Viral Hybrid Vectors for Somatic Integration - Are They the Better Solution?

Abstract
:1. Introduction
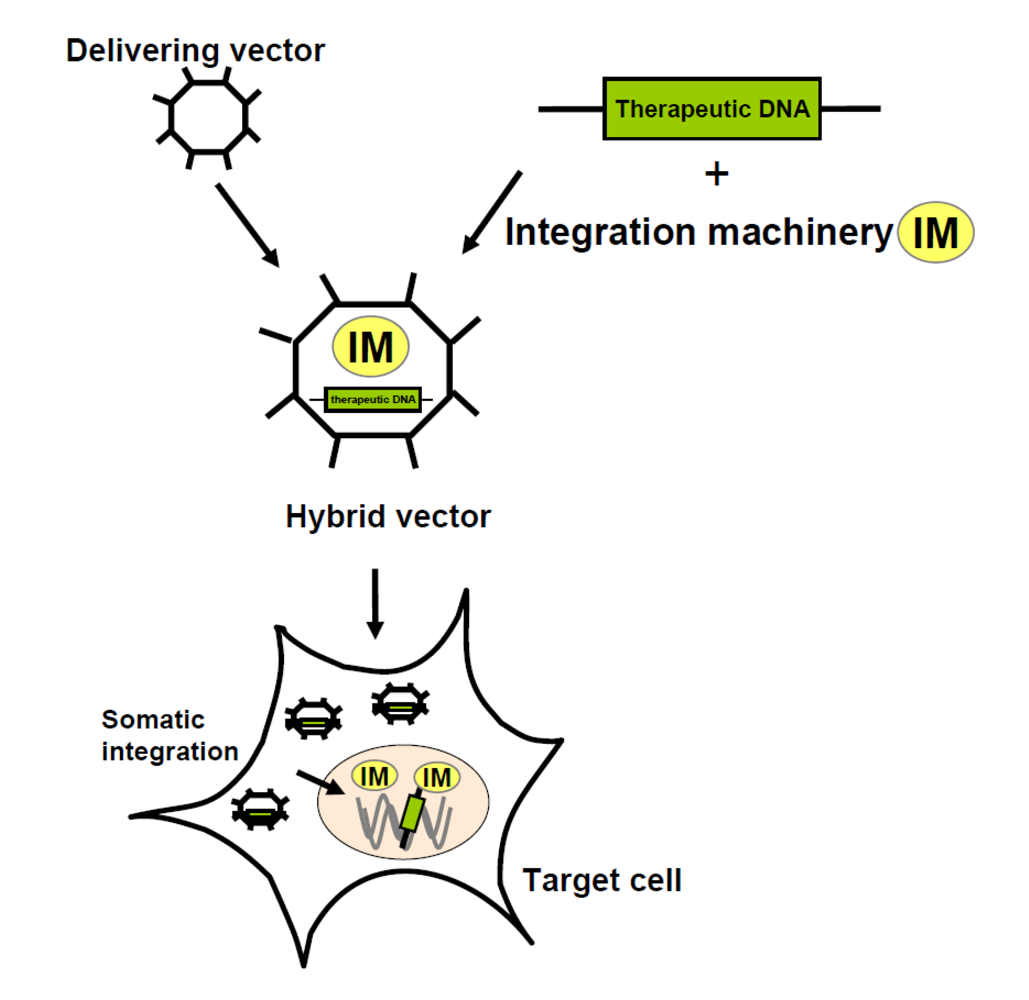

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Integration | SB | PhiC31 | Retro-transposon | AAV | RV | ZFN | DBD-IN-fusion proteins | |
|---|---|---|---|---|---|---|---|---|
| machinery | ||||||||
Delivering | ||||||||
| Vehicle | ||||||||
| HDAdV | + | + | + | + | + | - | - | |
| HSV-1 | + | - | - | + | + | - | - | |
| RV | - | - | - | - | / | - | + | |
| IDLV | + | - | - | - | - | + | - | |
| Hybrid vector | Integrating system | Integration pattern | Reference |
|---|---|---|---|
| HDAdV/AAV | AAV-ITR | random | [35,40-42] |
| HDAdV/AAVRep | AAV-Rep | site-specific | [30, 31, 43, 133] |
| HDAdV/SB | Sleeping Beauty transposase | random | [47] |
| HDAdV/PhiC31 | PhiC31 integrase | randomly into pseudo attP-sites | [51] |
| HDAdV/retrotransposon | Retrotransposon | consensus genomic target DNA | [54],[55] |
| HDAdV/RV | Integrase | randomly into active genes | [27], [28] |
| HDAdV/FV | Integrase | random | [59] |
| HSV-1/AAVRep | AAV-Rep | site-specific | [65-70] |
| HSV-1/SB | Sleeping Beauty tansposase | random | [71] |
| HSV-1/RV | Integrase | randomly into active genes | [74] |
| RV/IN | DBD-IN-fusion protein | site-specific | [9-97] |
| IDLV/ZFN | ZFN | site-specific | [101, 103] |
| IDLV/SB | Sleeping Beauty | random | [105, 106] |
2. Adenovirus-based hybrid vectors for somatic integration
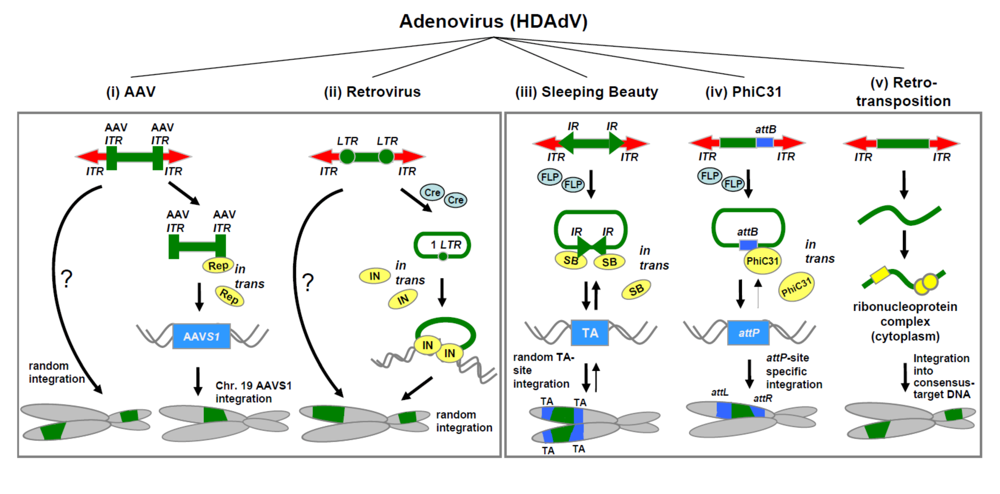
2.1. Adenovirus/AAV hybrid vectors.
2.2. Adenovirus/Sleeping Beauty transposase hybrid vectors.
2.3. Adenovirus/PhiC31 hybrid vectors.
2.4. Adenovirus/retrotransposon hybrid vectors.
2.5. Adenovirus/retrovirus hybrid vectors.
3. HSV-1 based hybrid vectors for somatic integration
3.1. HSV-1 amplicon/AAV hybrid vectors.
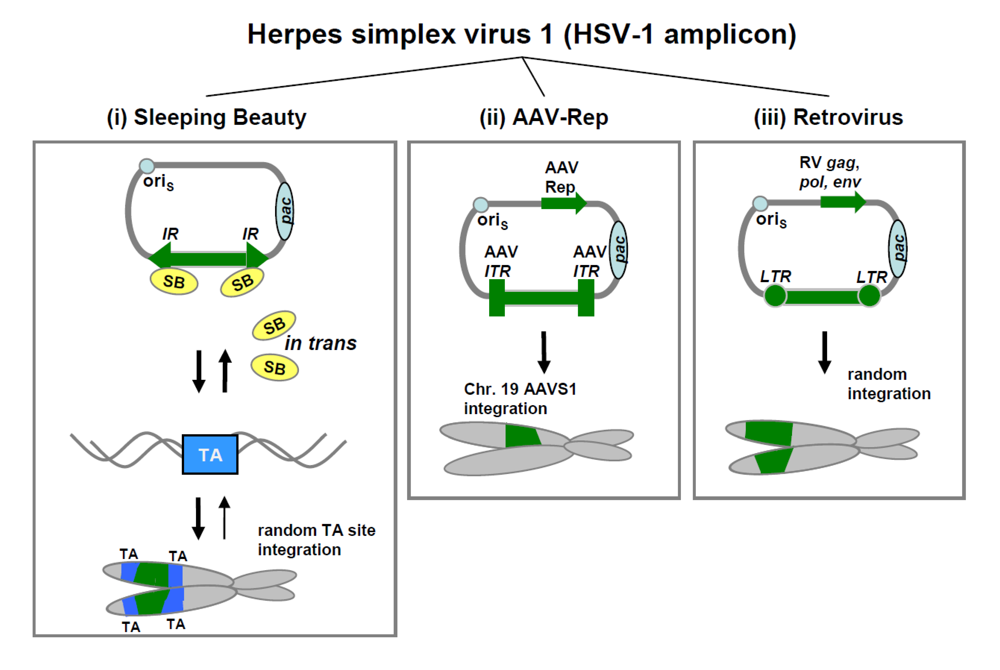
3.2. HSV-1 amplicon/SB transposase hybrid vectors.
3.3. HSV-1 amplicon/retrovirus (RV) hybrid vectors.
4. Retrovirus-based hybrid vectors for somatic integration
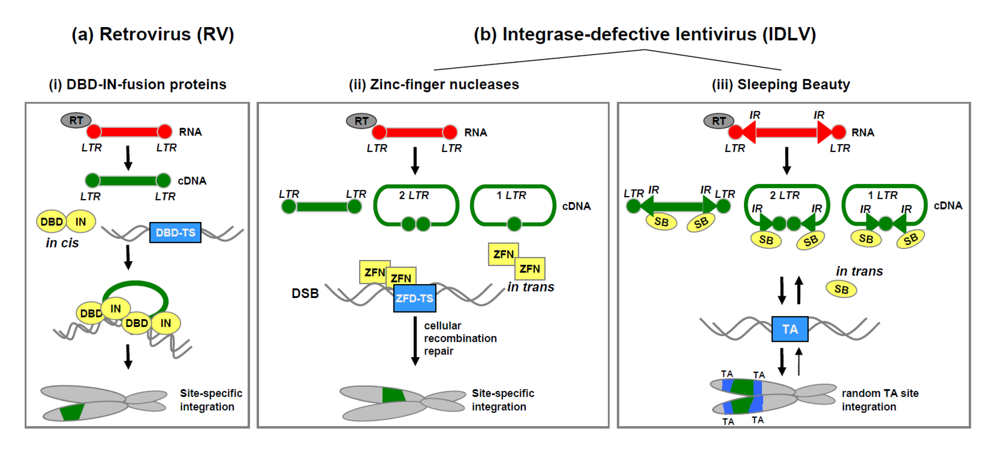
4.1. Retrovirus/integrase-fusion protein hybrid vectors.
4.2. Lentivirus/zinc-finger nuclease hybrid vectors.
4.3. Lentivirus/Sleeping Beauty transposase hybrid vectors.
5. Safety issues
5.1. Safety and genotoxicity of integration machineries used within hybrid vectors.
5.2. Assays to evaluate genotoxicity.
5.3. Safety of gene transfer vehicles utilized in the context of hybrid vectors.
6. Outlook
6.1. Towards a “one-vector system”.
6.2. Heading for targeted integration.
7. Conclusion
Acknowledgments
References
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvak, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.C.; Olivares, E.C.; Thyagarajan, B.; Calos, M.P. A phage integrase directs efficient site-specific integration in human cells. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 5995–6000. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.C.; Calos, M.P. Phage integrases: biology and applications. J. Mol. Biol. 2004, 335, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Barbas III, C.F. Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002, 20, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Progress and prospects: zinc-finger nucleases as gene therapy agents. Gene Ther. 2008, 15, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Howley P.M. 5th edLippincott Williams & Wilkins: Philadelphia, PA, USA, 2006.
- Jager, L.; Ehrhardt, A. Persistence of high-capacity adenoviral vectors as replication-defective monomeric genomes in vitro and in murine liver. Hum. Gene Ther. 2009, 20, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Stephen, S.L.; Sivanandam, V.G.; Kochanek, S. Homologous and heterologous recombination between adenovirus vector DNA and chromosomal DNA. J. Gene Med. 2008, 10, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Stabel, S.; Doerfler, W.; Friis, R.R. Integration sites of adenovirus type 12 DNA in transformed hamster cells and hamster tumor cells. J. Virol. 1980, 36, 22–40. [Google Scholar] [PubMed]
- Sutter, D.; Westphal, M.; Doerfler, W. Patterns of integration of viral DNA sequences in the genomes of adenovirus type 12-transformed hamster cells. Cell 1978, 14, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Rosahl, T.; Doerfler, W. Chromosomal localization of integrated adenovirus DNA in productively infected and in transformed mammalian cells. Virology 1986, 152, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, M.L.; Abedi, M.R.; Wixon, J. Gene therapy clinical trials worldwide to 2007--an update. J. Gene Med. 2007, 9, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Benihoud, K.; Yeh, P.; Perricaudet, M. Adenovirus vectors for gene delivery. Curr. Opin. Biotechnol. 1999, 10, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.L.; Kadan, M.J.; Gorziglia, M.I.; Kaleko, M.; Connelly, S. Generation and characterization of E1/E2a/E3/E4-deficient adenoviral vectors encoding human factor VIII. Mol. Ther. 2001, 3, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Lusky, M.; Christ, M.; Rittner, K.; Dieterle, A.; Dreyer, D.; Mourot, B.; Schultz, H.; Stoeckel, F.; Pavirani, A.; Mehtali, M. In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J. Virol. 1998, 72, 2022–2032. [Google Scholar] [PubMed]
- Yang, Y.; Ertl, H.C.; Wilson, J.M. MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity 1994, 1, 433–442. [Google Scholar] [CrossRef]
- Ehrhardt, A.; Kay, M.A. A new adenoviral helper-dependent vector results in long-term therapeutic levels of human coagulation factor IX at low doses in vivo. Blood 2002, 99, 3923–3930. [Google Scholar] [CrossRef] [PubMed]
- Parks, R.J.; Chen, L.; Anton, M.; Sankar, U.; Rudnicki, M.A.; Graham, F.L. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 13565–13570. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, A.P.; Fawcett, P.; Nakai, H.; McCaffrey, R.L.; Ehrhardt, A.; Pham, T.T.; Pandey, K.; Xu, H.; Feuss, S.; Storm, T.A.; Kay, M.A. The host response to adenovirus, helper-dependent adenovirus, and adeno-associated virus in mouse liver. Mol. Ther. 2008, 16, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Schiedner, G.; Hertel, S.; Johnston, M.; Biermann, V.; Dries, V.; Kochanek, S. Variables affecting in vivo performance of high-capacity adenovirus vectors. J. Virol. 2002, 76, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Grove, N.C.; Zuo, Y.; Edwards, R.; Palmer, D.; Cerullo, V.; Teruya, J.; Ng, P. Bioengineered Factor IX Molecules with Increased Catalytic Activity Improve the Therapeutic Index of Gene Therapy Vectors for Hemophilia B. Hum. Gene Ther. 2009, 20, 479–85. [Google Scholar] [CrossRef] [PubMed]
- Barr, D.; Tubb, J.; Ferguson, D.; Scaria, A.; Lieber, A.; Wilson, C.; Perkins, J.; Kay, M.A. Strain related variations in adenovirally mediated transgene expression from mouse hepatocytes in vivo: comparisons between immunocompetent and immunodeficient inbred strains. Gene Ther. 1995, 2, 151–155. [Google Scholar] [PubMed]
- Ehrhardt, A.; Xu, H.; Kay, M.A. Episomal persistence of recombinant adenoviral vector genomes during the cell cycle in vivo. J. Virol. 2003, 77, 7689–7695. [Google Scholar] [CrossRef] [PubMed]
- Soifer, H.S.; Kasahara, N. Retrotransposon-adenovirus hybrid vectors: efficient delivery and stable integration of transgenes via a two-stage mechanism. Curr. Gene Ther. 2004, 4, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Yant, S.R.; Ehrhardt, A.; Mikkelsen, J.G.; Meuse, L.; Pham, T.; Kay, M.A. Transposition from a gutless adeno-transposon vector stabilizes transgene expression in vivo. Nat. Biotechnol. 2002, 20, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, A.; Yant, S.R.; Giering, J.C.; Xu, H.; Engler, J.A.; Kay, M.A. Somatic integration from an adenoviral hybrid vector into a hot spot in mouse liver results in persistent transgene expression levels in vivo. Mol. Ther. 2007, 15, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.J.; Chong, H.; Bell, S.; Diaz, R.M.; Vile, R.G. Novel integrating adenoviral/retroviral hybrid vector for gene therapy. Hum. Gene Ther. 2002, 13, 745–760. [Google Scholar] [PubMed]
- Zheng, C.; Baum, B.J.; Iadarola, M.J.; O'Connell, B.C. Genomic integration and gene expression by a modified adenoviral vector. Nat. Biotechnol. 2000, 18, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.A.; Holkers, M.; Cudre-Mauroux, C.; van Nierop, G.P.; Knaan-Shanzer, S.; van, d. V.; Valerio, D.; de Vries, A.A. Mol. Ther. 2006, 13, 976–986. [CrossRef] [PubMed]
- Goncalves, M.A.; Holkers, M.; van Nierop, G.P.; Wieringa, R.; Pau, M.G.; de Vries, A.A. Targeted chromosomal insertion of large DNA into the human genome by a fiber-modified high-capacity adenovirus-based vector system. PLoS One 2008, 3, e3084. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.; Parks, R.J.; Lamartina, S.; Toniatti, C.; Pieroni, L.; Palombo, F.; Ciliberto, G.; Graham, F.L.; Cortese, R.; La, M.N.; Colloca, S. Site-specific integration mediated by a hybrid adenovirus/adeno-associated virus vector. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Kelley, W.M.; Burda, J.F.; Wilson, J.M. A novel adenovirus-adeno-associated virus hybrid vector that displays efficient rescue and delivery of the AAV genome. Hum. Gene Ther. 1996, 7, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Linden, R.M.; Ward, P.; Giraud, C.; Winocour, E.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 11288–11294. [Google Scholar] [CrossRef] [PubMed]
- Samulski, R.J.; Zhu, X.; Xiao, X.; Brook, J.D.; Housman, D.E.; Epstein, N.; Hunter, L.A. Targeted integration of adeno-associated virus(AAV) into human chromosome 19. EMBO J. 1991, 10, 3941–3950. [Google Scholar] [PubMed]
- Nakai, H.; Montini, E.; Fuess, S.; Storm, T.A.; Grompe, M.; Kay, M.A. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat. Genet. 2003, 34, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Nakai, H.; Thomas, C.E.; Storm, T.A.; Fuess, S.; Powell, S.; Wright, J.F.; Kay, M.A. A limited number of transducible hepatocytes restricts a wide-range linear vector dose response in recombinant adeno-associated virus-mediated liver transduction. J. Virol. 2002, 76, 11343–11349. [Google Scholar] [CrossRef] [PubMed]
- Lieber, A.; Steinwaerder, D.S.; Carlson, C.A.; Kay, M.A. Integrating adenovirus-adeno-associated virus hybrid vectors devoid of all viral genes. J. Virol. 1999, 73, 9314–9324. [Google Scholar] [PubMed]
- Sandalon, Z.; Gnatenko, D.V.; Bahou, W.F.; Hearing, P. Adeno-associated virus(AAV) Rep protein enhances the generation of a recombinant mini-adenovirus(Ad) utilizing an Ad/AAV hybrid virus. J. Virol. 2000, 74, 10381–10389. [Google Scholar] [CrossRef] [PubMed]
- Shayakhmetov, D.M.; Carlson, C.A.; Stecher, H.; Li, Q.; Stamatoyannopoulos, G.; Lieber, A. A high-capacity, capsid-modified hybrid adenovirus/adeno-associated virus vector for stable transduction of human hematopoietic cells. J. Virol. 2002, 76, 1135–1143. [Google Scholar] [PubMed]
- Goncalves, M.A.; Pau, M.G.; de Vries, A.A.; Valerio, D. Generation of a high-capacity hybrid vector: packaging of recombinant adenoassociated virus replicative intermediates in adenovirus capsids overcomes the limited cloning capacity of adenoassociated virus vectors. Virology 2001, 288, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.A.; van, d. V.; Janssen, J.M.; Maassen, B.T.; Heemskerk, E.H.; Opstelten, D.J.; Knaan-Shanzer, S.; Valerio, D.; de Vries, A.A. J. Virol. 2002, 76, 10734–10744. [CrossRef] [PubMed]
- Recchia, A.; Perani, L.; Sartori, D.; Olgiati, C.; Mavilio, F. Site-specific integration of functional transgenes into the human genome by adeno/AAV hybrid vectors. Mol. Ther. 2004, 10, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Izsvak, Z. Transposons for gene therapy! Curr. Gene Ther. 2006, 6, 593–607. [Google Scholar] [CrossRef]
- Yant, S.R.; Wu, X.; Huang, Y.; Garrison, B.; Burgess, S.M.; Kay, M.A. High-resolution genome-wide mapping of transposon integration in mammals. Mol. Cell Biol. 2005, 25, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, J.G.; Yant, S.R.; Meuse, L.; Huang, Z.; Xu, H.; Kay, M.A. Helper-Independent Sleeping Beauty transposon-transposase vectors for efficient nonviral gene delivery and persistent gene expression in vivo. Mol. Ther. 2003, 8, 654–665. [Google Scholar] [CrossRef]
- Yant, S.R.; Meuse, L.; Chiu, W.; Ivics, Z.; Izsvak, Z.; Kay, M.A. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat. Genet. 2000, 25, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Mates, L.; Chuah, M.K.; Belay, E.; Jerchow, B.; Manoj, N.; costa-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; Ma, L.; Samara-Kuko, E.; Gysemans, C.; Pryputniewicz, D.; Miskey, C.; Fletcher, B.; Vandendriessche, T.; Ivics, Z.; Izsvak, Z. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Kuhstoss, S.; Rao, R.N. Analysis of the integration function of the streptomycete bacteriophage phi C31. J. Mol. Biol. 1991, 222, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Chalberg, T.W.; Portlock, J.L.; Olivares, E.C.; Thyagarajan, B.; Kirby, P.J.; Hillman, R.T.; Hoelters, J.; Calos, M.P. Integration specificity of phage PhiC31 integrase in the human genome. J Mol. Biol. 2006, 357, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, A.; Engler, J.A.; Xu, H.; Cherry, A.M.; Kay, M.A. Molecular analysis of chromosomal rearrangements in mammalian cells after PhiC31-mediated integration. Hum. Gene Ther. 2006, 17, 1077–1094. [Google Scholar] [CrossRef] [PubMed]
- Olivares, E.C.; Hollis, R.P.; Chalberg, T.W.; Meuse, L.; Kay, M.A.; Calos, M.P. Site-specific genomic integration produces therapeutic Factor IX levels in mice. Nat. Biotechnol. 2002, 20, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Thyagarajan, B.; Olivares, E.C.; Hollis, R.P.; Ginsburg, D.S.; Calos, M.P. Site-specific genomic integration in mammalian cells mediated by phage PhiC31 integrase. Mol. Cell Biol. 2001, 21, 3926–3934. [Google Scholar] [CrossRef] [PubMed]
- Soifer, H.; Higo, C.; Kazazian Jr., H.H.; Moran, J.V.; Mitani, K.; Kasahara, N. Stable integration of transgenes delivered by a retrotransposon-adenovirus hybrid vector. Hum. Gene Ther. 2001, 12, 1417–1428. [Google Scholar] [PubMed]
- Kubo, S.; Seleme, M.C.; Soifer, H.S.; Perez, J.L.; Moran, J.V.; Kazazian Jr., H.H.; Kasahara, N. L1 retrotransposition in nondividing and primary human somatic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8036–8041. [Google Scholar] [CrossRef]
- Caplen, N.J.; Higginbotham, J.N.; Scheel, J.R.; Vahanian, N.; Yoshida, Y.; Hamada, H.; Blaese, R.M.; Ramsey, W.J. Adeno-retroviral chimeric viruses as in vivo transducing agents. Gene Ther. 1999, 6, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Duisit, G.; Salvetti, A.; Moullier, P.; Cosset, F.L. Functional characterization of adenoviral/retroviral chimeric vectors and their use for efficient screening of retroviral producer cell lines. Hum. Gene Ther. 1999, 10, 189–200. [Google Scholar] [PubMed]
- Feng, M.; Jackson Jr., W.H.; Goldman, C.K.; Rancourt, C.; Wang, M.; Dusing, S.K.; Siegal, G.; Curiel, D.T. Stable in vivo gene transduction via a novel adenoviral/retroviral chimeric vector. Nat. Biotechnol. 1997, 15, 866–870. [Google Scholar] [CrossRef] [PubMed]
- Picard-Maureau, M.; Kreppel, F.; Lindemann, D.; Juretzek, T.; Herchenroder, O.; Rethwilm, A.; Kochanek, S.; Heinkelein, M. Foamy virus--adenovirus hybrid vectors. Gene Ther. 2004, 11, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Nawa, A.; Luo, C.; Zhang, L.; Ushjima, Y.; Ishida, D.; Kamakura, M.; Fujimoto, Y.; Goshima, F.; Kikkawa, F.; Nishiyama, Y. Non-engineered, naturally oncolytic herpes simplex virus HSV1 HF-10: applications for cancer gene therapy. Curr. Gene Ther. 2008, 8, 208–221. [Google Scholar] [CrossRef]
- Spaete, R.R.; Frenkel, N. The herpes simplex virus amplicon: a new eucaryotic defective-virus cloning-amplifying vector. Cell 1982, 30, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Sena-Esteves, M.; Saeki, Y.; Fraefel, C.; Breakefield, X.O. HSV-1 amplicon vectors--simplicity and versatility. Mol. Ther. 2000, 2, 9–15. [Google Scholar] [CrossRef]
- Wade-Martins, R.; Smith, E.R.; Tyminski, E.; Chiocca, E.A.; Saeki, Y. An infectious transfer and expression system for genomic DNA loci in human and mouse cells. Nat. Biotechnol. 2001, 19, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.L. HSV-1-derived amplicon vectors: recent technological improvements and remaining difficulties--a review. Mem. Inst. Oswaldo Cruz 2009, 104, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Heister, T.; Heid, I.; Ackermann, M.; Fraefel, C. Herpes simplex virus type 1/adeno-associated virus hybrid vectors mediate site-specific integration at the adeno-associated virus preintegration site, AAVS1, on human chromosome 19. J. Virol. 2002, 76, 7163–7173. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Camp, S.M.; Niwano, M.; Shen, X.; Bakowska, J.C.; Breakefield, X.O.; Allen, P.D. Herpes simplex virus type 1/adeno-associated virus rep(+) hybrid amplicon vector improves the stability of transgene expression in human cells by site-specific integration. J. Virol. 2002, 76, 7150–7162. [Google Scholar] [CrossRef] [PubMed]
- Johnston, K.M.; Jacoby, D.; Pechan, P.A.; Fraefel, C.; Borghesani, P.; Schuback, D.; Dunn, R.J.; Smith, F.I.; Breakefield, X.O. HSV/AAV hybrid amplicon vectors extend transgene expression in human glioma cells. Hum. Gene Ther. 1997, 8, 359–370. [Google Scholar] [CrossRef]
- Cortes, M.L.; Oehmig, A.; Saydam, O.; Sanford, J.D.; Perry, K.F.; Fraefel, C.; Breakefield, X.O. Targeted integration of functional human ATM cDNA into genome mediated by HSV/AAV hybrid amplicon vector. Mol. Ther. 2008, 16, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jeppesen, I.; Nielsen, K.; Jensen, T.G. Phi c31 integrase induces chromosomal aberrations in primary human fibroblasts. Gene Ther. 2006, 13, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Oehmig, A.; Cortes, M.L.; Perry, K.F.; Sena-Esteves, M.; Fraefel, C.; Breakefield, X.O. Integration of active human beta-galactosidase gene (100 kb) into genome using HSV/AAV amplicon vector. Gene Ther. 2007, 14, 1078–1091. [Google Scholar] [CrossRef] [PubMed]
- Bowers, W.J.; Mastrangelo, M.A.; Howard, D.F.; Southerland, H.A.; Maguire-Zeiss, K.A.; Federoff, H.J. Neuronal precursor-restricted transduction via in utero CNS gene delivery of a novel bipartite HSV amplicon/transposase hybrid vector. Mol. Ther. 2006, 13, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Savard, N.; Cosset, F.L.; Epstein, A.L. Defective herpes simplex virus type 1 vectors harboring gag, pol, and env genes can be used to rescue defective retrovirus vectors. J. Virol. 1997, 71, 4111–4117. [Google Scholar] [PubMed]
- Sena-Esteves, M.; Saeki, Y.; Camp, S.M.; Chiocca, E.A.; Breakefield, X.O. Single-step conversion of cells to retrovirus vector producers with herpes simplex virus-Epstein-Barr virus hybrid amplicons. J. Virol. 1999, 73, 10426–10439. [Google Scholar] [PubMed]
- de Felipe, P.; Izquierdo, M.; Wandosell, F.; Lim, F. Integrating retroviral cassette extends gene delivery of HSV-1 expression vectors to dividing cells. Biotechniques 2001, 31, 394–395. [Google Scholar] [PubMed]
- Blaese, R.M.; Culver, K.W.; Miller, A.D.; Carter, C.S.; Fleisher, T.; Clerici, M.; Shearer, G.; Chang, L.; Chiang, Y.; Tolstoshev, P.; Greenblatt, J.J.; Rosenberg, S.A.; Klein, H.; Berger, M.; Mullen, C.A.; Ramsey, W.J.; Muul, L.; Morgan, R.A.; Anderson, W.F. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science 1995, 270, 475–480. [Google Scholar] [PubMed]
- Arrigo, S.J.; Weitsman, S.; Zack, J.A.; Chen, I.S. Characterization and expression of novel singly spliced RNA species of human immunodeficiency virus type 1. J. Virol. 1990, 64, 4585–4588. [Google Scholar] [PubMed]
- Coffin, J.M. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol. 1979, 42, 1–26. [Google Scholar] [CrossRef]
- Coffin, J.M. Retroviral DNA integration. Dev. Biol. Stand. 1992, 76, 141–151. [Google Scholar] [PubMed]
- Daniel, R.; Katz, R.A.; Skalka, A.M. A role for DNA-PK in retroviral DNA integration. Science 1999, 284, 644–647. [Google Scholar] [CrossRef] [PubMed]
- Skalka, A.M.; Katz, R.A. Retroviral DNA integration and the DNA damage response. Cell Death Differ. 2005, 12, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Daniel, R. Following the path of the virus: the exploitation of host DNA repair mechanisms by retroviruses. ACS Chem. Biol. 2006, 1, 217–226. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Beitzel, B.F.; Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS. Biol. 2004, 2, E234. [Google Scholar] [CrossRef] [PubMed]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS. Pathog. 2006, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Crise, B.; Burgess, S.M. Transcription start regions in the human genome are favored targets for MLV integration. Science 2003, 300, 1749–1751. [Google Scholar] [CrossRef] [PubMed]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Derse, D.; Crise, B.; Li, Y.; Princler, G.; Lum, N.; Stewart, C.; McGrath, C.F.; Hughes, S.H.; Munroe, D.J.; Wu, X. Human T-cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses. J. Virol. 2007, 81, 6731–6741. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von, K.C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; Sorensen, R.; Forster, A.; Fraser, P.; Cohen, J. I.; de Saint, B.G.; Alexander, I.; Wintergerst, U.; Frebourg, T.; Aurias, A.; Stoppa-Lyonnet, D.; Romana, S.; Radford-Weiss, I.; Gross, F.; Valensi, F.; Delabesse, E.; Macintyre, E.; Sigaux, F.; Soulier, J.; Leiva, L.E.; Wissler, M.; Prinz, C.; Rabbitts, T.H.; Le, D.F.; Fischer, A.; Cavazzana-Calvo, M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dullmann, J.; Schiedlmeier, B.; Schmidt, M.; Von, K.C.; Meyer, J.; Forster, M.; Stocking, C.; Wahlers, A.; Frank, O.; Ostertag, W.; Kuhlcke, K.; Eckert, H.G.; Fehse, B.; Baum, C. Murine leukemia induced by retroviral gene marking. Science 2002, 296, 497. [Google Scholar] [CrossRef] [PubMed]
- Tomanin, R.; Scarpa, M. Why do we need new gene therapy viral vectors? Characteristics, limitations and future perspectives of viral vector transduction. Curr. Gene Ther. 2004, 4, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D. Tethering human immunodeficiency virus 1 integrase to a DNA site directs integration to nearby sequences. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 9233–9237. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D.; Miller, M.D. Tethering human immunodeficiency virus type 1 preintegration complexes to target DNA promotes integration at nearby sites. J. Virol. 1997, 71, 458–464. [Google Scholar] [PubMed]
- Goulaouic, H.; Chow, S.A. Directed integration of viral DNA mediated by fusion proteins consisting of human immunodeficiency virus type 1 integrase and Escherichia coli LexA protein. J. Virol. 1996, 70, 37–46. [Google Scholar] [PubMed]
- Katz, R.A.; Merkel, G.; Skalka, A.M. Targeting of retroviral integrase by fusion to a heterologous DNA binding domain: in vitro activities and incorporation of a fusion protein into viral particles. Virology 1996, 217, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Dong, Z.; Wilkinson, T.A.; Barbas III, C.F.; Chow, S.A. Human immunodeficiency virus type 1 incorporated with fusion proteins consisting of integrase and the designed polydactyl zinc finger protein E2C can bias integration of viral DNA into a predetermined chromosomal region in human cells. J. Virol. 2006, 80, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Segal, D.J.; Dreier, B.; Barbas III, C.F. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 14628–14633. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Dreier, B.; Barbas III, C.F. Positive and negative regulation of endogenous genes by designed transcription factors. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Vargas Jr., J.; Gusella, G.L.; Najfeld, V.; Klotman, M.E.; Cara, A. Novel integrase-defective lentiviral episomal vectors for gene transfer. Hum. Gene Ther. 2004, 15, 361–372. [Google Scholar] [PubMed]
- Moldt, B.; Staunstrup, N.H.; Jakobsen, M.; Yanez-Munoz, R.J.; Mikkelsen, J.G. Genomic insertion of lentiviral DNA circles directed by the yeast Flp recombinase. BMC. Biotechnol. 2008, 8, 60. [Google Scholar] [CrossRef]
- Nightingale, S.J.; Hollis, R.P.; Pepper, K.A.; Petersen, D.; Yu, X.J.; Yang, C.; Bahner, I.; Kohn, D.B. Transient gene expression by nonintegrating lentiviral vectors. Mol. Ther. 2006, 13, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, A.; Genovese, P.; Beausejour, C.M.; Colleoni, S.; Lee, Y.L.; Kim, K.A.; Ando, D.; Urnov, F.D.; Galli, C.; Gregory, P.D.; Holmes, M.C.; Naldini, L. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007, 25, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Bitinaite, J.; Wah, D.A.; Aggarwal, A.K.; Schildkraut, I. FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 10570–10575. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; Thibodeau-Beganny, S.; Guhl, E.; Alwin, S.; Eichtinger, M.; Joung, J.K.; Cathomen, T. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol. Ther. 2008, 16, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.; Connor, B.; Carney, J.P. Mechanisms of eukaryotic DNA double strand break repair. Front Biosci. 2006, 11, 1958–1976. [Google Scholar] [CrossRef] [PubMed]
- Staunstrup, N.H.; Moldt, B.; Mates, L.; Villesen, P.; Jakobsen, M.; Ivics, Z.; Izsvak, Z.; Mikkelsen, J.G. Hybrid lentivirus-transposon vectors with a random integration profile in human cells. Mol. Ther. 2009, 17, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
- Vink, C.A.; Gaspar, H.B.; Gabriel, R.; Schmidt, M.; McIvor, R.S.; Thrasher, A.J.; Qasim, W. Sleeping beauty transposition from nonintegrating lentivirus. Mol. Ther. 2009, 17, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.A. Sleeping beauty vector system moves toward human trials in the United States. Mol. Ther. 2008, 16, 1515–1516. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Skjorringe, T.; Gjetting, T.; Jensen, T.G. PhiC31 integrase induces a DNA damage response and chromosomal rearrangements in human adult fibroblasts. BMC. Biotechnol. 2009, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Alwin, S.; Gere, M.B.; Guhl, E.; Effertz, K.; Barbas III, C.F.; Segal, D.J.; Weitzman, M.D.; Cathomen, T. Custom zinc-finger nucleases for use in human cells. Mol. Ther. 2005, 12, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003, 300, 763. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; Gregory, P.D.; Pabo, C.O.; Rebar, E.J. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Szczepek, M.; Brondani, V.; Buchel, J.; Serrano, L.; Segal, D.J.; Cathomen, T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 2007, 25, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, H.; Gomos, J.; Berns, K.I.; Falck-Pedersen, E. Adeno-associated virus site-specific integration and AAVS1 disruption. J. Virol. 2004, 78, 7874–7882. [Google Scholar] [CrossRef] [PubMed]
- Tan, I.; Ng, C.H.; Lim, L.; Leung, T. Phosphorylation of a novel myosin binding subunit of protein phosphatase 1 reveals a conserved mechanism in the regulation of actin cytoskeleton. J. Biol. Chem. 2001, 276, 21209–21216. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Miller, A.D.; McNamara, S.; Emerson, J.; Gibson, R.L.; Ramsey, B.; Aitken, M.L. Prevalence of neutralizing antibodies against adeno-associated virus(AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: Implications for gene therapy using AAV vectors. Hum. Gene Ther. 2006, 17, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Antoni, B.A.; Rabson, A.B.; Miller, I.L.; Trempe, J.P.; Chejanovsky, N.; Carter, B.J. Adeno-associated virus Rep protein inhibits human immunodeficiency virus type 1 production in human cells. J. Virol. 1991, 65, 396–404. [Google Scholar] [PubMed]
- Schmidt, M.; Afione, S.; Kotin, R.M. Adeno-associated virus type 2 Rep78 induces apoptosis through caspase activation independently of p53. J. Virol. 2000, 74, 9441–9450. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chen, F.; Trempe, J.P. Characterization of cell lines that inducibly express the adeno-associated virus Rep proteins. J. Virol. 1994, 68, 4847–4856. [Google Scholar] [PubMed]
- Modlich, U.; Bohne, J.; Schmidt, M.; Von, K.C.; Knoss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef] [PubMed]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi, S.L.; Benedicenti, F.; Ambrosi, A.; Di, S.C.; Doglioni, C.; Von, K.C.; Naldini, L. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Allocca, M.; Doria, M.; Petrillo, M.; Colella, P.; Garcia-Hoyos, M.; Gibbs, D.; Kim, S.R.; Maguire, A.; Rex, T.S.; Di, V.U.; Cutillo, L.; Sparrow, J.R.; Williams, D.S.; Bennett, J.; Auricchio, A. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J. Clin. Invest. 2008, 118, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; Meng, X.; Miller, J.C.; Zhang, L.; Rebar, E.J.; Gregory, P.D.; Urnov, F.D.; Jaenisch, R. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Katzer, A.; Stuwe, E.E.; Fiedler, D.; Knespel, S.; Izsvak, Z. Targeted Sleeping Beauty transposition in human cells. Mol. Ther. 2007, 15, 1137–1144. [Google Scholar] [PubMed]
- Yant, S.R.; Huang, Y.; Akache, B.; Kay, M.A. Site-directed transposon integration in human cells. Nucleic Acids Res. 2007, 35, e50. [Google Scholar] [CrossRef] [PubMed]
- Keravala, A.; Lee, S.; Thyagarajan, B.; Olivares, E.C.; Gabrovsky, V.E.; Woodard, L.E.; Calos, M.P. Mutational derivatives of PhiC31 integrase with increased efficiency and specificity. Mol. Ther. 2009, 17, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Naldini, L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet. 2009, 10, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, C.J.; Muhlebach, M.D.; Cichutek, K. Lentiviral vectors with measles virus glycoproteins - dream team for gene transfer? Trends Biotechnol. 2009, 27, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Frampton Jr., A.R.; Goins, W.F.; Nakano, K.; Burton, E.A.; Glorioso, J.C. HSV trafficking and development of gene therapy vectors with applications in the nervous system. Gene Ther. 2005, 12, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Schaffer, D.V. Designer gene delivery vectors: molecular engineering and evolution of adeno-associated viral vectors for enhanced gene transfer. Pharm. Res. 2008, 25, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Van, V.K.; Mohiuddin, Y.; McClung, S.; Blouin, V.; Rolling, F.; Moullier, P.; gbandje-McKenna, M.; Snyder, R.O. Adeno-associated virus capsid serotype identification: Analytical methods development and application. J. Virol. Methods 2009, 159, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Schaffer, D.V. Advanced targeting strategies for murine retroviral and adeno-associated viral vectors. Adv. Biochem. Eng. Biotechnol. 2005, 99, 147–167. [Google Scholar] [PubMed]
- Costantini, L.C.; Jacoby, D.R.; Wang, S.; Fraefel, C.; Breakefield, X.O.; Isacson, O. Gene transfer to the nigrostriatal system by hybrid herpes simplex virus/adeno- associated virus amplicon vectors. Hum Gene Ther. 1999, 10, 2441–2443. [Google Scholar] [PubMed]
- Ueno, T.; Matsumura, H.; Tanaka, K.; Iwasaki, T.; Ueno, M.; Fujinaga, K.; Asada, K.; Kato, I. Site-specific integration of a transgene mediated by a hybrid adenovirus/adeno-associated virus vector using the Cre/loxP-expression-switching system. Biochem. Biophys. Res. Commun. 2000, 273, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Broberg, E.K.; Hukkanen, V. Immune response to herpes simplex virus and gamma134.5 deleted HSV vectors. Curr. Gene Ther. 2005, 5, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Follenzi, A.; Santambrogio, L.; Annoni, A. Immune responses to lentiviral vectors. Curr. Gene Ther. 2007, 7, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.P.; Cerullo, V.; Lee, B. Immune response to helper dependent adenoviral mediated liver gene therapy: challenges and prospects. Curr. Gene Ther. 2007, 7, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Yant, S.R.; Park, J.; Huang, Y.; Mikkelsen, J.G.; Kay, M.A. Mutational analysis of the N-terminal DNA-binding domain of sleeping beauty transposase: critical residues for DNA binding and hyperactivity in mammalian cells. Mol. Cell Biol. 2004, 24, 9239–9247. [Google Scholar] [CrossRef] [PubMed]
- Baum, C.; Von, K.C.; Staal, F.J.; Li, Z.; Fehse, B.; Schmidt, M.; Weerkamp, F.; Karlsson, S.; Wagemaker, G.; Williams, D.A. Chance or necessity? Insertional mutagenesis in gene therapy and its consequences. Mol. Ther. 2004, 9, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Walisko, O.; Schorn, A.; Rolfs, F.; Devaraj, A.; Miskey, C.; Izsvak, Z.; Ivics, Z. Transcriptional activities of the Sleeping Beauty transposon and shielding its genetic cargo with insulators. Mol. Ther. 2008, 16, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Kuate, S.; Stefanou, D.; Hoffmann, D.; Wildner, O.; Uberla, K. Production of lentiviral vectors by transient expression of minimal packaging genes from recombinant adenoviruses. J. Gene Med. 2004, 6, 1197–1205. [Google Scholar] [CrossRef]
- Kubo, S.; Mitani, K. A new hybrid system capable of efficient lentiviral vector production and stable gene transfer mediated by a single helper-dependent adenoviral vector. J. Virol. 2003, 77, 2964–2971. [Google Scholar] [CrossRef] [PubMed]
- Soifer, H.; Higo, C.; Logg, C.R.; Jih, L.J.; Shichinohe, T.; Harboe-Schmidt, E.; Mitani, K.; Kasahara, N. A novel, helper-dependent, adenovirus-retrovirus hybrid vector: stable transduction by a two-stage mechanism. Mol. Ther. 2002, 5, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Geurts, A.M.; Yang, Y.; Clark, K.J.; Liu, G.; Cui, Z.; Dupuy, A.J.; Bell, J.B.; Largaespada, D.A.; Hackett, P.B. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol. Ther. 2003, 8, 108–117. [Google Scholar] [CrossRef]
- Robert, M.A.; Benoit, R.; Desfosse, L.; Mairey, E.; Tremblay, J.P. ; Massie, B.; Gilbert, R. Integration site mapping of linear and circular gutless adenovirus mediated by the phage phiC31 integrase. Abstract book of American Society of Gene Therapy S46.
- Niemeyer, G.P.; Herzog, R.W.; Mount, J.; Arruda, V.R.; Tillson, D.M.; Hathcock, J.; van Ginkel, F.W.; High, K.A.; Lothrop, C.D. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood 2009, 113, 797–806. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Müther, N.; Noske, N.; Ehrhardt, A. Viral Hybrid Vectors for Somatic Integration - Are They the Better Solution? Viruses 2009, 1, 1295-1324. https://doi.org/10.3390/v1031295
Müther N, Noske N, Ehrhardt A. Viral Hybrid Vectors for Somatic Integration - Are They the Better Solution? Viruses. 2009; 1(3):1295-1324. https://doi.org/10.3390/v1031295
Chicago/Turabian StyleMüther, Nadine, Nadja Noske, and Anja Ehrhardt. 2009. "Viral Hybrid Vectors for Somatic Integration - Are They the Better Solution?" Viruses 1, no. 3: 1295-1324. https://doi.org/10.3390/v1031295
APA StyleMüther, N., Noske, N., & Ehrhardt, A. (2009). Viral Hybrid Vectors for Somatic Integration - Are They the Better Solution? Viruses, 1(3), 1295-1324. https://doi.org/10.3390/v1031295