Plasmacytoid Dendritic Cells and the Control of Herpesvirus Infections

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Impact on pDC biology | |||||
|---|---|---|---|---|---|
| Herpesviruses | natural host | IFN-I production | Infection | Maturation$ | |
| Herpes Simplex virus 1 | HSV-1 | human | Yes* [2] | ND | ND |
| Herpes Simplex virus 2 | HSV-2 | human | Yes*±[3] | No [4] | ND |
| Varicella-zooster virus | VZV | human | ND | ND | ND |
| Human cytomegalovirus | HCMV | human | Yes [5,6,7] | No (blood pDCs) [6] | Partial [8] |
| Yes (tonsil pDCs) [7] | |||||
| Human Herpesvirus 6 | HHV-6 | human | ND | Yes [9] | ND |
| Human Herpesvirus 7 | HHV-7 | human | ND | Yes [10] | ND |
| Epstein-Barr virus | EBV | human | Yes [11] | ND | Yes [11] |
| Human Herpesvirus 8 | HHV-8 | human | ND | ND | ND |
| Murine cytomegalovirus | MCMV | mouse | Yes*±[12] | No±[13] | Yes±[13] |
2. pDCs Contribute to IFN-I Production, Viral Control during Herpesvirus Infections
2.1. pDCs produce high levels of all IFN-I in vitro in response to herpesviruses
2.2. pDCs are the major producers of IFN-I in vivo in mice infected with MCMV or HSV-2
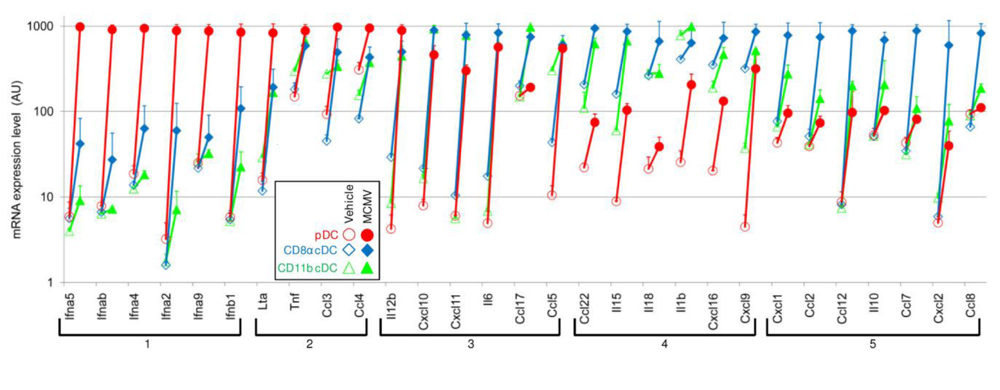
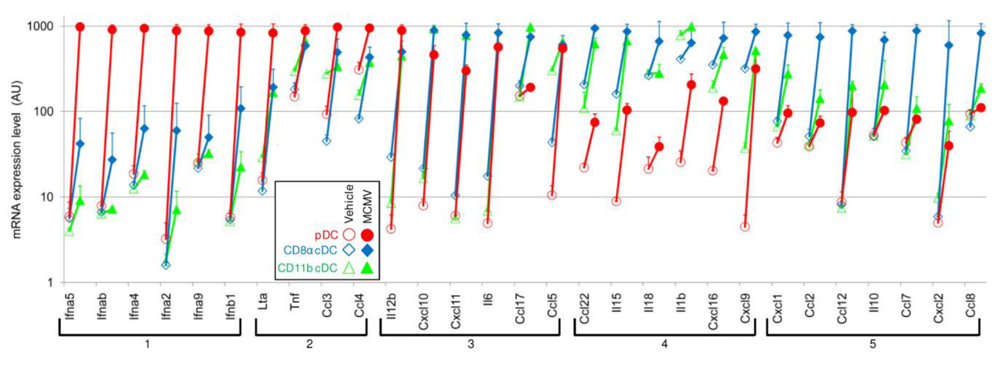
3. pDC Innate Antiviral Functions Are Not Restricted to IFN-I Production but Globally Contribute to the Orchestration of Inflammation, to the Activation of Other Innate Cells
3.1. pDCs produce multiple cytokines, chemokines in response to viruses
3.2. pDC-derived cytokineschemokines contribute to the activation of other innate immune effectors
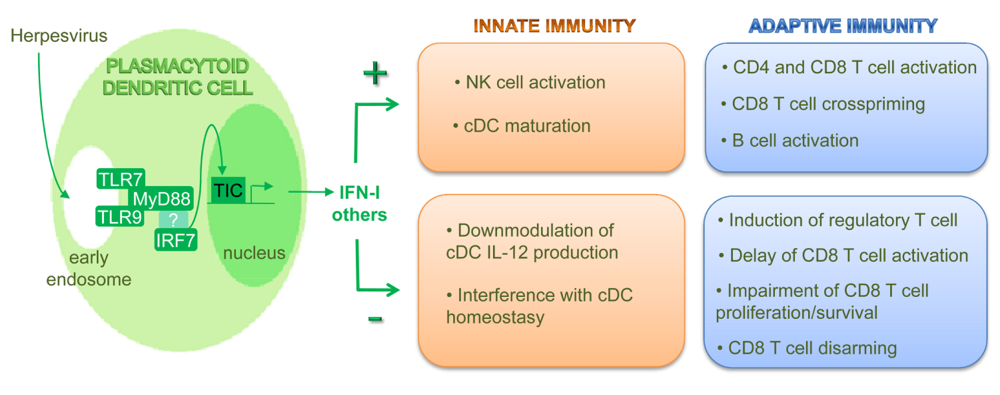
4. pDCs Are Equipped with a Unique Molecular Machinery for Herpesvirus Recognition, Downstream High Level IFN-I Production
4.2. pDC unique ability to rapidly produce high level IFN-I results from constitutive expression of IRF7 together with TLR7/9, MyD88 in endosomal multimolecular complexes
5. Multiple Mechanisms Dampen pDC Responses to Viral Stimuli
5.1. pDCs express several endocytic receptors which signal through a specific ITAM-dependent pathway, inhibit TLR-induced cytokine production
5.2. Other intrinsic inhibitory mechanisms contribute to dampen pDC activation
6. pDCs Constitute a Link between Innate, Adaptive Immunity
6.2. pDCs can contribute to the negative regulation of adaptive immunity
7. pDC response to Herpesvirus Infections Is a Double-Edged Sword: It Contributes to the Control of Viral Replication but Can also Take Part in the Induction of Immunosuppression or Immunopathology
| The goods of IFN-I responses | Ref. | The bads of IFN-I responses | Ref. |
|---|---|---|---|
| Direct antiviral effects | [15] | Development of autoimmunity or immunopathology | [15,143] |
| Promotion of cDC maturation | [13,23] | Induction of DC apoptosis | [144] |
| promotion of cDC cross-presentation | [24] | Prevention of DC renewal | [145] |
| promotion of NK cell activation | [19,22] | General inhibition of hematopoiesis | [146,147] |
| Help to CD8 T lymphocytes | [17,18,20] | Anti-proliferative or pro-apoptotic effects on CD8 T cells | [15,148,149] |
| Help to B lymphocytes | [150,151] | Susceptibility to bacterial surinfections | [152,153] |
7.1. Herpesviruses can exploit pDC functions to promote their replication
7.3. Is-there a role for herpesvirus-dependent pDC activation in the triggering of certain autoimmune diseases?
8. From the Bench to the Bedside: Exploiting pDC Responses to Herpesvirus Infections for Therapeutic Purposes
8.1. Boosting pDC antiviral responses

8.2. Dampening pDC activation
8.3. Exploiting herpesviruses as potent vectors for vaccination against other intracellular pathogens
9. Conclusion
| Major concepts, outstanding questions regarding pDC responses to herpesviruses |
|---|
| IFN-I are innate cytokines endowed with potent direct, indirect antiviral activities. |
| The expression of the receptor for IFN-I is ubiquitous, allows widespread systemic effects of the cytokines. |
| IFN-I responses are complex, can induce protective antiviral responses or immunopathology depending on the timing, level, anatomical site of their production. |
| Herpesviruses can interfere with the induction of, or the responses to, IFN-I to escape immunity. |
| pDCs are the main IFN-I producers in response to many viruses including all the herpesviruses tested. |
| pDCs are able to sense herpesvirus infections through the TLR7/9 receptors in a MyD88 dependant manner. |
| pDCs produce a large panel of cytokines/chemokines, thus must play a major role in the orchestration of early inflammation, downstream activation of innate, adaptive immune effectors. |
| Mature pDCs can cross-present viral antigens for cognate CD8 T cell activation. |
| Excessive pDC activation during viral infections can contribute to immunopathology. |
| It is not known whether pDC responses to common herpesvirus infections could contribute to the development of certain autoimmune diseases in susceptible individuals. |
| It is not known whether,, how, pDCs are required for the induction, polarization of T cell responses during herpesvirus infections in vivo. |
| To rigorously evaluate the multiple roles of pDCs in vivo in the orchestration of antiviral immune responses, the genetic engineering of novel mouse models specifically devoid of pDCs or selectively affected in their functions will be crucial. |
| pDCs are interesting targets for the design of novel immunotherapeutic approaches against viral infections. |
Acknowledgments
References and Notes
- Schleiss, M.R. Persistent, recurring viral infections: the human herpesviruses. Curr. Probl. Pediatr. Adolesc. Health Care 2009, 39, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Stout-Delgado, H.W.; Yang, X.; Walker, W.E.; Tesar, B.M.; Goldstein, D.R. Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J. Immunol. 2008, 181, 6747–6756. [Google Scholar] [PubMed]
- Donaghy, H.; Bosnjak, L.; Harman, A.N.; Marsden, V.; Tyring, S.K.; Meng, T.C.; Cunningham, A.L. Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes simplex virus infection. J. Virol. 2009, 83, 1952–1961. [Google Scholar] [CrossRef] [PubMed]
- Cederarv, M.; Soderberg-Naucler, C.; Odeberg, J. HCMV infection of PDCs deviates the NK cell response into cytokine-producing cells unable to perform cytotoxicity. Immunobiology 2009, 214, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kvale, E.O.; Dalgaard, J.; Lund-Johansen, F.; Rollag, H.; Farkas, L.; Midtvedt, K.; Jahnsen, F.L.; Brinchmann, J.E.; Olweus, J. CD11c+ dendritic cells, plasmacytoid DCs are activated by human cytomegalovirus, retain efficient T cell-stimulatory capability upon infection. Blood 2006, 107, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.; Meyer-Koenig, U.; Hufert, F.T. Human cytomegalovirus impairs the function of plasmacytoid dendritic cells in lymphoid organs. PLoS One 2008, 3, e3482. [Google Scholar] [CrossRef] [PubMed]
- Varani, S.; Cederarv, M.; Feld, S.; Tammik, C.; Frascaroli, G.; Landini, M.P.; Soderberg-Naucler, C. Human cytomegalovirus differentially controls B cell, T cell responses through effects on plasmacytoid dendritic cells. J. Immunol. 2007, 179, 7767–7776. [Google Scholar] [PubMed]
- Takemoto, M.; Imasawa, T.; Yamanishi, K.; Mori, Y. Role of dendritic cells infected with human herpesvirus 6 in virus transmission to CD4(+) T cells. Virology 2009, 385, 294–302. [Google Scholar] [CrossRef] [PubMed]
- de Vries, H.J.; Teunissen, M.B.; Zorgdrager, F.; Picavet, D.; Cornelissen, M. Lichen planus remission is associated with a decrease of human herpes virus type 7 protein expression in plasmacytoid dendritic cells. Arch. Dermatol. Res. 2007, 299, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.H.; Kireta, S.; Russ, G.R.; Coates, P.T. Human plasmacytoid dendritic cells regulate immune responses to Epstein-Barr virus (EBV) infection, delay EBV-related mortality in humanized NOD-SCID mice. Blood 2007, 109, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Dalod, M.; Salazar-Mather, T.P.; Malmgaard, L.; Lewis, C.; Asselin-Paturel, C.; Briere, F.; Trinchieri, G.; Biron, C.A. Interferon alpha/beta, interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 2002, 195, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Dalod, M.; Hamilton, T.; Salomon, R.; Salazar-Mather, T.P.; Henry, S.C.; Hamilton, J.D.; Biron, C.A. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization, differential regulation by interferon alpha/beta. J. Exp. Med. 2003, 197, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Baccala, R.; Beutler, B.; Kono, D.H. Type I interferons (alpha/beta) in immunity, autoimmunity. Annu. Rev. Immunol. 2005, 23, 307–336. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sastre, A.; Biron, C.A. Type 1 interferons, the virus-host relationship: a lesson in detente. Science 2006, 312, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Aichele, P.; Unsoeld, H.; Koschella, M.; Schweier, O.; Kalinke, U.; Vucikuja, S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J. Immunol. 2006, 176, 4525–4529. [Google Scholar] [PubMed]
- Le Bon, A.; Durand, V.; Kamphuis, E.; Thompson, C.; Bulfone-Paus, S.; Rossmann, C.; Kalinke, U.; Tough, D.F. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 2006, 176, 4682–4689. [Google Scholar] [PubMed]
- Martinez, J.; Huang, X.; Yang, Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J. Immunol. 2008, 180, 1592–1597. [Google Scholar] [PubMed]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion, memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.B.; Salazar-Mather, T.P.; Dalod, M.Y.; Van Deusen, J.B.; Wei, X.Q.; Liew, F.Y.; Caligiuri, M.A.; Durbin, J.E.; Biron, C.A. Coordinated, distinct roles for IFN-alpha beta, IL-12,, IL-15 regulation of NK cell responses to viral infection. J. Immunol. 2002, 169, 4279–4287. [Google Scholar] [PubMed]
- Honda, K.; Sakaguchi, S.; Nakajima, C.; Watanabe, A.; Yanai, H.; Matsumoto, M.; Ohteki, T.; Kaisho, T.; Takaoka, A.; Akira, S.; Seya, T.; Taniguchi, T. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. U S A 2003, 100, 10872–10877. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Tough, D.F. Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev. 2008, 19, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines,, their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Blasius, A.L.; Barchet, W.; Cella, M.; Colonna, M. Development, function of murine B220+CD11c+NK1.1+ cells identify them as a subset of NK cells. J. Exp. Med. 2007, 204, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Caminschi, I.; Ahmet, F.; Heger, K.; Brady, J.; Nutt, S.L.; Vremec, D.; Pietersz, S.; Lahoud, M.H.; Schofield, L.; Hansen, D.S.; O'Keeffe, M.; Smyth, M.J.; Bedoui, S.; Davey, G.M.; Villadangos, J.A.; Heath, W.R.; Shortman, K. Putative IKDCs are functionally, developmentally similar to natural killer cells, but not to dendritic cells. J. Exp. Med. 2007, 204, 2579–2590. [Google Scholar] [CrossRef] [PubMed]
- Robbins, S.H.; Walzer, T.; Dembele, D.; Thibault, C.; Defays, A.; Bessou, G.; Xu, H.; Vivier, E.; Sellars, M.; Pierre, P.; Sharp, F.R.; Chan, S.; Kastner, P.; Dalod, M. Novel insights into the relationships between dendritic cell subsets in human, mouse revealed by genome-wide expression profiling. Genome Biol. 2008, 9, R17. [Google Scholar] [CrossRef] [PubMed]
- Vosshenrich, C.A.; Lesjean-Pottier, S.; Hasan, M.; Richard-Le Goff, O.; Corcuff, E.; Mandelboim, O.; Di Santo, J.P. CD11cloB220+ interferon-producing killer dendritic cells are activated natural killer cells. J. Exp. Med. 2007, 204, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Wong, J.; Villadangos, J.A. Cutting edge: B220+CCR9- dendritic cells are not plasmacytoid dendritic cells but are precursors of conventional dendritic cells. J. Immunol. 2009, 183, 1514–1517. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. IPC: professional type 1 interferon-producing cells, plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Asselin-Paturel, C.; Boonstra, A.; Dalod, M.; Durand, I.; Yessaad, N.; Dezutter-Dambuyant, C.; Vicari, A.; O'Garra, A.; Biron, C.; Briere, F.; Trinchieri, G. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2001, 2, 1144–1150. [Google Scholar] [CrossRef]
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid monocytes migrate to inflamed lymph nodes, produce large amounts of type I interferon. Nat. Med. 1999, 5, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Yanagita, M.; Gunn, M.D. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes, spleen display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 2001, 194, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Bjorck, P. Isolation, characterization of plasmacytoid dendritic cells from Flt3 ligand, granulocyte-macrophage colony-stimulating factor-treated mice. Blood 2001, 98, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Chehimi, J.; Starr, S.E.; Kawashima, H.; Miller, D.S.; Trinchieri, G.; Perussia, B.; Bandyopadhyay, S. Dendritic cells, IFN-alpha-producing cells are two functionally distinct non-B, non-monocytic HLA-DR+ cell subsets in human peripheral blood. Immunology 1989, 68, 486–490. [Google Scholar] [PubMed]
- Fitzgerald-Bocarsly, P.; Feldman, M.; Mendelsohn, M.; Curl, S.; Lopez, C. Human mononuclear cells which produce interferon-alpha during NK(HSV-FS) assays are HLA-DR positive cells distinct from cytolytic natural killer effectors. J. Leukoc. Biol. 1988, 43, 323–334. [Google Scholar] [PubMed]
- Ito, T.; Kanzler, H.; Duramad, O.; Cao, W.; Liu, Y.J. Specialization, kinetics,, repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 2006, 107, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; French, A.R.; Barchet, W.; Fischer, J.A.; Dzionek, A.; Pingel, J.T.; Orihuela, M.M.; Akira, S.; Yokoyama, W.M.; Colonna, M. TLR9-dependent recognition of MCMV by IPC, DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 2004, 21, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.B.; Sorensen, L.N.; Malmgaard, L.; Ank, N.; Baines, J.D.; Chen, Z.J.; Paludan, S.R. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway,, novel recognition systems. J. Virol. 2007, 81, 13315–13324. [Google Scholar] [CrossRef] [PubMed]
- Bozza, S.; Gaziano, R.; Bonifazi, P.; Zelante, T.; Pitzurra, L.; Montagnoli, C.; Moretti, S.; Castronari, R.; Sinibaldi, P.; Rasi, G.; Garaci, E.; Bistoni, F.; Romani, L. Thymosin alpha1 activates the TLR9/MyD88/IRF7-dependent murine cytomegalovirus sensing for induction of anti-viral responses in vivo. Int. Immunol. 2007, 19, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Andoniou, C.E.; van Dommelen, S.L.; Voigt, V.; Andrews, D.M.; Brizard, G.; Asselin-Paturel, C.; Delale, T.; Stacey, K.J.; Trinchieri, G.; Degli-Esposti, M.A. Interaction between conventional dendritic cells, natural killer cells is integral to the activation of effective antiviral immunity. Nat. Immunol. 2005, 6, 1011–1019. [Google Scholar] [CrossRef]
- Zucchini, N.; Bessou, G.; Robbins, S.H.; Chasson, L.; Raper, A.; Crocker, P.R.; Dalod, M. Individual plasmacytoid dendritic cells are major contributors to the production of multiple innate cytokines in an organ-specific manner during viral infection. Int. Immunol. 2008, 20, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Scheu, S.; Dresing, P.; Locksley, R.M. Visualization of IFNbeta production by plasmacytoid versus conventional dendritic cells under specific stimulation conditions in vivo. Proc. Natl. Acad. Sci. U S A 2008, 105, 20416–20421. [Google Scholar] [CrossRef] [PubMed]
- Allman, D.; Dalod, M.; Asselin-Paturel, C.; Delale, T.; Robbins, S.H.; Trinchieri, G.; Biron, C.A.; Kastner, P.; Chan, S. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood 2006, 108, 4025–4034. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Linehan, M.M.; Iijima, N.; Iwasaki, A. Cutting Edge: Plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J. Immunol. 2006, 177, 7510–7514. [Google Scholar] [PubMed]
- Kumagai, Y.; Takeuchi, O.; Kato, H.; Kumar, H.; Matsui, K.; Morii, E.; Aozasa, K.; Kawai, T.; Akira, S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 2007, 27, 240–252. [Google Scholar] [CrossRef] [PubMed]
- GeurtsvanKessel, C.H.; Willart, M.A.; van Rijt, L.S.; Muskens, F.; Kool, M.; Baas, C.; Thielemans, K.; Bennett, C.; Clausen, B.E.; Hoogsteden, H.C.; Osterhaus, A.D.; Rimmelzwaan, G.F.; Lambrecht, B.N. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J. Exp. Med. 2008, 205, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.I.; Buehler, D.; Hensley, S.E.; Cavanagh, L.L.; Wherry, E.J.; Kastner, P.; Chan, S.; Weninger, W. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J. Immunol. 2009, 182, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Wetzel, J.D.; He, J.; Mikacenic, C.; Dermody, T.S.; Kelsall, B.L. Type I interferons produced by hematopoietic cells protect mice against lethal infection by mammalian reovirus. J. Exp. Med. 2007, 204, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Contractor, N.; Louten, J.; Kim, L.; Biron, C.A.; Kelsall, B.L. Cutting edge: Peyer's patch plasmacytoid dendritic cells (pDCs) produce low levels of type I interferons: possible role for IL-10, TGFbeta,, prostaglandin E2 in conditioning a unique mucosal pDC phenotype. J. Immunol. 2007, 179, 2690–2694. [Google Scholar] [PubMed]
- Steinberg, C.; Eisenacher, K.; Gross, O.; Reindl, W.; Schmitz, F.; Ruland, J.; Krug, A. The IFN regulatory factor 7-dependent type I IFN response is not essential for early resistance against murine cytomegalovirus infection. Eur. J. Immunol. 2009, 39, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Scalzo, A.A.; Yokoyama, W.M. Cmv1, natural killer cell responses to murine cytomegalovirus infection. Curr. Top. Microbiol. Immunol. 2008, 321, 101–122. [Google Scholar] [PubMed]
- Abbo, L.; Vincek, V.; Dickinson, G.; Shrestha, N.; Doblecki, S.; Haslett, P.A. Selective defect in plasmacyoid dendritic cell function in a patient with AIDS-associated atypical genital herpes simplex vegetans treated with imiquimod. Clin. Infect. Dis. 2007, 44, e25–e27. [Google Scholar] [CrossRef] [PubMed]
- Dalloul, A.; Oksenhendler, E.; Chosidow, O.; Ribaud, P.; Carcelain, G.; Louvet, S.; Massip, P.; Lebon, P.; Autran, B. Severe herpes virus (HSV-2) infection in two patients with myelodysplasia, undetectable NK cells, plasmacytoid dendritic cells in the blood. J. Clin. Virol. 2004, 30, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Kittan, N.A.; Bergua, A.; Haupt, S.; Donhauser, N.; Schuster, P.; Korn, K.; Harrer, T.; Schmidt, B. Impaired plasmacytoid dendritic cell innate immune responses in patients with herpes virus-associated acute retinal necrosis. J. Immunol. 2007, 179, 4219–4230. [Google Scholar] [PubMed]
- Zuniga, E.I.; Liou, L.Y.; Mack, L.; Mendoza, M.; Oldstone, M.B. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe 2008, 4, 374–386. [Google Scholar] [CrossRef]
- Baranek, T.; Dalod, M. How opportunistic agents benefit from viral infections: the plasmacytoid dendritic cell connection. Cell Host Microbe 2008, 4, 305–307. [Google Scholar] [CrossRef]
- Nascimbeni, M.; Perie, L.; Chorro, L.; Diocou, S.; Kreitmann, L.; Louis, S.; Garderet, L.; Fabiani, B.; Berger, A.; Schmitz, J.; Marie, J.P.; Molina, T.J.; Pacanowski, J.; Viard, J.P.; Oksenhendler, E.; Beq, S.; Abehsira-Amar, O.; Cheynier, R.; Hosmalin, A. Plasmacytoid dendritic cells accumulate in spleens from chronically HIV-infected patients but barely participate in interferon-alpha expression. Blood 2009, 113, 6112–6119. [Google Scholar] [CrossRef] [PubMed]
- Slyker, J.A.; Lohman-Payne, B.L.; John-Stewart, G.C.; Maleche-Obimbo, E.; Emery, S.; Richardson, B.; Dong, T.; Iversen, A.K.; Mbori-Ngacha, D.; Overbaugh, J.; Emery, V.C.; Rowland-Jones, S.L. Acute cytomegalovirus infection in Kenyan HIV-infected infants. AIDS 2009, 23, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.E.; Moore, R.D.; Richman, D.D.; Keruly, J.; Chaisson, R.E. Incidence, natural history of cytomegalovirus disease in patients with advanced human immunodeficiency virus disease treated with zidovudine. The Zidovudine Epidemiology Study Group. J. Infect. Dis. 1992, 166, 1223–1227. [Google Scholar] [PubMed]
- Van de Perre, P.; Segondy, M.; Foulongne, V.; Ouedraogo, A.; Konate, I.; Huraux, J.M.; Mayaud, P.; Nagot, N. Herpes simplex virus, HIV-1: deciphering viral synergy. Lancet Infect. Dis. 2008, 8, 490–497. [Google Scholar] [CrossRef]
- Hosmalin, A.; Lebon, P. Type I interferon production in HIV-infected patients. J. Leukoc. Biol. 2006, 80, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Britt, W. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute, chronic disease. Curr. Top. Microbiol. Immunol. 2008, 325, 417–470. [Google Scholar] [PubMed]
- Giraud, S.; Dhedin, N.; Gary-Gouy, H.; Lebon, P.; Vernant, J.P.; Dalloul, A. Plasmacytoid dendritic cell reconstitution following bone marrow transplantation: subnormal recovery, functional deficit of IFN-alpha/beta production in response to herpes simplex virus. J. Interferon Cytokine Res. 2005, 25, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Thomson, A.W. Dexamethasone preferentially suppresses plasmacytoid dendritic cell differentiation, enhances their apoptotic death. Clin. Immunol. 2006, 118, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Boor, P.P.; Metselaar, H.J.; Mancham, S.; Tilanus, H.W.; Kusters, J.G.; Kwekkeboom, J. Prednisolone suppresses the function, promotes apoptosis of plasmacytoid dendritic cells. Am. J. Transplant. 2006, 6, 2332–2341. [Google Scholar] [CrossRef] [PubMed]
- Cisse, B.; Caton, M.L.; Lehner, M.; Maeda, T.; Scheu, S.; Locksley, R.; Holmberg, D.; Zweier, C.; den Hollander, N.S.; Kant, S.G.; Holter, W.; Rauch, A.; Zhuang, Y.; Reizis, B. Transcription factor E2-2 is an essential, specific regulator of plasmacytoid dendritic cell development. Cell 2008, 135, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Megjugorac, N.J.; Young, H.A.; Amrute, S.B.; Olshalsky, S.L.; Fitzgerald-Bocarsly, P. Virally stimulated plasmacytoid dendritic cells produce chemokines, induce migration of T, NK cells. J. Leukoc. Biol. 2004, 75, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Mather, T.P.; Hokeness, K.L. Cytokine, chemokine networks: pathways to antiviral defense. Curr. Top. Microbiol. Immunol. 2006, 303, 29–46. [Google Scholar] [PubMed]
- Fehniger, T.A.; Cai, S.F.; Cao, X.; Bredemeyer, A.J.; Presti, R.M.; French, A.R.; Ley, T.J. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B, perforin mRNAs. Immunity 2007, 26, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Mortier, E.; Woo, T.; Advincula, R.; Gozalo, S.; Ma, A. IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J. Exp. Med. 2008, 205, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Barr, D.P.; Belz, G.T.; Reading, P.C.; Wojtasiak, M.; Whitney, P.G.; Heath, W.R.; Carbone, F.R.; Brooks, A.G. A role for plasmacytoid dendritic cells in the rapid IL-18-dependent activation of NK cells following HSV-1 infection. Eur. J. Immunol. 2007, 37, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Decalf, J.; Fernandes, S.; Longman, R.; Ahloulay, M.; Audat, F.; Lefrerre, F.; Rice, C.M.; Pol, S.; Albert, M.L. Plasmacytoid dendritic cells initiate a complex chemokine, cytokine network, are a viable drug target in chronic HCV patients. J. Exp. Med. 2007, 204, 2423–2437. [Google Scholar] [CrossRef] [PubMed]
- Piqueras, B.; Connolly, J.; Freitas, H.; Palucka, A.K.; Banchereau, J. Upon viral exposure, myeloid, plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood 2006, 107, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Hanabuchi, S.; Watanabe, N.; Wang, Y.H.; Ito, T.; Shaw, J.; Cao, W.; Qin, F.X.; Liu, Y.J. Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL). Blood 2006, 107, 3617–3623. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Reis e Sousa, C. Innate recognition of viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Zust, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Kalinke, U.; Zust, R.; Konig, M.; Reizis, B.; Lopez-Macias, C.; Thiel, V.; Ludewig, B. Type I IFN-mediated protection of macrophages, dendritic cells secures control of murine coronavirus infection. J. Immunol. 2009, 182, 1099–1106. [Google Scholar] [PubMed]
- Crozat, K.; Vivier, E.; Dalod, M. Crosstalk between components of the innate immune system: promoting anti-microbial defenses, avoiding immunopathologies. Immunol. Rev. 2009, 227, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M.; Kagan, J.C.; Medzhitov, R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 2006, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Megjugorac, N.J.; Jacobs, E.S.; Izaguirre, A.G.; George, T.C.; Gupta, G.; Fitzgerald-Bocarsly, P. Image-based study of interferongenic interactions between plasmacytoid dendritic cells, HSV-infected monocyte-derived dendritic cells. Immunol. Invest. 2007, 36, 739–761. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Sato, A.; Akira, S.; Medzhitov, R.; Iwasaki, A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 2003, 198, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Delale, T.; Paquin, A.; Asselin-Paturel, C.; Dalod, M.; Brizard, G.; Bates, E.E.; Kastner, P.; Chan, S.; Akira, S.; Vicari, A.; Biron, C.A.; Trinchieri, G.; Briere, F. MyD88-dependent, -independent murine cytomegalovirus sensing for IFN-alpha release, initiation of immune responses in vivo. J. Immunol. 2005, 175, 6723–6732. [Google Scholar] [PubMed]
- Krug, A.; Luker, G.D.; Barchet, W.; Leib, D.A.; Akira, S.; Colonna, M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2004, 103, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.; Loewendorf, A.; De Trez, C.; Fulton, J.; Rhode, A.; Shumway, H.; Ha, S.; Patterson, G.; Pfeffer, K.; Nedospasov, S.A.; Ware, C.F.; Benedict, C.A. Lymphotoxin-mediated crosstalk between B cells, splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe 2008, 3, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Rebsamen, M.; Heinz, L.X.; Meylan, E.; Michallet, M.C.; Schroder, K.; Hofmann, K.; Vazquez, J.; Benedict, C.A.; Tschopp, J. DAI/ZBP1 recruits RIP1, RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009, 10, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I, activate signaling to induce type I IFN. EMBO J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef] [PubMed]
- Zucchini, N.; Bessou, G.; Traub, S.; Robbins, S.H.; Uematsu, S.; Akira, S.; Alexopoulou, L.; Dalod, M. Cutting edge: Overlapping functions of TLR7, TLR9 for innate defense against a herpesvirus infection. J. Immunol. 2008, 180, 5799–5803. [Google Scholar] [PubMed]
- Mancuso, G.; Gambuzza, M.; Midiri, A.; Biondo, C.; Papasergi, S.; Akira, S.; Teti, G.; Beninati, C. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat. Immunol. 2009, 10, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Fukui, R.; Saitoh, S.; Matsumoto, F.; Kozuka-Hata, H.; Oyama, M.; Tabeta, K.; Beutler, B.; Miyake, K. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J. Exp. Med. 2009, 206, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; Taniguchi, T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marie, I.; Smith, E.; Prakash, A. Enhancement, diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 2002, 22, 87–93. [Google Scholar] [PubMed]
- Guiducci, C.; Ott, G.; Chan, J.H.; Damon, E.; Calacsan, C.; Matray, T.; Lee, K.D.; Coffman, R.L.; Barrat, F.J. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J. Exp. Med. 2006, 203, 1999–2008. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Ohba, Y.; Yanai, H.; Negishi, H.; Mizutani, T.; Takaoka, A.; Taya, C.; Taniguchi, T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 2005, 434, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Barchet, W.; Cella, M.; Odermatt, B.; Asselin-Paturel, C.; Colonna, M.; Kalinke, U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J. Exp. Med. 2002, 195, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, Y.; Kumar, H.; Koyama, S.; Kawai, T.; Takeuchi, O.; Akira, S. Cutting Edge: TLR-Dependent viral recognition along with type I IFN positive feedback signaling masks the requirement of viral replication for IFN-{alpha} production in plasmacytoid dendritic cells. J. Immunol. 2009, 182, 3960–3964. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Montoya, M.; Unger, H.; Alexopoulou, L.; Roy, P.; Haswell, L.E.; Al-Shamkhani, A.; Flavell, R.; Borrow, P.; Reis e Sousa, C. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003, 424, 324–328. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, V.R. Induction, evasion of the type I interferon response by cytomegaloviruses. Adv. Exp. Med. Biol. 2007, 598, 309–324. [Google Scholar] [PubMed]
- Hengel, H.; Koszinowski, U.H.; Conzelmann, K.K. Viruses know it all: new insights into IFN networks. Trends Immunol. 2005, 26, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.G.; Unterholzner, L. Viral evasion, subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.L.; Barry, P.A.; Szubin, R.; Wang, D.; Baumgarth, N. Human cytomegalovirus suppresses type I interferon secretion by plasmacytoid dendritic cells through its interleukin 10 homolog. Virology 2009, 390, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Pascual, V. Type I interferon in systemic lupus erythematosus, other autoimmune diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Conrad, C.; Meller, S.; Gilliet, M. Plasmacytoid dendritic cells in the skin: to sense or not to sense nucleic acids. Semin. Immunol. 2009, 21, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Rosen, D.B.; Ito, T.; Bover, L.; Bao, M.; Watanabe, G.; Yao, Z.; Zhang, L.; Lanier, L.L.; Liu, Y.J. Plasmacytoid dendritic cell-specific receptor ILT7-Fc epsilonRI gamma inhibits Toll-like receptor-induced interferon production. J. Exp. Med. 2006, 203, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.; Schneider, E.; Grun, J.R.; Grutzkau, A.; Kuppers, R.; Schmitz, J.; Winkels, G. CD303 (BDCA-2) signals in plasmacytoid dendritic cells via a BCR-like signalosome involving Syk, Slp65, PLCgamma2. Eur. J. Immunol. 2007, 37, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zhang, L.; Rosen, D.B.; Bover, L.; Watanabe, G.; Bao, M.; Lanier, L.L.; Liu, Y.J. BDCA2/Fc epsilon RI gamma complex signals through a novel BCR-like pathway in human plasmacytoid dendritic cells. PLoS Biol. 2007, 5, e248. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.; Vermi, W.; Krug, A.; Facchetti, F.; Cella, M.; Colonna, M. A cell-surface molecule selectively expressed on murine natural interferon-producing cells that blocks secretion of interferon-alpha. Blood 2004, 103, 4201–4206. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Cella, M.; Maldonado, J.; Takai, T.; Colonna, M. Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood 2006, 107, 2474–2476. [Google Scholar] [CrossRef] [PubMed]
- Sjolin, H.; Robbins, S.H.; Bessou, G.; Hidmark, A.; Tomasello, E.; Johansson, M.; Hall, H.; Charifi, F.; Karlsson Hedestam, G.B.; Biron, C.A.; Karre, K.; Hoglund, P.; Vivier, E.; Dalod, M. DAP12 signaling regulates plasmacytoid dendritic cell homeostasis, down-modulates their function during viral infection. J. Immunol. 2006, 177, 2908–2916. [Google Scholar] [PubMed]
- Blasius, A.L.; Giurisato, E.; Cella, M.; Schreiber, R.D.; Shaw, A.S.; Colonna, M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 2006, 177, 3260–3265. [Google Scholar] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Wentrup, F.; Benitez-Ribas, D.; Tacken, P.J.; Punt, C.J.; Figdor, C.G.; de Vries, I.J.; Adema, G.J. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation, inhibits IFN-alpha production. Blood 2008, 111, 4245–4253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Raper, A.; Sugita, N.; Hingorani, R.; Salio, M.; Palmowski, M.J.; Cerundolo, V.; Crocker, P.R. Characterization of Siglec-H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood 2006, 107, 3600–3608. [Google Scholar] [CrossRef] [PubMed]
- Robbins, S.H.; Bessou, G.; Cornillon, A.; Zucchini, N.; Rupp, B.; Ruzsics, Z.; Sacher, T.; Tomasello, E.; Vivier, E.; Koszinowski, U.H.; Dalod, M. Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog. 2007, 3, e123. [Google Scholar] [CrossRef] [PubMed]
- Villadangos, J.A.; Young, L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity 2008, 29, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Young, L.J.; Wilson, N.S.; Schnorrer, P.; Proietto, A.; ten Broeke, T.; Matsuki, Y.; Mount, A.M.; Belz, G.T.; O'Keeffe, M.; Ohmura-Hoshino, M.; Ishido, S.; Stoorvogel, W.; Heath, W.R.; Shortman, K.; Villadangos, J.A. Differential MHC class II synthesis, ubiquitination confers distinct antigen-presenting properties on conventional, plasmacytoid dendritic cells. Nat. Immunol. 2008, 9, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Sadaka, C.; Marloie-Provost, M.A.; Soumelis, V.; Benaroch, P. Developmental regulation of MHC II expression, transport in human plasmacytoid-derived dendritic cells. Blood 2009, 113, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Hoeffel, G.; Ripoche, A.C.; Matheoud, D.; Nascimbeni, M.; Escriou, N.; Lebon, P.; Heshmati, F.; Guillet, J.G.; Gannage, M.; Caillat-Zucman, S.; Casartelli, N.; Schwartz, O.; De la Salle, H.; Hanau, D.; Hosmalin, A.; Maranon, C. Antigen crosspresentation by human plasmacytoid dendritic cells. Immunity 2007, 27, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Mouries, J.; Moron, G.; Schlecht, G.; Escriou, N.; Dadaglio, G.; Leclerc, C. Plasmacytoid dendritic cells efficiently cross-prime naive T cells in vivo after TLR activation. Blood 2008, 112, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Di Pucchio, T.; Chatterjee, B.; Smed-Sorensen, A.; Clayton, S.; Palazzo, A.; Montes, M.; Xue, Y.; Mellman, I.; Banchereau, J.; Connolly, J.E. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat. Immunol. 2008, 9, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, H.; Matsuno, K.; Toda, E.; Nishiwaki, T.; Matsuo, N.; Nakano, A.; Narumi, S.; Lu, B.; Gerard, C.; Ishikawa, S.; Matsushima, K. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. J. Exp. Med. 2005, 202, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, H.; Matsuno, K.; Zhang, Y.; Nishiwaki, T.; Kitabatake, M.; Ueha, S.; Narumi, S.; Morikawa, S.; Ezaki, T.; Lu, B.; Gerard, C.; Ishikawa, S.; Matsushima, K. Evidence for recruitment of plasmacytoid dendritic cell precursors to inflamed lymph nodes through high endothelial venules. Int. Immunol. 2004, 16, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Palucka, A.K.; Blanck, J.P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon, interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Kadowaki, N.; Kitawaki, T.; Uchiyama, T. Virus-stimulated plasmacytoid dendritic cells induce CD4+ cytotoxic regulatory T cells. Blood 2006, 107, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Kvale, E.O.; Floisand, Y.; Lund-Johansen, F.; Rollag, H.; Farkas, L.; Ghanekar, S.; Brandtzaeg, P.; Jahnsen, F.L.; Olweus, J. Plasmacytoid DCs regulate recall responses by rapid induction of IL-10 in memory T cells. Blood 2007, 109, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Asselin-Paturel, C.; Vacca, C.; Bianchi, R.; Gizzi, S.; Fioretti, M.C.; Trinchieri, G.; Grohmann, U.; Puccetti, P. Murine plasmacytoid dendritic cells initiate the immunosuppressive pathway of tryptophan catabolism in response to CD200 receptor engagement. J. Immunol. 2004, 173, 3748–3754. [Google Scholar] [PubMed]
- Mellor, A.L.; Baban, B.; Chandler, P.R.; Manlapat, A.; Kahler, D.J.; Munn, D.H. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J. Immunol. 2005, 175, 5601–5605. [Google Scholar] [PubMed]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest. 2007, 117, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Herbeuval, J.P.; Hardy, A.W.; Anderson, S.A.; Dolan, M.J.; Fuchs, D.; Shearer, G.M. HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood 2007, 109, 3351–3359. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Munn, D.; Fallahi, A.; Lifson, J.; Chaperot, L.; Plumas, J.; Bhardwaj, N. HIV-activated human plasmacytoid DCs induce Tregs through an indoleamine 2,3-dioxygenase-dependent mechanism. J. Clin. Invest. 2008, 118, 3431–3439. [Google Scholar] [CrossRef] [PubMed]
- Rolle, A.; Olweus, J. Dendritic cells in cytomegalovirus infection: viral evasion, host countermeasures. APMIS 2009, 117, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Hsing, L.; Pham, T.T.; Rudensky, A.Y. Coordination of early protective immunity to viral infection by regulatory T cells. Science 2008, 320, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Ronnblom, L.; Eloranta, M.L.; Alm, G.V. Role of natural interferon-alpha producing cells (plasmacytoid dendritic cells) in autoimmunity. Autoimmunity 2003, 36, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; Self, S.G.; Borrow, P. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest, delayed responses in acute hepatitis B, C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Hyrcza, M.D.; Kovacs, C.; Loutfy, M.; Halpenny, R.; Heisler, L.; Yang, S.; Wilkins, O.; Ostrowski, M.; Der, S.D. Distinct transcriptional profiles in ex vivo CD4+, CD8+ T cells are established early in human immunodeficiency virus type 1 infection, are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J. Virol. 2007, 81, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- Mandl, J.N.; Barry, A.P.; Vanderford, T.H.; Kozyr, N.; Chavan, R.; Klucking, S.; Barrat, F.J.; Coffman, R.L.; Staprans, S.I.; Feinberg, M.B. Divergent TLR7, TLR9 signaling, type I interferon production distinguish pathogenic, nonpathogenic AIDS virus infections. Nat. Med. 2008, 14, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Shearer, G.M.; Chougnet, C. Immune dysregulation in human immunodeficiency virus infection: know it, fix it, prevent it? J. Intern. Med. 2009, 265, 78–96. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Harper, J.M.; Taubert, D.; Hartmann, P.; Fatkenheuer, G.; Jung, N.; van Lunzen, J.; Stellbrink, H.J.; Gallo, R.C.; Romerio, F. Increased interferon alpha expression in circulating plasmacytoid dendritic cells of HIV-1-infected patients. J. Acquir. Immune Defic. Syndr. 2008, 48, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Chang, J.J.; Chan, E.S.; Pollard, R.B.; Sidhu, H.K.; Kulkarni, S.; Wen, T.F.; Lindsay, R.J.; Orellana, L.; Mildvan, D.; Bazner, S.; Streeck, H.; Alter, G.; Lifson, J.D.; Carrington, M.; Bosch, R.J.; Robbins, G.K.; Altfeld, M. Sex differences in the Toll-like receptor-mediated response of plasmacytoid dendritic cells to HIV-1. Nat. Med. 2009. [Google Scholar]
- Diop, O.M.; Ploquin, M.J.; Mortara, L.; Faye, A.; Jacquelin, B.; Kunkel, D.; Lebon, P.; Butor, C.; Hosmalin, A.; Barre-Sinoussi, F.; Muller-Trutwin, M.C. Plasmacytoid dendritic cell dynamics, alpha interferon production during Simian immunodeficiency virus infection with a nonpathogenic outcome. J. Virol. 2008, 82, 5145–5152. [Google Scholar] [CrossRef] [PubMed]
- Riviere, Y.; Gresser, I.; Guillon, J.C.; Tovey, M.G. Inhibition by anti-interferon serum of lymphocytic choriomeningitis virus disease in suckling mice. Proc. Natl. Acad. Sci. U S A 1977, 74, 2135–2139. [Google Scholar] [CrossRef] [PubMed]
- Yen, J.H.; Ganea, D. Interferon beta induces mature dendritic cell apoptosis through caspase-11 / caspase-3 activation. Blood 2009, 114, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Hahm, B.; Trifilo, M.J.; Zuniga, E.I.; Oldstone, M.B. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity 2005, 22, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.; Fehr, J.; Hengartner, H.; Zinkernagel, R.M. Virus-induced transient bone marrow aplasia: major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J. Exp. Med. 1997, 185, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Dong, C.; Cooper, M.D. Impairment of T, B cell development by treatment with a type I interferon. J. Exp. Med. 1998, 187, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Bahl, K.; Kim, S.K.; Calcagno, C.; Ghersi, D.; Puzone, R.; Celada, F.; Selin, L.K.; Welsh, R.M. IFN-induced attrition of CD8 T cells in the presence or absence of cognate antigen during the early stages of viral infections. J. Immunol. 2006, 176, 4284–4295. [Google Scholar] [PubMed]
- Gil, M.P.; Salomon, R.; Louten, J.; Biron, C.A. Modulation of STAT1 protein levels: a mechanism shaping CD8 T-cell responses in vivo. Blood 2006, 107, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Schiavoni, G.; D'Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type i interferons potently enhance humoral immunity, can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Thompson, C.; Kamphuis, E.; Durand, V.; Rossmann, C.; Kalinke, U.; Tough, D.F. Cutting edge: enhancement of antibody responses through direct stimulation of B, T cells by type I IFN. J. Immunol. 2006, 176, 2074–2078. [Google Scholar] [PubMed]
- Navarini, A.A.; Recher, M.; Lang, K.S.; Georgiev, P.; Meury, S.; Bergthaler, A.; Flatz, L.; Bille, J.; Landmann, R.; Odermatt, B.; Hengartner, H.; Zinkernagel, R.M. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc. Natl. Acad. Sci. U S A 2006, 103, 15535–15539. [Google Scholar] [CrossRef] [PubMed]
- Shahangian, A.; Chow, E.K.; Tian, X.; Kang, J.R.; Ghaffari, A.; Liu, S.Y.; Belperio, J.A.; Cheng, G.; Deng, J.C. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 2009, 119, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Iversen, A.C.; Steinkjer, B.; Nilsen, N.; Bohnhorst, J.; Moen, S.H.; Vik, R.; Stephens, P.; Thomas, D.W.; Benedict, C.A.; Espevik, T. A proviral role for CpG in cytomegalovirus infection. J. Immunol. 2009, 182, 5672–5681. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Skinner, P.J.; Ha, S.J.; Duan, L.; Mattila, T.L.; Hage, A.; White, C.; Barber, D.L.; O'Mara, L.; Southern, P.J.; Reilly, C.S.; Carlis, J.V.; Miller, C.J.; Ahmed, R.; Haase, A.T. Visualizing antigen-specific, infected cells in situ predicts outcomes in early viral infection. Science 2009, 323, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Orange, J.S.; Salazar-Mather, T.P.; Opal, S.M.; Biron, C.A. Mechanisms for virus-induced liver disease: tumor necrosis factor-mediated pathology independent of natural killer, T cells during murine cytomegalovirus infection. J. Virol. 1997, 71, 9248–9258. [Google Scholar] [PubMed]
- Banchereau, J.; Pascual, V.; Palucka, A.K. Autoimmunity through cytokine-induced dendritic cell activation. Immunity 2004, 20, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.B.; Harley, I.T.; Guthridge, J.M.; James, J.A. The curiously suspicious: a role for Epstein-Barr virus in lupus. Lupus 2006, 15, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Posnett, D.N. Herpesviruses, autoimmunity. Curr. Opin. Investig. Drugs 2008, 9, 505–514. [Google Scholar] [PubMed]
- Shen, H.; Iwasaki, A. A crucial role for plasmacytoid dendritic cells in antiviral protection by CpG ODN-based vaginal microbicide. J. Clin. Invest. 2006, 116, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.; Molina, J.M.; Scieux, C.; Ribaud, P.; Morfin, F. Topical imiquimod for recurrent acyclovir-resistant HSV infection. Am. J. Med. 2006, 119, e9–e11. [Google Scholar] [CrossRef] [PubMed]
- Vollstedt, S.; Franchini, M.; Hefti, H.P.; Odermatt, B.; O'Keeffe, M.; Alber, G.; Glanzmann, B.; Riesen, M.; Ackermann, M.; Suter, M. Flt3 ligand-treated neonatal mice have increased innate immunity against intracellular pathogens, efficiently control virus infections. J. Exp. Med. 2003, 197, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Estes, J.D.; Schlievert, P.M.; Duan, L.; Brosnahan, A.J.; Southern, P.J.; Reilly, C.S.; Peterson, M.L.; Schultz-Darken, N.; Brunner, K.G.; Nephew, K.R.; Pambuccian, S.; Lifson, J.D.; Carlis, J.V.; Haase, A.T. Glycerol monolaurate prevents mucosal SIV transmission. Nature 2009, 458, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Guiducci, C.; Coffman, R.L.; Barrat, F.J. Signalling pathways leading to IFN-alpha production in human plasmacytoid dendritic cell, the possible use of agonists or antagonists of TLR7, TLR9 in clinical indications. J. Intern. Med. 2009, 265, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.R.; Moseley, C.E.; Boltz, D.A.; Negovetich, N.J.; Reynolds, C.; Franks, J.; Brown, S.A.; Doherty, P.C.; Webster, R.G.; Thomas, P.G. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc. Natl. Acad. Sci. U S A 2009, 106, 5306–5311. [Google Scholar] [CrossRef] [PubMed]
- Querec, T.; Bennouna, S.; Alkan, S.; Laouar, Y.; Gorden, K.; Flavell, R.; Akira, S.; Ahmed, R.; Pulendran, B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8,, 9 to stimulate polyvalent immunity. J. Exp. Med. 2006, 203, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Karrer, U.; Sierro, S.; Wagner, M.; Oxenius, A.; Hengel, H.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J. Immunol. 2003, 170, 2022–2029. [Google Scholar] [PubMed]
- Simon, C.O.; Holtappels, R.; Tervo, H.M.; Bohm, V.; Daubner, T.; Oehrlein-Karpi, S.A.; Kuhnapfel, B.; Renzaho, A.; Strand, D.; Podlech, J.; Reddehase, M.J.; Grzimek, N.K. CD8 T cells control cytomegalovirus latency by epitope-specific sensing of transcriptional reactivation. J. Virol. 2006, 80, 10436–10456. [Google Scholar] [CrossRef] [PubMed]
- Karrer, U.; Wagner, M.; Sierro, S.; Oxenius, A.; Hengel, H.; Dumrese, T.; Freigang, S.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Expansion of protective CD8+ T-cell responses driven by recombinant cytomegaloviruses. J. Virol. 2004, 78, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Vieville, C.; Whizin, N.; Coyne-Johnson, L.; Siess, D.C.; Drummond, D.D.; Legasse, A.W.; Axthelm, M.K.; Oswald, K.; Trubey, C.M.; Piatak, M.; Lifson, J.D.; Nelson, J.A.; Jarvis, M.A.; Picker, L.J. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 2009, 15, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; Lorenzo, L.; Plancoulaine, S.; Senechal, B.; Geissmann, F.; Tabeta, K.; Hoebe, K.; Du, X.; Miller, R.L.; Heron, B.; Mignot, C.; de Villemeur, T.B.; Lebon, P.; Dulac, O.; Rozenberg, F.; Beutler, B.; Tardieu, M.; Abel, L.; Casanova, J.L. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Puel, A.; Zhang, S.; Eidenschenk, C.; Ku, C.L.; Casrouge, A.; Picard, C.; von Bernuth, H.; Senechal, B.; Plancoulaine, S.; Al-Hajjar, S.; Al-Ghonaium, A.; Marodi, L.; Davidson, D.; Speert, D.; Roifman, C.; Garty, B.Z.; Ozinsky, A.; Barrat, F.J.; Coffman, R.L.; Miller, R.L.; Li, X.; Lebon, P.; Rodriguez-Gallego, C.; Chapel, H.; Geissmann, F.; Jouanguy, E.; Casanova, J.L. Human TLR-7-, -8-,, -9-mediated induction of IFN-alpha/beta, -lambda Is IRAK-4 dependent, redundant for protective immunity to viruses. Immunity 2005, 23, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, L.B.; Ben-Ali, M.; Quach, H.; Laval, G.; Patin, E.; Pickrell, J.K.; Bouchier, C.; Tichit, M.; Neyrolles, O.; Gicquel, B.; Kidd, J.R.; Kidd, K.K.; Alcais, A.; Ragimbeau, J.; Pellegrini, S.; Abel, L.; Casanova, J.L.; Quintana-Murci, L. Evolutionary dynamics of human Toll-like receptors, their different contributions to host defense. PLoS Genet. 2009, 5, e1000562. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Baranek, T.; Zucchini, N.; Dalod, M. Plasmacytoid Dendritic Cells and the Control of Herpesvirus Infections. Viruses 2009, 1, 383-419. https://doi.org/10.3390/v1030383
Baranek T, Zucchini N, Dalod M. Plasmacytoid Dendritic Cells and the Control of Herpesvirus Infections. Viruses. 2009; 1(3):383-419. https://doi.org/10.3390/v1030383
Chicago/Turabian StyleBaranek, Thomas, Nicolas Zucchini, and Marc Dalod. 2009. "Plasmacytoid Dendritic Cells and the Control of Herpesvirus Infections" Viruses 1, no. 3: 383-419. https://doi.org/10.3390/v1030383
APA StyleBaranek, T., Zucchini, N., & Dalod, M. (2009). Plasmacytoid Dendritic Cells and the Control of Herpesvirus Infections. Viruses, 1(3), 383-419. https://doi.org/10.3390/v1030383