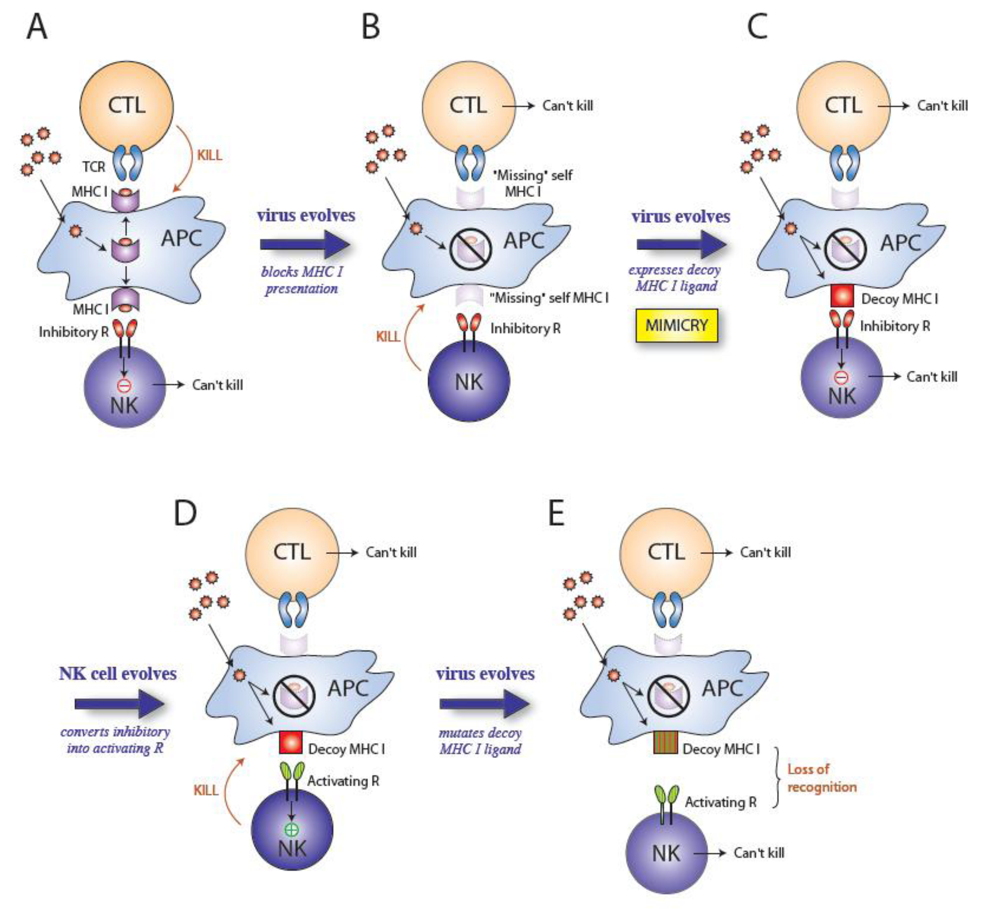
2. Cytomegaloviruses evade the immune system
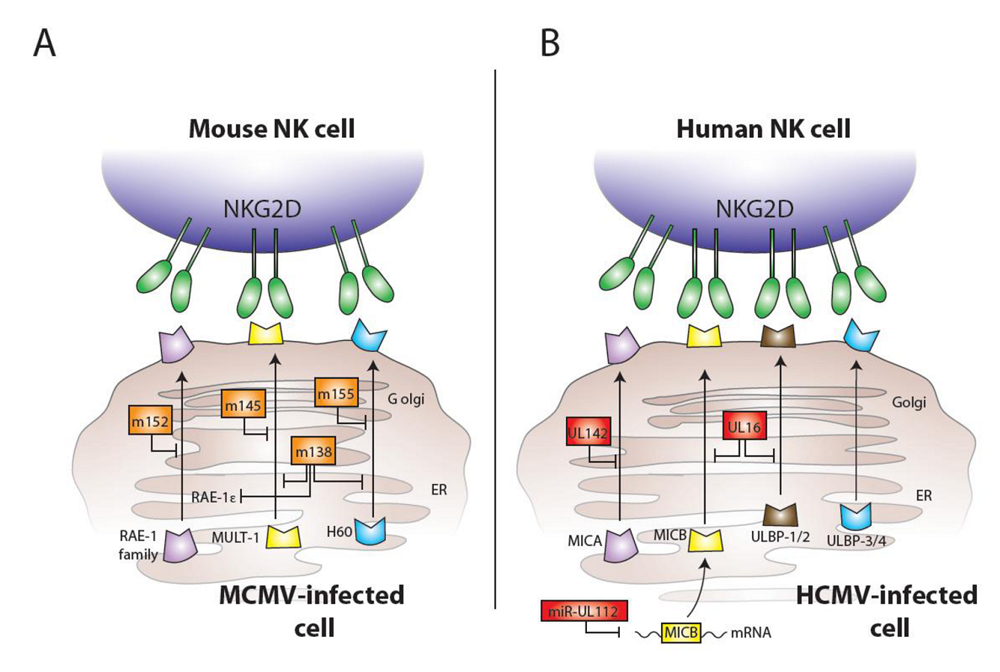
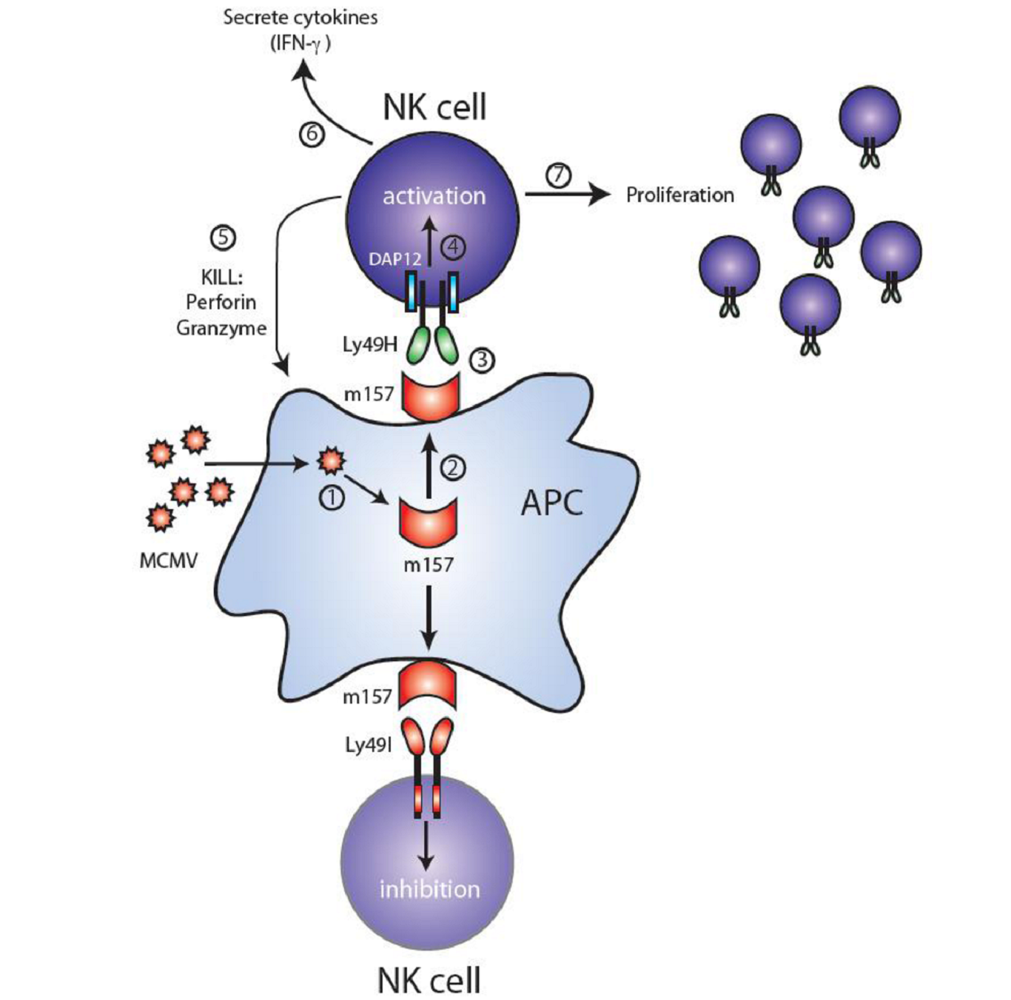
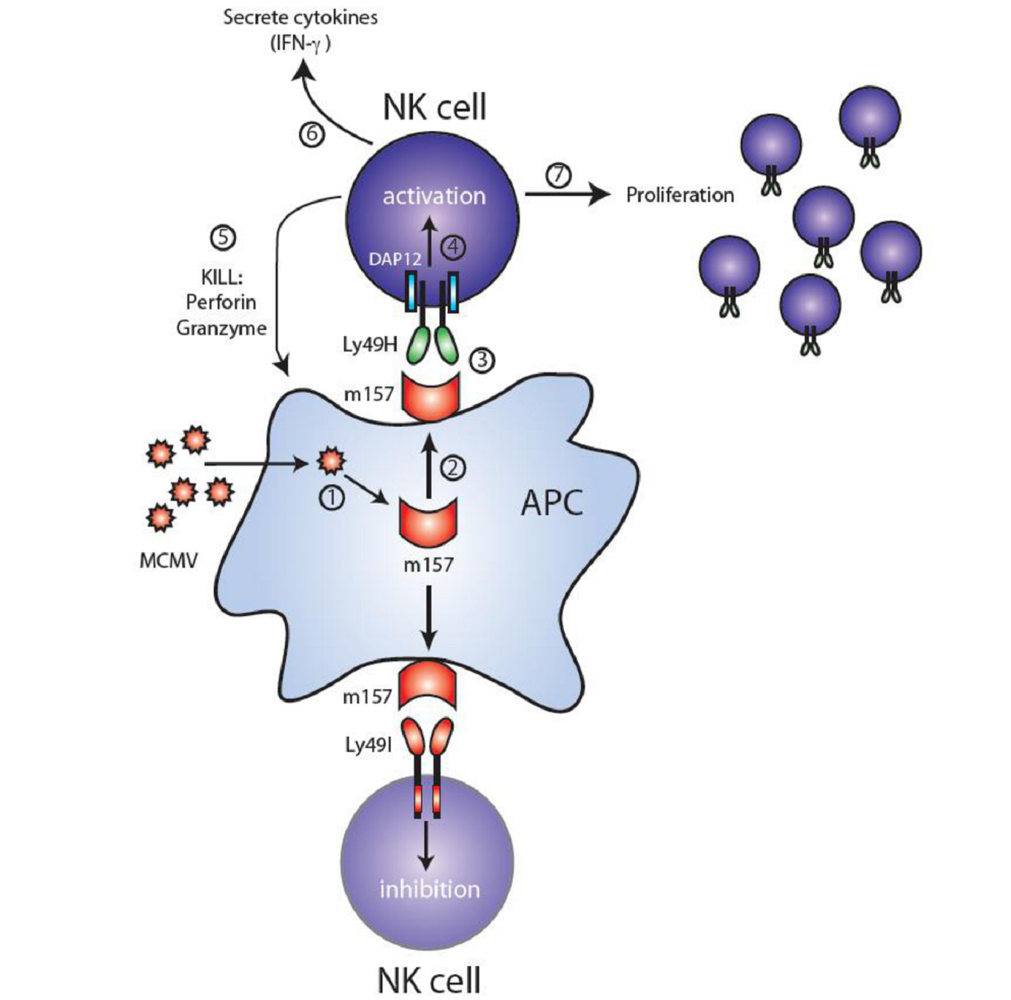
3. Ly49H and MCMV
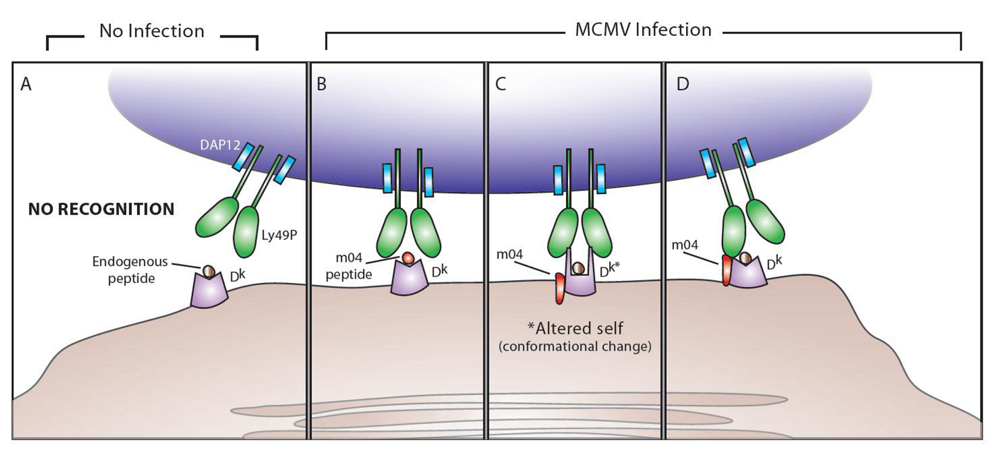
4. Ly49P and MCMV
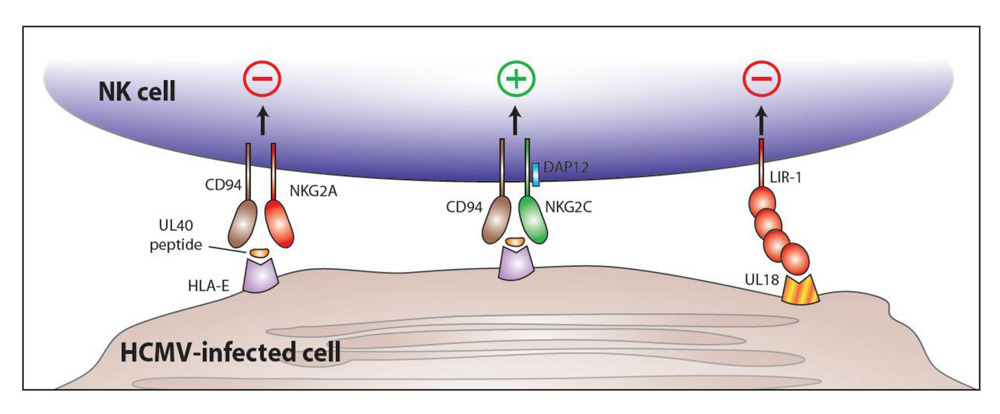
5. CD94-NKG2C and HCMV

6. Concluding Remarks
Acknowledgments
References and Notes
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 2003, 3, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.C.; Plebanski, M.; Gupta, S.; Morris, J.; Cox, M.; Aidoo, M.; Kwiatkowski, D.; Greenwood, B.M.; Whittle, H.C.; Hill, A.V. Association of malaria parasite population structure, HLA, and immunological antagonism. Science 1998, 279, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.V.; Allsopp, C.E.; Kwiatkowski, D.; Anstey, N.M.; Twumasi, P.; Rowe, P.A.; Bennett, S.; Brewster, D.; McMichael, A.J.; Greenwood, B.M. Common west African HLA antigens are associated with protection from severe malaria. Nature 1991, 352, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, Jr., E.S. Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol. 2002, 10, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Lanier, L.L. Evolutionary struggles between NK cells and viruses. Nat. Rev. Immunol. 2008, 8, 259–268. [Google Scholar] [CrossRef]
- Kavanagh, D.G.; Gold, M.C.; Wagner, M.; Koszinowski, U.H.; Hill, A.B. The multiple immune-evasion genes of murine cytomegalovirus are not redundant: m4 and m152 inhibit antigen presentation in a complementary and cooperative fashion. J. Exp. Med. 2001, 194, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, D.G.; Koszinowski, U.H.; Hill, A.B. The murine cytomegalovirus immune evasion protein m4/gp34 forms biochemically distinct complexes with class I MHC at the cell surface and in a pre-Golgi compartment. J. Immunol. 2001, 167, 3894–3902. [Google Scholar] [PubMed]
- Kleijnen, M.F.; Huppa, J.B.; Lucin, P.; Mukherjee, S.; Farrell, H.; Campbell, A.E.; Koszinowski, U.H.; Hill, A.B.; Ploegh, H.L. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 1997, 16, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.A.; Park, S.H.; Lee, P.; Bendelac, A.; Shenk, T.E. Murine cytomegalovirus m02 gene family protects against natural killer cell-mediated immune surveillance. J. Virol. 2002, 76, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Muranyi, W.; Lucin, P.; Burgert, H.G.; Hengel, H.; Koszinowski, U.H. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 1999, 18, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, H.; Thale, R.; Lucin, P.; Muranyi, W.; Flohr, T.; Hengel, H.; Farrell, H.; Rawlinson, W.; Koszinowski, U.H. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 1997, 6, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Gruhler, A.; Galocha, B.; Jones, T.R.; Wiertz, E.J.; Ploegh, H.L.; Peterson, P.A.; Yang, Y.; Fruh, K. The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 1997, 6, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Barel, M.T.; Ressing, M.; Pizzato, N.; van Leeuwen, D.; Le Bouteiller, P.; Lenfant, F.; Wiertz, E.J. Human cytomegalovirus-encoded US2 differentially affects surface expression of MHC class I locus products and targets membrane-bound, but not soluble HLA-G1 for degradation. J. Immunol. 2003, 171, 6757–6765. [Google Scholar] [PubMed]
- Gruhler, A.; Peterson, P.A.; Fruh, K. Human cytomegalovirus immediate early glycoprotein US3 retains MHC class I molecules by transient association. Traffic 2000, 1, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Hengel, H.; Koopmann, J.O.; Flohr, T.; Muranyi, W.; Goulmy, E.; Hammerling, G.J.; Koszinowski, U.H.; Momburg, F. A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 1997, 6, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.R.; Wiertz, E.J.; Sun, L.; Fish, K.N.; Nelson, J.A.; Ploegh, H.L. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc. Natl. Acad. Sci. U S A 1996, 93, 11327–11333. [Google Scholar] [CrossRef] [PubMed]
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Wiertz, E.J.; Tortorella, D.; Bogyo, M.; Yu, J.; Mothes, W.; Jones, T.R.; Rapoport, T.A.; Ploegh, H.L. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 1996, 384, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Bahram, S.; Bresnahan, M.; Geraghty, D.E.; Spies, T. A second lineage of mammalian major histocompatibility complex class I genes. Proc. Natl. Acad. Sci. U S A 1994, 91, 6259–6263. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Chalupny, N.J.; Sutherland, C.L.; Lawrence, W.A.; Rein-Weston, A.; Cosman, D. ULBP4 is a novel ligand for human NKG2D. Biochem. Biophys. Res. Commun. 2003, 305, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Cosman, D.; Mullberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Carayannopoulos, L.N.; Naidenko, O.V.; Fremont, D.H.; Yokoyama, W.M. Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J. Immunol. 2002, 169, 4079–4083. [Google Scholar] [PubMed]
- Cerwenka, A.; Bakker, A.B.; McClanahan, T.; Wagner, J.; Wu, J.; Phillips, J.H.; Lanier, L.L. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 2000, 12, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, A.; Hsia, J.K.; Hsiung, M.Y.; Raulet, D.H. A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur. J. Immunol. 2003, 33, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Lodoen, M.; Ogasawara, K.; Hamerman, J.A.; Arase, H.; Houchins, J.P.; Mocarski, E.S.; Lanier, L.L. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J. Exp. Med. 2003, 197, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Krmpotic, A.; Ruzsics, Z.; Bubic, I.; Lenac, T.; Halenius, A.; Loewendorf, A.; Messerle, M.; Hengel, H.; Jonjic, S.; Koszinowski, U.H. Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J. Virol. 2005, 79, 2920–2930. [Google Scholar] [CrossRef] [PubMed]
- Krmpotic, A.; Hasan, M.; Loewendorf, A.; Saulig, T.; Halenius, A.; Lenac, T.; Polic, B.; Bubic, I.; Kriegeskorte, A.; Pernjak-Pugel, E.; Messerle, M.; Hengel, H.; Busch, D.H.; Koszinowski, U.H.; Jonjic, S. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 2005, 201, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Lodoen, M.B.; Abenes, G.; Umamoto, S.; Houchins, J.P.; Liu, F.; Lanier, L.L. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J. Exp. Med. 2004, 200, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Arapovic, J.; Lenac Rovis, T.; Reddy, A.B.; Krmpotic, A.; Jonjic, S. Promiscuity of MCMV immunoevasin of NKG2D: m138/fcr-1 down-modulates RAE-1varepsilon in addition to MULT-1 and H60. Mol. Immunol. 2009. [Google Scholar]
- Lenac, T.; Budt, M.; Arapovic, J.; Hasan, M.; Zimmermann, A.; Simic, H.; Krmpotic, A.; Messerle, M.; Ruzsics, Z.; Koszinowski, U.H.; Hengel, H.; Jonjic, S. The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J. Exp. Med. 2006, 203, 1843–1850. [Google Scholar] [CrossRef] [PubMed]
- Mintern, J.D.; Klemm, E.J.; Wagner, M.; Paquet, M.E.; Napier, M.D.; Kim, Y.M.; Koszinowski, U.H.; Ploegh, H.L. Viral interference with B7-1 costimulation: a new role for murine cytomegalovirus fc receptor-1. J. Immunol. 2006, 177, 8422–8431. [Google Scholar] [PubMed]
- Bacon, L.; Eagle, R.A.; Meyer, M.; Easom, N.; Young, N.T.; Trowsdale, J. Two human ULBP/RAET1 molecules with transmembrane regions are ligands for NKG2D. J. Immunol. 2004, 173, 1078–1084. [Google Scholar] [PubMed]
- Chalupny, N.J.; Rein-Weston, A.; Dosch, S.; Cosman, D. Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem. Biophys. Res. Commun. 2006, 346, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Ashiru, O.; Reeves, M.B.; Okecha, G.; Trowsdale, J.; Tomasec, P.; Wilkinson, G.W.; Sinclair, J.; Sissons, J.G. Human cytomegalovirus encodes an MHC class I-like molecule (UL142) that functions to inhibit NK cell lysis. J. Immunol. 2005, 175, 7457–7465. [Google Scholar] [PubMed]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; Goldman-Wohl, D.; Greenfield, C.; Yagel, S.; Hengel, H.; Altuvia, Y.; Margalit, H.; Mandelboim, O. Host immune system gene targeting by a viral miRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Desrosiers, M.P.; Kielczewska, A.; Loredo-Osti, J.C.; Adam, S.G.; Makrigiannis, A.P.; Lemieux, S.; Pham, T.; Lodoen, M.B.; Morgan, K.; Lanier, L.L.; Vidal, S.M. Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat. Genet. 2005, 37, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Girard, S.; Macina, D.; Busa, M.; Zafer, A.; Belouchi, A.; Gros, P.; Vidal, S.M. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat. Genet. 2001, 28, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, A.A.; Fitzgerald, N.A.; Simmons, A.; La Vista, A.B.; Shellam, G.R. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J. Exp. Med. 1990, 171, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, A.A.; Lyons, P.A.; Fitzgerald, N.A.; Forbes, C.A.; Yokoyama, W.M.; Shellam, G.R. Genetic mapping of Cmv1 in the region of mouse chromosome 6 encoding the NK gene complex-associated loci Ly49 and musNKR-P1. Genomics 1995, 27, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.G.; Fulmek, S.; Matsumoto, K.; Cho, R.; Lyons, P.A.; Levy, E.R.; Scalzo, A.A.; Yokoyama, W.M. A 2-Mb YAC contig and physical map of the natural killer gene complex on mouse chromosome 6. Genomics 1997, 42, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Depatie, C.; Muise, E.; Lepage, P.; Gros, P.; Vidal, S.M. High-resolution linkage map in the proximity of the host resistance locus Cmv1. Genomics 1997, 39, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Forbes, C.A.; Brown, M.G.; Cho, R.; Shellam, G.R.; Yokoyama, W.M.; Scalzo, A.A. The Cmv1 host resistance locus is closely linked to the Ly49 multigene family within the natural killer cell gene complex on mouse chromosome 6. Genomics 1997, 41, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, W.M.; Plougastel, B.F. Immune functions encoded by the natural killer gene complex. Nat. Rev. Immunol. 2003, 3, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, A.A.; Fitzgerald, N.A.; Wallace, C.R.; Gibbons, A.E.; Smart, Y.C.; Burton, R.C.; Shellam, G.R. The effect of the Cmv-1 resistance gene, which is linked to the natural killer cell gene complex, is mediated by natural killer cells. J. Immunol. 1992, 149, 581–589. [Google Scholar] [PubMed]
- Vidal, S.M.; Lanier, L.L. NK cell recognition of mouse cytomegalovirus-infected cells. Curr. Top. Microbiol. Immunol. 2006, 298, 183–206. [Google Scholar] [PubMed]
- Brown, M.G.; Dokun, A.O.; Heusel, J.W.; Smith, H.R.; Beckman, D.L.; Blattenberger, E.A.; Dubbelde, C.E.; Stone, L.R.; Scalzo, A.A.; Yokoyama, W.M. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 2001, 292, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Daniels, K.A.; Devora, G.; Lai, W.C.; O'Donnell, C.L.; Bennett, M.; Welsh, R.M. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J. Exp. Med. 2001, 194, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.M.; Wu, J.; Bakker, A.B.; Phillips, J.H.; Lanier, L.L. Ly-49D and Ly-49H associate with mouse DAP12 and form activating receptors. J. Immunol. 1998, 161, 7–10. [Google Scholar] [PubMed]
- Coudert, J.D.; Scarpellino, L.; Gros, F.; Vivier, E.; Held, W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood 2008, 111, 3571–3578. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Sun, J.C.; Hesslein, D.G.; Arase, H.; Phillips, J.H.; Takai, T.; Lanier, L.L. Ly49H signaling through DAP10 is essential for optimal natural killer cell responses to mouse cytomegalovirus infection. J. Exp. Med. 2009, 206, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Tassi, I.; Le Friec, G.; Gilfillan, S.; Takai, T.; Yokoyama, W.M.; Colonna, M. DAP10 associates with Ly49 receptors but contributes minimally to their expression and function in vivo. Eur. J. Immunol. 2009, 39, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Sjolin, H.; Tomasello, E.; Mousavi-Jazi, M.; Bartolazzi, A.; Karre, K.; Vivier, E.; Cerboni, C. Pivotal role of KARAP/DAP12 adaptor molecule in the natural killer cell-mediated resistance to murine cytomegalovirus infection. J. Exp. Med. 2002, 195, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.P.; French, A.R.; Plougastel, B.F.; Pingel, J.T.; Orihuela, M.M.; Buller, M.L.; Yokoyama, W.M. Ly49h is necessary for genetic resistance to murine cytomegalovirus. Immunogenetics 2008, 60, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Fodil-Cornu, N.; Lee, S.H.; Belanger, S.; Makrigiannis, A.P.; Biron, C.A.; Buller, R.M.; Vidal, S.M. Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J. Immunol. 2008, 181, 6394–6405. [Google Scholar] [PubMed]
- Lee, S.H.; Zafer, A.; de Repentigny, Y.; Kothary, R.; Tremblay, M.L.; Gros, P.; Duplay, P.; Webb, J.R.; Vidal, S.M. Transgenic expression of the activating natural killer receptor Ly49H confers resistance to cytomegalovirus in genetically susceptible mice. J. Exp. Med. 2003, 197, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Arase, H.; Mocarski, E.S.; Campbell, A.E.; Hill, A.B.; Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002, 296, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.R.; Heusel, J.W.; Mehta, I.K.; Kim, S.; Dorner, B.G.; Naidenko, O.V.; Iizuka, K.; Furukawa, H.; Beckman, D.L.; Pingel, J.T.; Scalzo, A.A.; Fremont, D.H.; Yokoyama, W.M. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. U S A 2002, 99, 8826–8831. [Google Scholar] [PubMed]
- Adams, E.J.; Juo, Z.S.; Venook, R.T.; Boulanger, M.J.; Arase, H.; Lanier, L.L.; Garcia, K.C. Structural elucidation of the m157 mouse cytomegalovirus ligand for Ly49 natural killer cell receptors. Proc. Natl. Acad. Sci. U S A 2007, 104, 10128–10133. [Google Scholar] [CrossRef] [PubMed]
- Dokun, A.O.; Kim, S.; Smith, H.R.; Kang, H.S.; Chu, D.T.; Yokoyama, W.M. Specific and nonspecific NK cell activation during virus infection. Nat. Immunol. 2001, 2, 951–956. [Google Scholar] [CrossRef]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Bubic, I.; Wagner, M.; Krmpotic, A.; Saulig, T.; Kim, S.; Yokoyama, W.M.; Jonjic, S.; Koszinowski, U.H. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J. Virol. 2004, 78, 7536–7544. [Google Scholar] [CrossRef] [PubMed]
- Lodoen, M.B.; Lanier, L.L. Viral modulation of NK cell immunity. Nat. Rev. Microbiol. 2005, 3, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Karre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, H.G.; Karre, K. In search of the 'missing self': MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Barrell, B.G. Human cytomegalovirus encodes a glycoprotein homologous to MHC class-I antigens. Nature 1988, 331, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.L.; Johnson, J.L.; Feldman, R.M.; Neveu, J.M.; Lane, W.S.; Bjorkman, P.J. The MHC class I homolog encoded by human cytomegalovirus binds endogenous peptides. Immunity 1995, 3, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Abi-Rached, L.; Parham, P. Natural selection drives recurrent formation of activating killer cell immunoglobulin-like receptor and Ly49 from inhibitory homologues. J. Exp. Med. 2005, 201, 1319–1332. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.G.; Scalzo, A.A. NK gene complex dynamics and selection for NK cell receptors. Semin. Immunol. 2008, 20, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.T.; Gagnier, L.; Mager, D.L. Sequence analysis of the ly49 cluster in C57BL/6 mice: a rapidly evolving multigene family in the immune system. Genomics 2002, 80, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Voigt, V.; Forbes, C.A.; Tonkin, J.N.; Degli-Esposti, M.A.; Smith, H.R.; Yokoyama, W.M.; Scalzo, A.A. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl. Acad. Sci. U S A 2003, 100, 13483–13488. [Google Scholar] [CrossRef] [PubMed]
- French, A.R.; Pingel, J.T.; Wagner, M.; Bubic, I.; Yang, L.; Kim, S.; Koszinowski, U.; Jonjic, S.; Yokoyama, W.M. Escape of mutant double-stranded DNA virus from innate immune control. Immunity 2004, 20, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Kielczewska, A.; Pyzik, M.; Sun, T.; Krmpotic, A.; Lodoen, M.B.; Munks, M.W.; Babic, M.; Hill, A.B.; Koszinowski, U.H.; Jonjic, S.; Lanier, L.L.; Vidal, S.M. Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J. Exp. Med. 2009, 206, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kavanagh, D.G.; Hill, A.B. Cellular and molecular requirements for association of the murine cytomegalovirus protein m4/gp34 with major histocompatibility complex class I molecules. J. Virol. 2006, 80, 6048–6055. [Google Scholar] [CrossRef] [PubMed]
- LoPiccolo, D.M.; Gold, M.C.; Kavanagh, D.G.; Wagner, M.; Koszinowski, U.H.; Hill, A.B. Effective inhibition of K(b)- and D(b)-restricted antigen presentation in primary macrophages by murine cytomegalovirus. J. Virol. 2003, 77, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Gutermann, A.; Podlech, J.; Reddehase, M.J.; Koszinowski, U.H. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 2002, 196, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Gillert-Marien, D.; Thomas, D.; Podlech, J.; Deegen, P.; Herter, S.; Oehrlein-Karpi, S.A.; Strand, D.; Wagner, M.; Reddehase, M.J. Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 2006, 80, 7613–7624. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.J.; Forbes, C.A.; Moro, D.; Scalzo, A.A. Extensive sequence variation exists among isolates of murine cytomegalovirus within members of the m02 family of genes. J. Gen. Virol. 2007, 88, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Dimasi, N.; Biassoni, R. Structural and functional aspects of the Ly49 natural killer cell receptors. Immunol. Cell. Biol. 2005, 83, 1–8. [Google Scholar] [CrossRef]
- Hanke, T.; Takizawa, H.; McMahon, C.W.; Busch, D.H.; Pamer, E.G.; Miller, J.D.; Altman, J.D.; Liu, Y.; Cado, D.; Lemonnier, F.A.; Bjorkman, P.J.; Raulet, D.H. Direct assessment of MHC class I binding by seven Ly49 inhibitory NK cell receptors. Immunity 1999, 11, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Makrigiannis, A.P.; Pau, A.T.; Saleh, A.; Winkler-Pickett, R.; Ortaldo, J.R.; Anderson, S.K. Class I MHC-binding characteristics of the 129/J Ly49 repertoire. J. Immunol. 2001, 166, 5034–5043. [Google Scholar] [PubMed]
- Michaelsson, J.; Achour, A.; Salcedo, M.; Kase-Sjostrom, A.; Sundback, J.; Harris, R.A.; Karre, K. Visualization of inhibitory Ly49 receptor specificity with soluble major histocompatibility complex class I tetramers. Eur. J. Immunol. 2000, 30, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Bohm, V.; Simon, C.O.; Podlech, J.; Seckert, C.K.; Gendig, D.; Deegen, P.; Gillert-Marien, D.; Lemmermann, N.A.; Holtappels, R.; Reddehase, M.J. The immune evasion paradox: immunoevasins of murine cytomegalovirus enhance priming of CD8 T cells by preventing negative feedback regulation. J. Virol. 2008, 82, 11637–11650. [Google Scholar] [CrossRef] [PubMed]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [PubMed]
- Guma, M.; Angulo, A.; Vilches, C.; Gomez-Lozano, N.; Malats, N.; Lopez-Botet, M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004, 104, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Cabrera, C.; Erkizia, I.; Bofill, M.; Clotet, B.; Ruiz, L.; Lopez-Botet, M. Human cytomegalovirus infection is associated with increased proportions of NK cells that express the CD94/NKG2C receptor in aviremic HIV-1-positive patients. J. Infect. Dis. 2006, 194, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; Baars, P.A.; Dantin, C.; van den Burg, M.; van Lier, R.A.; Roosnek, E. Human NK cells can control CMV infection in the absence of T cells. Blood 2008, 112, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.G.; Posch, P.E.; Scorzelli, C.J.; Borrego, F.; Coligan, J.E. NKG2A complexed with CD94 defines a novel inhibitory natural killer cell receptor. J. Exp. Med. 1997, 185, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.; Rodriguez, A.; Carretero, M.; Lopez-Botet, M.; Phillips, J.H.; Lanier, L.L. Molecular characterization of human CD94: a type II membrane glycoprotein related to the C-type lectin superfamily. Eur. J. Immunol. 1995, 25, 2433–2437. [Google Scholar] [CrossRef] [PubMed]
- Houchins, J.P.; Yabe, T.; McSherry, C.; Bach, F.H. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 1991, 173, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Lazetic, S.; Chang, C.; Houchins, J.P.; Lanier, L.L.; Phillips, J.H. Human natural killer cell receptors involved in MHC class I recognition are disulfide-linked heterodimers of CD94 and NKG2 subunits. J. Immunol. 1996, 157, 4741–4745. [Google Scholar] [PubMed]
- Vance, R.E.; Jamieson, A.M.; Raulet, D.H. Recognition of the class Ib molecule Qa-1(b) by putative activating receptors CD94/NKG2C and CD94/NKG2E on mouse natural killer cells. J. Exp. Med. 1999, 190, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Carretero, M.; Cantoni, C.; Bellon, T.; Bottino, C.; Biassoni, R.; Rodriguez, A.; Perez-Villar, J.J.; Moretta, L.; Moretta, A.; Lopez-Botet, M. The CD94 and NKG2-A C-type lectins covalently assemble to form a natural killer cell inhibitory receptor for HLA class I molecules. Eur. J. Immunol. 1997, 27, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Houchins, J.P.; Lanier, L.L.; Niemi, E.C.; Phillips, J.H.; Ryan, J.C. Natural killer cell cytolytic activity is inhibited by NKG2-A and activated by NKG2-C. J. Immunol. 1997, 158, 3603–3609. [Google Scholar] [PubMed]
- Le Drean, E.; Vely, F.; Olcese, L.; Cambiaggi, A.; Guia, S.; Krystal, G.; Gervois, N.; Moretta, A.; Jotereau, F.; Vivier, E. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. Eur. J. Immunol. 1998, 28, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Tullio, V.; Zingoni, A.; Piccoli, M.; Frati, L.; Lopez-Botet, M.; Santoni, A. CD94/NKG2-A inhibitory complex blocks CD16-triggered Syk and extracellular regulated kinase activation, leading to cytotoxic function of human NK cells. J. Immunol. 1999, 162, 7181–7188. [Google Scholar] [PubMed]
- Lanier, L.L.; Corliss, B.; Wu, J.; Phillips, J.H. Association of DAP12 with activating CD94/NKG2C NK cell receptors. Immunity 1998, 8, 693–701. [Google Scholar] [CrossRef]
- Borrego, F.; Ulbrecht, M.; Weiss, E.H.; Coligan, J.E.; Brooks, A.G. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J. Exp. Med. 1998, 187, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Braud, V.M.; Allan, D.S.; O'Callaghan, C.A.; Soderstrom, K.; D'Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; Lanier, L.L.; McMichael, A.J. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Llano, M.; Carretero, M.; Ishitani, A.; Navarro, F.; Lopez-Botet, M.; Geraghty, D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci. U S A 1998, 95, 5199–5204. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, C.J.; DeCloux, A.; Woods, A.S.; Cotter, R.J.; Soloski, M.J.; Forman, J. Identification of a Tap-dependent leader peptide recognized by alloreactive T cells specific for a class Ib antigen. Cell 1994, 79, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Braud, V.; Jones, E.Y.; McMichael, A. The human major histocompatibility complex class Ib molecule HLA-E binds signal sequence-derived peptides with primary anchor residues at positions 2 and 9. Eur. J. Immunol. 1997, 27, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Goodlett, D.R.; Ishitani, A.; Marquardt, H.; Geraghty, D.E. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J. Immunol. 1998, 160, 4951–4960. [Google Scholar] [PubMed]
- Llano, M.; Lee, N.; Navarro, F.; Garcia, P.; Albar, J.P.; Geraghty, D.E.; Lopez-Botet, M. HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: preferential response to an HLA-G-derived nonamer. Eur. J. Immunol. 1998, 28, 2854–2863. [Google Scholar] [CrossRef] [PubMed]
- Tomasec, P.; Braud, V.M.; Rickards, C.; Powell, M.B.; McSharry, B.P.; Gadola, S.; Cerundolo, V.; Borysiewicz, L.K.; McMichael, A.J.; Wilkinson, G.W. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science 2000, 287, 1031. [Google Scholar] [CrossRef] [PubMed]
- Ulbrecht, M.; Martinozzi, S.; Grzeschik, M.; Hengel, H.; Ellwart, J.W.; Pla, M.; Weiss, E.H. Cutting edge: the human cytomegalovirus UL40 gene product contains a ligand for HLA-E and prevents NK cell-mediated lysis. J. Immunol. 2000, 164, 5019–5022. [Google Scholar] [PubMed]
- Wang, E.C.; McSharry, B.; Retiere, C.; Tomasec, P.; Williams, S.; Borysiewicz, L.K.; Braud, V.M.; Wilkinson, G.W. UL40-mediated NK evasion during productive infection with human cytomegalovirus. Proc. Natl. Acad. Sci. U S A 2002, 99, 7570–7575. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, B.K.; Pizarro, J.C.; Kerns, J.; Strong, R.K. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc. Natl. Acad. Sci. U S A 2008, 105, 6696–6701. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.; Smith, G.; Beck, S.; Minson, T. A complex between the MHC class I homologue encoded by human cytomegalovirus and beta 2 microglobulin. Nature 1990, 347, 770–772. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Navarro, F.; Bellon, T.; Llano, M.; Garcia, P.; Samaridis, J.; Angman, L.; Cella, M.; Lopez-Botet, M. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J. Exp. Med. 1997, 186, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Cosman, D.; Fanger, N.; Borges, L.; Kubin, M.; Chin, W.; Peterson, L.; Hsu, M.L. A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 1997, 7, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.L.; Heikeman, A.P.; Bjorkman, P.J. The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity 1999, 11, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Budt, M.; Saez, A.; Brckalo, T.; Hengel, H.; Angulo, A.; Lopez-Botet, M. Expansion of CD94/NKG2C+ NK cells in response to human cytomegalovirus-infected fibroblasts. Blood 2006, 107, 3624–3631. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Sun, J.C.; Lanier, L.L. The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors. Viruses 2009, 1, 362-382. https://doi.org/10.3390/v1030362
Sun JC, Lanier LL. The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors. Viruses. 2009; 1(3):362-382. https://doi.org/10.3390/v1030362
Chicago/Turabian StyleSun, Joseph C., and Lewis L. Lanier. 2009. "The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors" Viruses 1, no. 3: 362-382. https://doi.org/10.3390/v1030362
APA StyleSun, J. C., & Lanier, L. L. (2009). The Natural Selection of Herpesviruses and Virus-Specific NK Cell Receptors. Viruses, 1(3), 362-382. https://doi.org/10.3390/v1030362