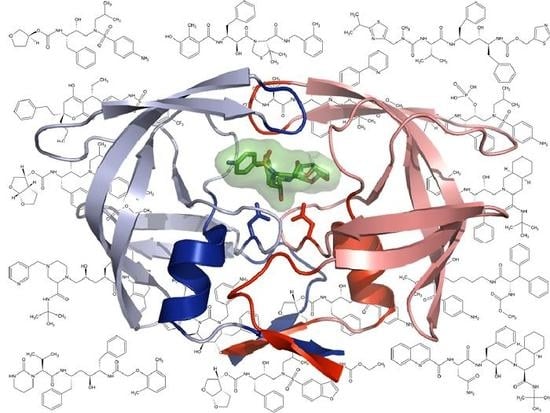
Current and Novel Inhibitors of HIV Protease

Abstract
Abbreviations
1.Introduction

{kind=link}
{kind=link}
{kind=link}
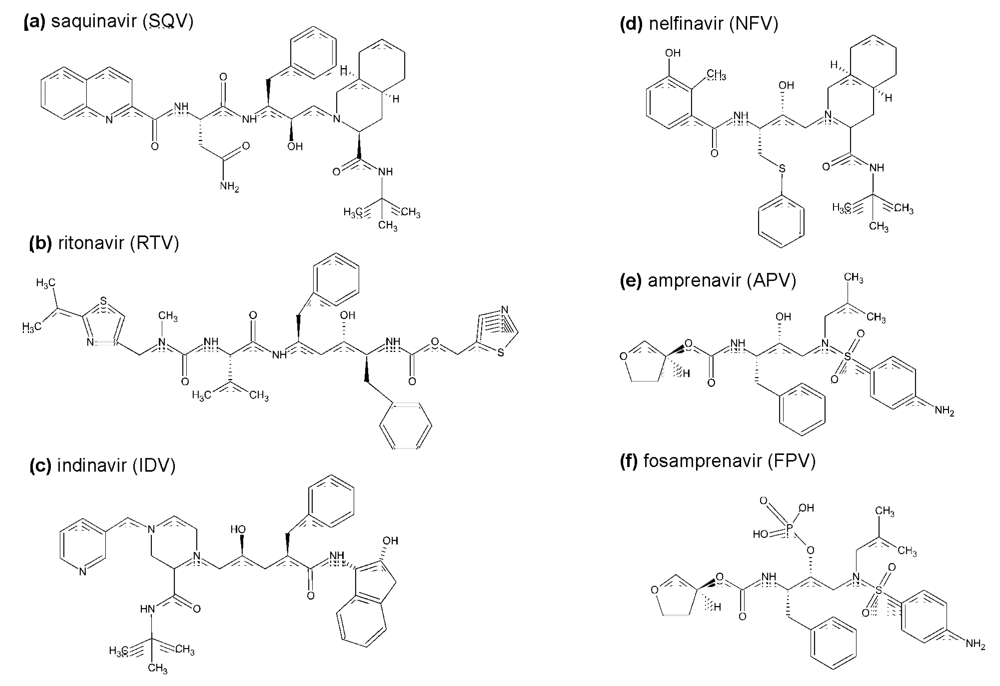{kind=link}
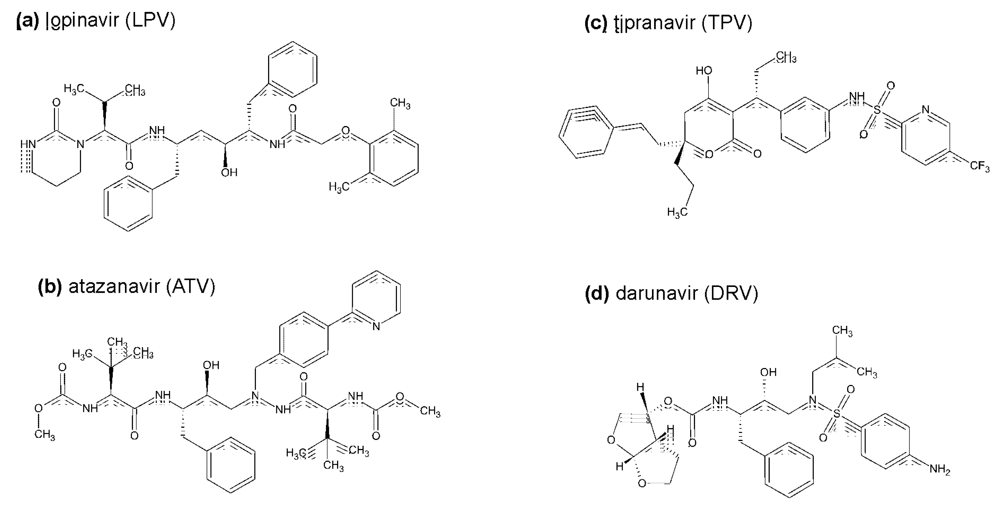{kind=link}
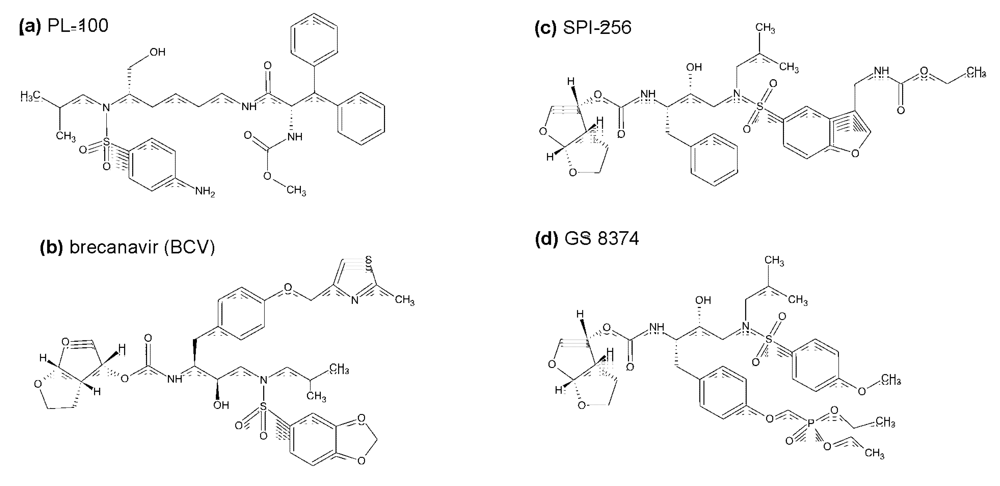{kind=link}
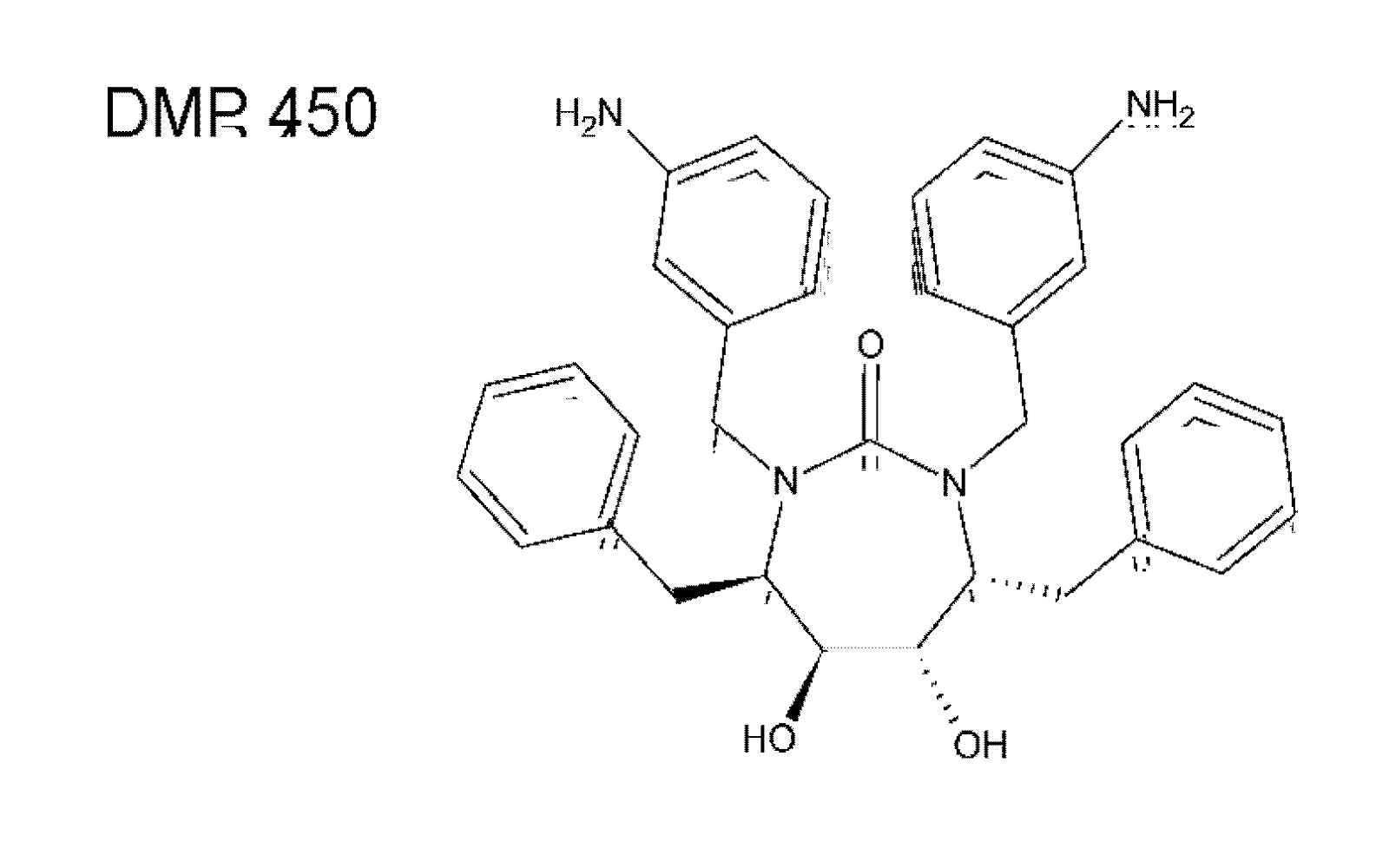{kind=link}
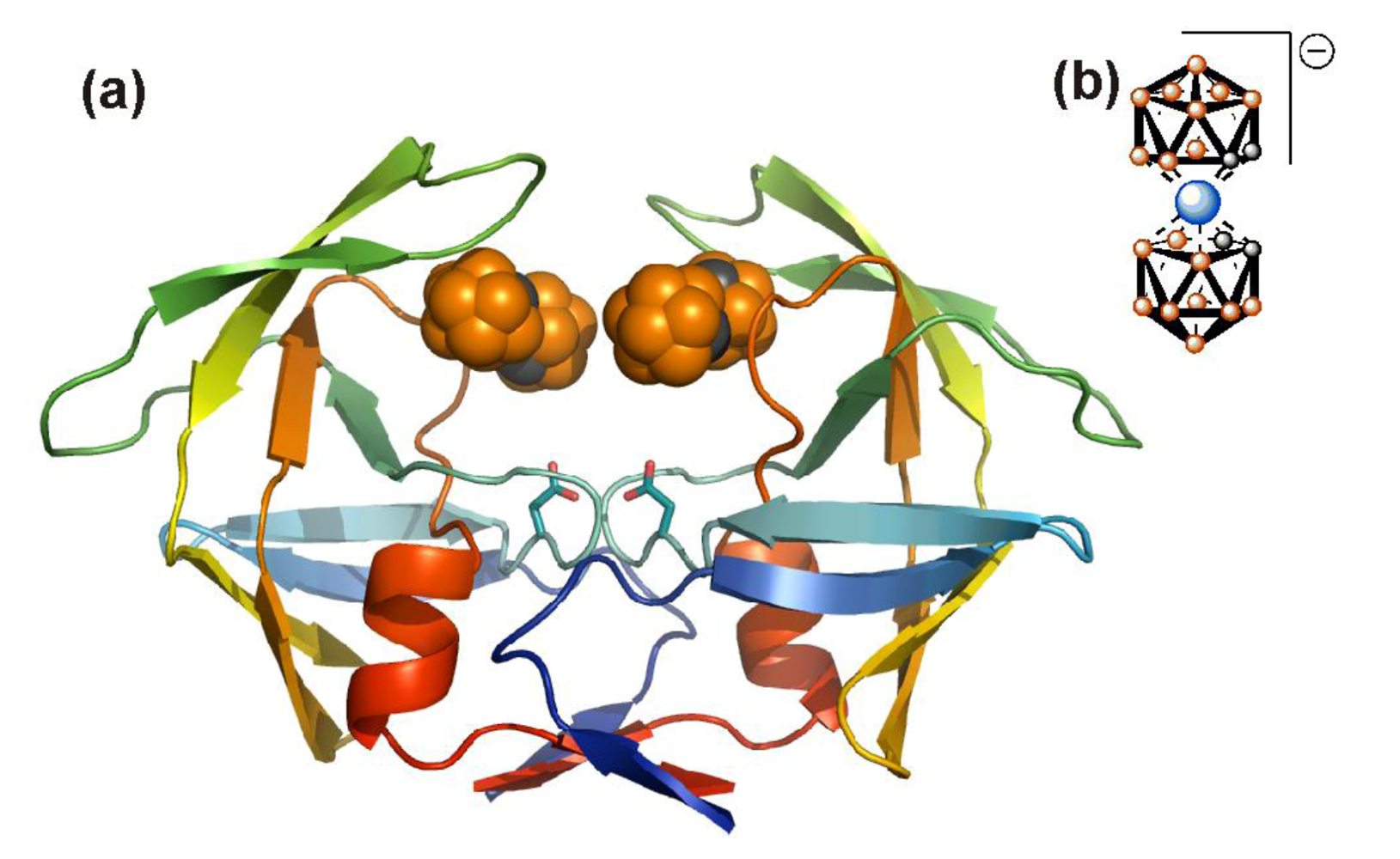{kind=link}
{kind=link}
{kind=link}
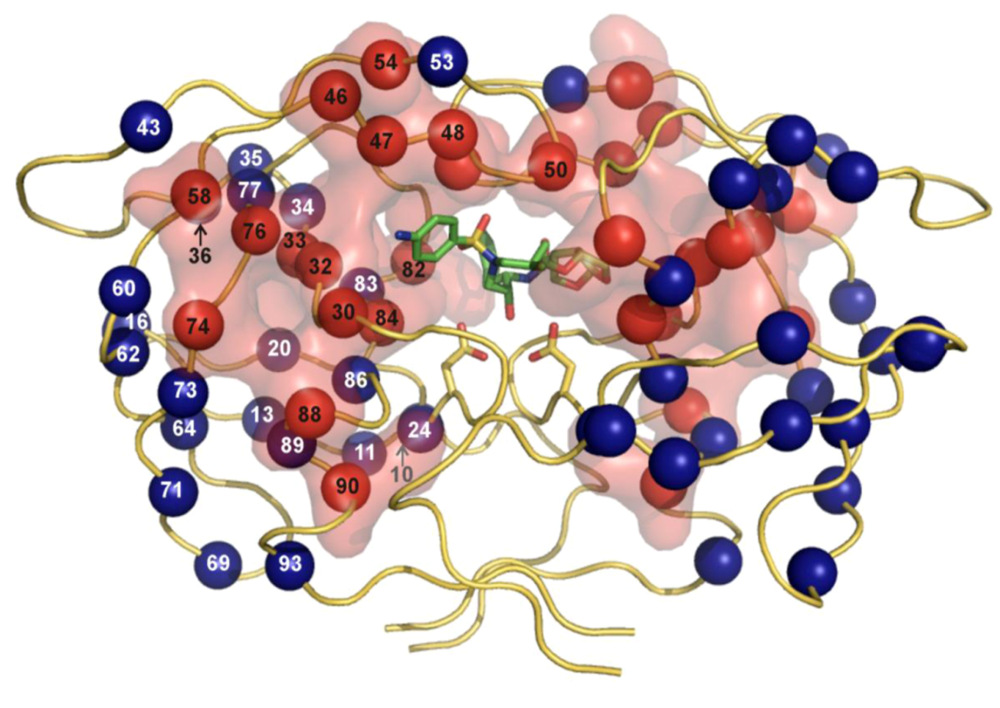
| PIb | Major mutationsc | Minor mutationsd |
|---|---|---|
| Atazanavir +/- ritonavir | 50, 84, 88 | 10, 16, 20, 24, 32, 33, 34, 36, 46, 48, 53, 54, 60, 62, 64, 71, 73, 82, 85, 90, 93 |
| Darunavire | 50, 54, 76, 84 | 11, 32, 33, 47, 74, 89 |
| Fosamprenavire | 50, 84 | 10, 32, 46, 47, 54, 73, 76, 82, 90 |
| Indinavire | 46, 82, 84 | 10, 20, 24, 32, 36, 54, 71, 73, 76, 77, 90 |
| Lopinavire | 32, 47, 82 | 10, 20, 24, 33, 46, 50, 53, 54, 63, 71, 73, 76, 84, 90 |
| Nelfinavir | 30, 90 | 10, 36, 46, 71, 77, 82, 84, 88 |
| Saquinavire | 48, 90 | 10, 24, 54, 62, 71, 73, 77, 82, 84 |
| Tipranavire | 33, 47, 58, 74, 82, 84 | 10, 13, 20, 35, 36, 43, 46, 54, 69, 83, 90 |
2. Inhibitors currently used in clinical practice: structure, antiviral activity, side-effects, and pharmaceutical boosting
| Generic name and abbreviation | Commonly recommended dosage | Most common side effects and off-target activities | Position in the present therapeutic arsenal |
|---|---|---|---|
| Ritonavir (RTV)a | 100–200 mg p.o. BID as pharmacokinetic booster of various PIs (original pharmacodynamic dose was 600 mg p.o. BID) | nausea, diarrhea, abdomenalgia, hyperlipidemia, lipodystrophy syndrome, inhibition of the cytochrome P450 3A4 | practically only pharmacoenhancing of various PIs |
| Saquinavir (SQV) | 1000 mg + RTV 100 mg p.o. BID | diarrhea, hyperlipidemia, lipodystrophy syndrome | second-line HAART therapy |
| Indinavir (IDV) | 800 mg + RTV 100 mg p.o. BID | nephrolithiasis, lipodystrophy syndrome, hyperlipideamia, hepatotoxicity | second/third-line HAART therapy in case of resistance or intolerance |
| Nelfinavir (NFV) | 1250 mg p.o. BID | diarrhea, hyperlipidemia, lipodystrophy syndrome | second/third-line HAART therapy in case of resistance or intolerance, approved for therapy of children |
| Lopinavir (LPV/r) b | 400 mg + RTV 100 mg p.o. BID | diarrhea, hyperlipideamia, lipodystrophy syndrome | first line option for PI based HAART regimen |
| Amprenavir (APV) | 600 mg + RTV 100 mg p.o. BID | diarrhea, toxoallergic rash, hyperlipidemia, lipodystrophy syndrome | replaced by its prodrug fos-amprenavir |
| Generic name and abbreviation | Commonly recommended dosage | Most common side effects and off-target activities | Position in the present therapeutic arsenal |
| Ritonavir (RTV)a | 100–200 mg p.o. BID as pharmacokinetic booster of various PIs (original pharmacodynamic dose was 600 mg p.o. BID) | nausea, diarrhea, abdomenalgia, hyperlipidemia, lipodystrophy syndrome, inhibition of the cytochrome P450 3A4 | practically only pharmacoenhancing of various PIs |
| Saquinavir (SQV) | 1000 mg + RTV 100 mg p.o. BID | diarrhea, hyperlipidemia,lipodystrophy syndrome | second-line HAART therapy |
| Indinavir (IDV) | 800 mg + RTV 100 mg p.o. BID | nephrolithiasis, lipodystrophy syndrome, hyperlipideamia, hepatotoxicity | second/third-line HAART therapy in case of resistance or intolerance |
| Nelfinavir (NFV) | 1250 mg p.o. BID | diarrhea, hyperlipidemia, lipodystrophy syndrome | second/third-line HAART therapy in case of resistance or intolerance, approved for therapy of children |
| Lopinavir (LPV/r) b | 400 mg + RTV 100 mg p.o. BID | diarrhea, hyperlipideamia, lipodystrophy syndrome | first line option for PI based HAART regimen |
| Amprenavir (APV) | 600 mg + RTV 100 mg p.o. BID | diarrhea, toxoallergic rash, hyperlipidemia, lipodystrophy syndrome | replaced by its prodrug fos-amprenavir |
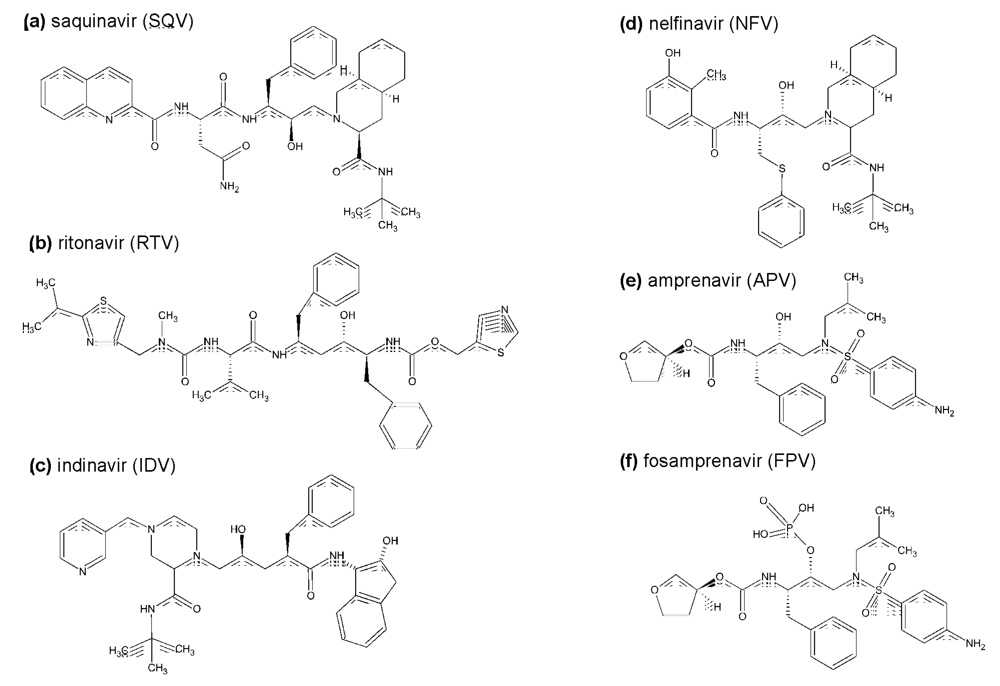
2.1. The first generation HIV protease inhibitors
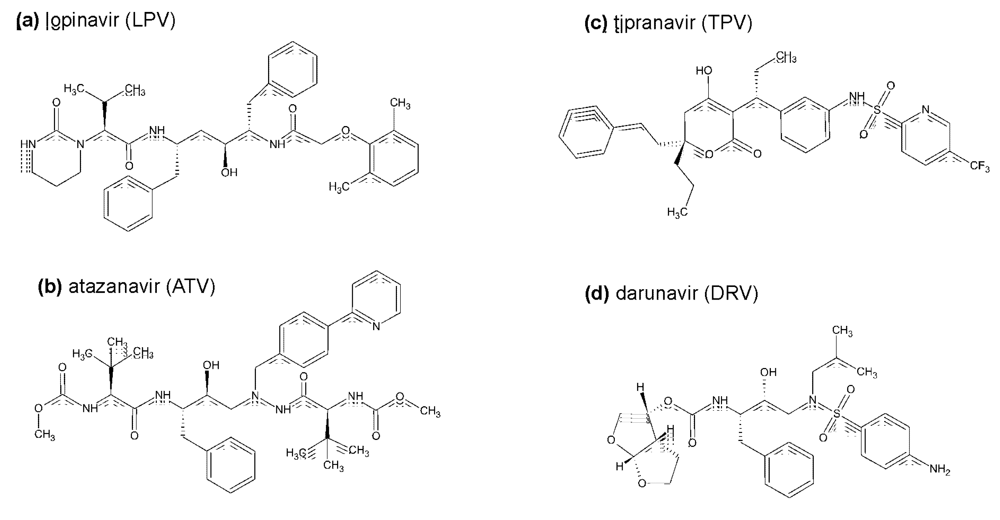
2.2. The second generation HIV protease inhibitors
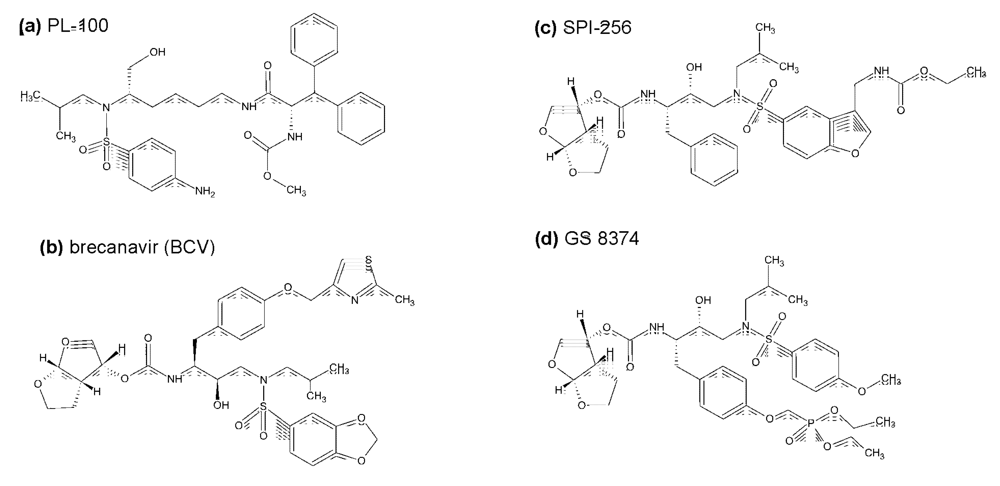
3. Inhibitors of HIV protease in the pipeline
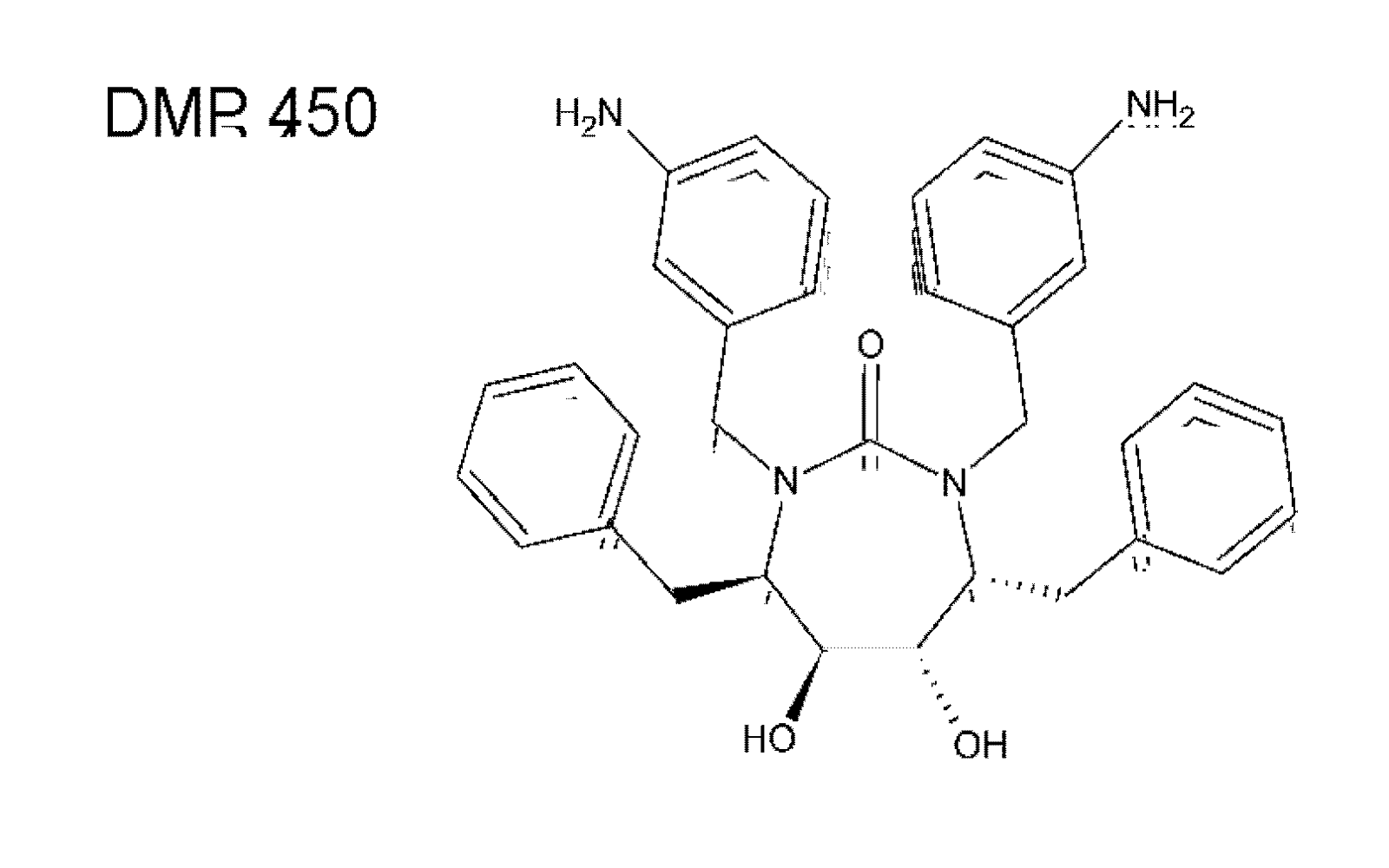
4. Other non-peptidic HIV protease active site inhibitors
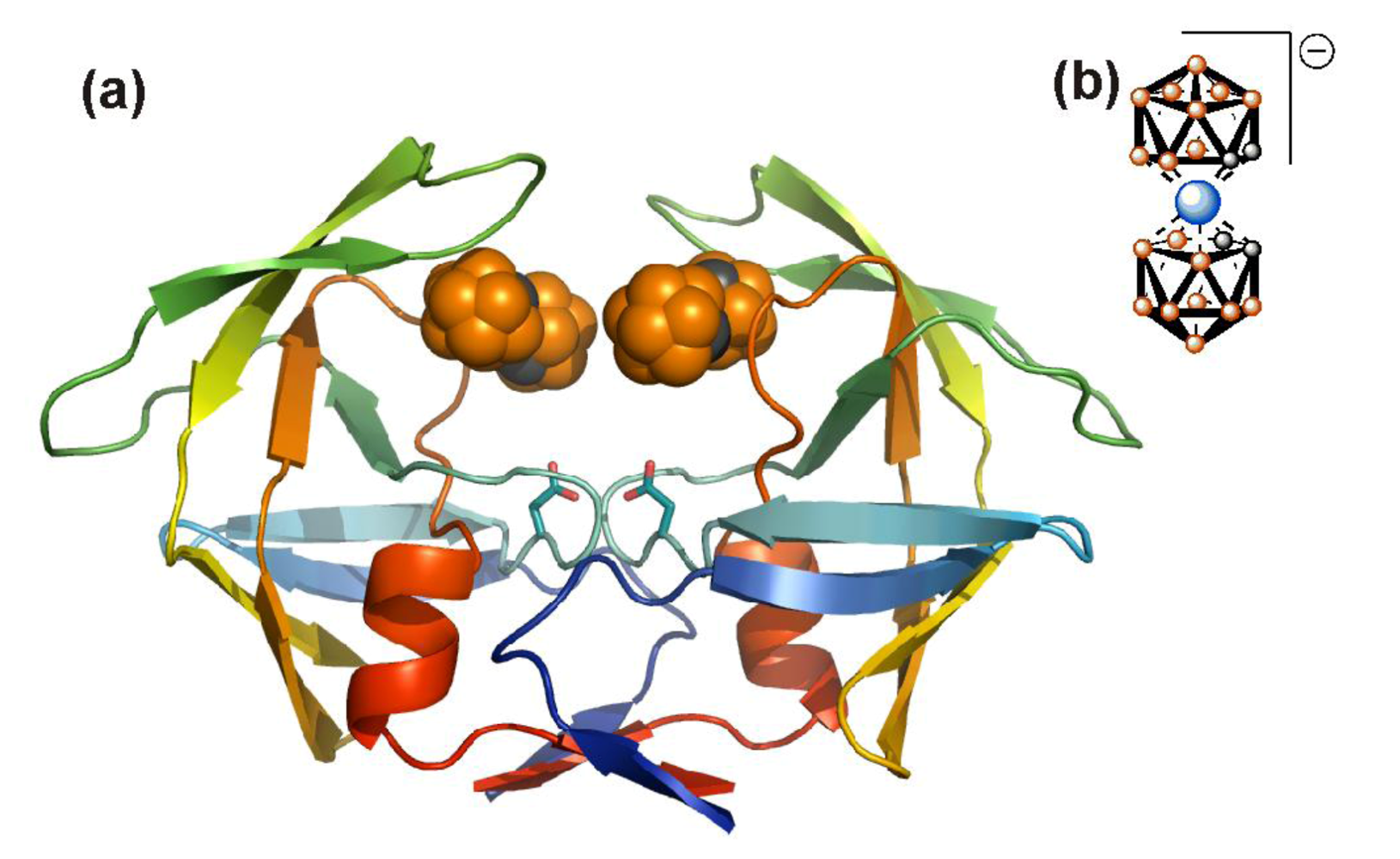
5. Alternative HIV protease inhibitors targeting functional domains outside the enzyme active site
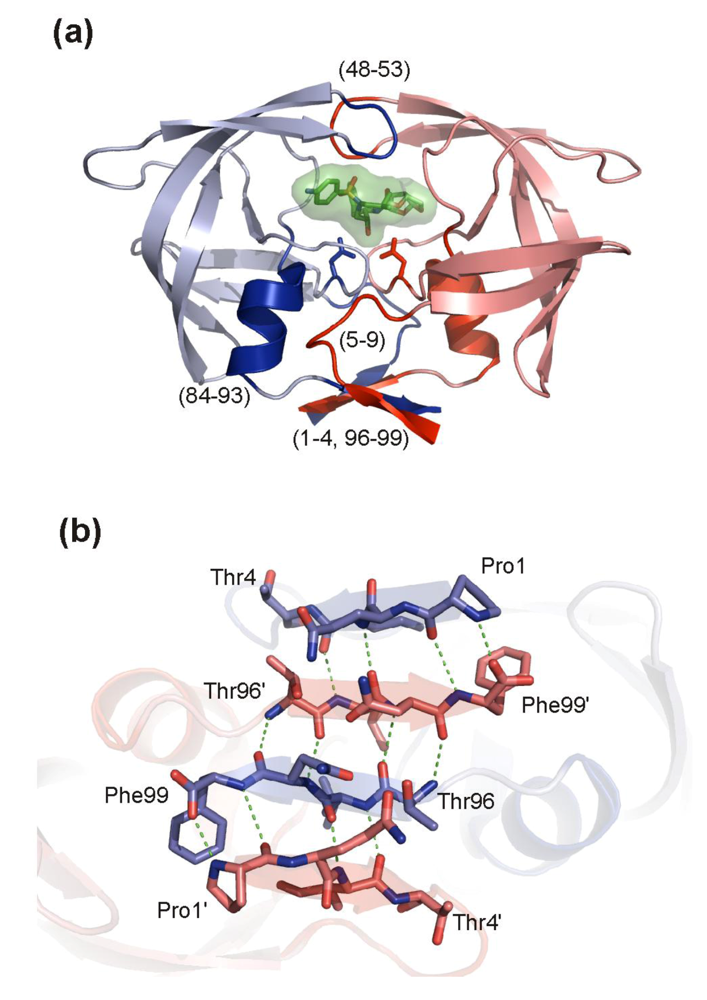
5.1. HIV PR dimerization inhibitors
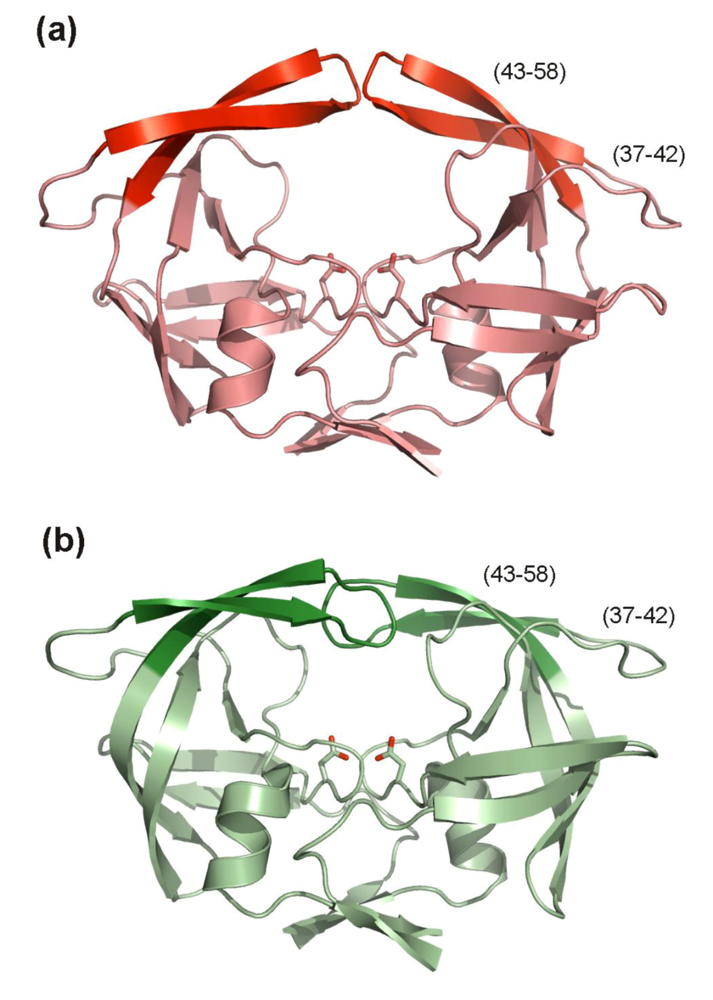
5.2. Inhibitors targeting HIV PR flaps
6. Conclusions
Acknowledgments
References
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. U S A 1988, 85, 4686–4690. [Google Scholar] [CrossRef] [PubMed]
- Winters, M.A.; Merigan, T.C. Insertions in the human immunodeficiency virus type 1 protease and reverse transcriptase genes: clinical impact and molecular mechanisms. Antimicrob. Agents Chemother. 2005, 49, 2575–2582. [Google Scholar] [CrossRef] [PubMed]
- Kozisek, M.; Saskova, K.G.; Rezacova, P.; Brynda, J.; van Maarseveen, N.M.; de Jong, D.; Boucher, C.A.; Kagan, R.M.; Nijhuis, M.; Konvalinka, J. Ninety-nine is not enough: molecular characterization of inhibitor-resistant human immunodeficiency virus type 1 protease mutants with insertions in the flap region. J. Virol. 2008, 82, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Doyon, L.; Croteau, G.; Thibeault, D.; Poulin, F.; Pilote, L.; Lamarre, D. Second locus involved in human immunodeficiency virus type 1 resistance to protease inhibitors. J. Virol. 1996, 70, 3763–3769. [Google Scholar] [PubMed]
- Mammano, F.; Petit, C.; Clavel, F. Resistance-associated loss of viral fitness in human immunodeficiency virus type 1: phenotypic analysis of protease and gag coevolution in protease inhibitor-treated patients. J. Virol. 1998, 72, 7632–7637. [Google Scholar] [PubMed]
- Nijhuis, M.; van Maarseveen, N.M.; Lastere, S.; Schipper, P.; Coakley, E.; Glass, B.; Rovenska, M.; de Jong, D.; Chappey, C.; Goedegebuure, I.W.; Heilek-Snyder, G.; Dulude, D.; Cammack, N.; Brakier-Gingras, L.; Konvalinka, J.; Parkin, N.; Krausslich, H.G.; Brun-Vezinet, F.; Boucher, C. A. A novel substrate-based HIV-1 protease inhibitor drug resistance mechanism. PLoS Med. 2007, 4, 152–163. [Google Scholar] [CrossRef]
- Johnson, V.A.; Brun-Vezinet, F.; Clotet, B.; Gunthard, H.F.; Kuritzkes, D.R.; Pillay, D.; Schapiro, J.M.; Richman, D.D. Update of the Drug Resistance Mutations in HIV-1. Top HIV Med. 2008, 16, 138–145. [Google Scholar] [PubMed]
- Saskova, K.G.; Kozisek, M.; Rezacova, P.; Brynda, J.; Yashina, T.; Kagan, R.M.; Konvalinka, J. Molecular characterization of clinical isolates of human immunodeficiency virus resistant to the protease inhibitor darunavir. J. Virol. 2009, 83, 8810–8818. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System . DeLano Scientific: Palo Alto, CA, USA, 2002. [Google Scholar]
- Nolan, D.; Reiss, P.; Mallal, S. Adverse effects of antiretroviral therapy for HIV infection: a review of selected topics. Expert Opin. Drug Saf. 2005, 4, 201–218. [Google Scholar]
- Shibuyama, S.; Gevorkyan, A.; Yoo, U.; Tim, S.; Dzhangiryan, K.; Scott, J. Understanding and avoiding antiretroviral adverse events. Curr. Pharm. Des. 2006, 12, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Wohl, D.; McComsey, G.; Tebas, P.; Brown, T.; Glesby, M.; Reeds, D.; Shikuma, C.; Mulligan, K.; Dube, M.; Wininger, D.; Huang, J.; Revuelta, M.; Currier, J.; Swindells, S.; Fichtenbaum, C.; Basar, M.; Tungsiripat, M.; Meyer, W.; Weihe, J.; Wanke, C. Current concepts in the diagnosis and management of metabolic complications of HIV infection and its therapy. Clin. Infect Dis. 2006, 43, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu. Rev. Biophys. Biomol. Struct. 1998, 27, 249–284. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A. Rational approach to AIDS drug design through structural biology. Annu. Rev. Med. 2002, 53, 595–614. [Google Scholar] [CrossRef] [PubMed]
- Prejdova, J.; Soucek, M.; Konvalinka, J. Determining and overcoming resistance to HIV protease inhibitors. Curr. Drug Targets Infect. Disord. 2004, 4, 137–152. [Google Scholar] [CrossRef]
- de Clercq, E. New approaches toward anti-HIV chemotherapy. J. Med. Chem. 2005, 48, 1297–1313. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.D.; Das, D.; Mitsuya, H. Overcoming HIV drug resistance through rational drug design based on molecular, biochemical, and structural profiles of HIV resistance. Cell Mol. Life Sci. 2006, 63, 1706–1724. [Google Scholar] [CrossRef] [PubMed]
- Mastrolorenzo, A.; Rusconi, S.; Scozzafava, A.; Barbaro, G.; Supuran, C.T. Inhibitors of HIV-1 protease: current state of the art 10 years after their introduction. From antiretroviral drugs to antifungal, antibacterial and antitumor agents based on aspartic protease inhibitors. Curr. Med. Chem. 2007, 14, 2734–2748. [Google Scholar] [CrossRef] [PubMed]
- de Clercq, E. The history of antiretrovirals: key discoveries over the past 25 years. Rev. Med. Virol. 2009, 19, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Schiffer, C.A.; Lee, S.K.; Swanstrom, R. Viral protease inhibitors. In Handbook of Experimental Pharmacology; Kräusslich, H.G., Bartenschlager, R., Eds.; Springer: Berlin, Heidelberg, Germany, 2009; Volume 189, pp. 85–110. [Google Scholar]
- Gulnik, S.V.; Afonina, E.; Eissenstaat, M. HIV-1 protease inhibitors as antiretroviral agents. Lu, C., Li, A.P., Eds.; John Wiley and Sons, Inc, 2009. [Google Scholar]
- Witvrouw, M.; Pannecouque, C.; Switzer, W.M.; Folks, T.M.; de Clercq, E.; Heneine, W. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antivir. Ther. 2004, 9, 57–65. [Google Scholar] [PubMed]
- Mallewa, J.E.; Wilkins, E.; Vilar, J.; Mallewa, M.; Doran, D.; Back, D.; Pirmohamed, M. HIV-associated lipodystrophy: a review of underlying mechanisms and therapeutic options. J. Antimicrob. Chemother. 2008, 62, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, G.; Iacobellis, G. Metabolic syndrome associated with HIV and highly active antiretroviral therapy. Curr. Diab. Rep. 2009, 9, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Flint, O.P.; Noor, M.A.; Hruz, P.W.; Hylemon, P.B.; Yarasheski, K.; Kotler, D.P.; Parker, R.A.; Bellamine, A. The role of protease inhibitors in the pathogenesis of HIV-associated lipodystrophy: cellular mechanisms and clinical implications. Toxicol. Pathol. 2009, 37, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Duvivier, C.; Kolta, S.; Assoumou, L.; Ghosn, J.; Rozenberg, S.; Murphy, R.L.; Katlama, C.; Costagliola, D. Greater decrease in bone mineral density with protease inhibitor regimens compared with nonnucleoside reverse transcriptase inhibitor regimens in HIV-1 infected naive patients. AIDS 2009, 27, 817–824. [Google Scholar] [CrossRef]
- Polli, J.W.; Jarrett, J.L.; Studenberg, S.D.; Humphreys, J.E.; Dennis, S.W.; Brouwer, K.R.; Woolley, J.L. Role of P-glycoprotein on the CNS disposition of amprenavir (141W94), an HIV protease inhibitor. Pharm. Res. 1999, 16, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Sacktor, N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr. Opin. Neurol. 2006, 19, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Kwara, A.; Delong, A.; Rezk, N.; Hogan, J.; Burtwell, H.; Chapman, S.; Moreira, C.C.; Kurpewski, J.; Ingersoll, J.; Caliendo, A.M.; Kashuba, A.; Cu-Uvin, S. Antiretroviral drug concentrations and HIV RNA in the genital tract of HIV-infected women receiving long-term highly active antiretroviral therapy. Clin. Infec. Dis. 2008, 46, 719–725. [Google Scholar] [CrossRef]
- Lowe, S.H.; Wensing, A.M.; Droste, J.A.; ten Kate, R.W.; Jurriaans, S.; Burger, D.M.; Borleffs, J.C.; Lange, J.M.; Prins, J.M. No virological failure in semen during properly suppressive antiretroviral therapy despite subtherapeutic local drug concentrations. HIV Clin Trials 2006, 7, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.E. Protease-inhibitor boosting in the treatment-experienced patient. AIDS Rev. 2004, 6, 226–233. [Google Scholar] [PubMed]
- Xu, L.; Desai, M.C. Pharmacokinetic enhancers for HIV drugs. Curr. Opin. Investig. Drugs 2009, 10, 775–786. [Google Scholar] [PubMed]
- Youle, M. Overview of boosted protease inhibitors in treatment-experienced HIV-infected patients. J. Antimicrob. Chemother. 2007, 60, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Winston, A.; Boffito, M. The management of HIV-1 protease inhibitor pharmacokinetic interactions. J. Antimicrob. Chemother. 2005, 56, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Roberts, N.A.; Martin, J.A.; Kinchington, D.; Broadhurst, A.V.; Craig, J.C.; Duncan, I.B.; Galpin, S.A.; Handa, B.K.; Kay, J.; Krohn, A.; et al. Rational design of peptide-based HIV proteinase inhibitors. Science 1990, 248, 358–361. [Google Scholar] [PubMed]
- Kilby, J.M.; Sfakianos, G.; Gizzi, N.; Siemon-Hryczyk, P.; Ehrensing, E.; Oo, C.; Buss, N.; Saag, M. S. Safety and pharmacokinetics of once-daily regimens of soft-gel capsule saquinavir plus minidose ritonavir in human immunodeficiency virus-negative adults. Antimicrob. Agents Chemother. 2000, 44, 2672–2678. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Noble, S. Saquinavir soft-gel capsule formulation. A review of its use in patients with HIV infection. Drugs 1998, 55, 461–486. [Google Scholar] [CrossRef] [PubMed]
- Kempf, D.J.; Marsh, K.C.; Fino, L.C.; Bryant, P.; Craig-Kennard, A.; Sham, H.L.; Zhao, C.; Vasavanonda, S.; Kohlbrenner, W.E.; Wideburg, N.E.; et al. Design of orally bioavailable, symmetry-based inhibitors of HIV protease. Bioorg. Med. Chem. 1994, 2, 847–858. [Google Scholar] [CrossRef]
- Moyle, G.J.; Back, D. Principles and practice of HIV-protease inhibitor pharmacoenhancement. HIV Med. 2001, 2, 105–113. [Google Scholar] [CrossRef]
- Dorsey, B.D.; Levin, R.B.; McDaniel, S.L.; Vacca, J.P.; Guare, J.P.; Darke, P.L.; Zugay, J.A.; Emini, E.A.; Schleif, W.A.; Quintero, J.C.; et al. L-735,524: the design of a potent and orally bioavailable HIV protease inhibitor. J. Med. Chem. 1994, 37, 3443–3451. [Google Scholar] [CrossRef] [PubMed]
- Nadler, R.B.; Rubenstein, J.N.; Eggener, S.E.; Loor, M.M.; Smith, N.D. The etiology of urolithiasis in HIV infected patients. J. Urol. 2003, 169, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Capaldini, L. Protease inhibitors' metabolic side effects: cholesterol, triglycerides, blood sugar, and "Crix belly." Interview with Lisa Capaldini, Interview by John S. James. AIDS Treat. News 1997, 277, 1–4. [Google Scholar] [PubMed]
- Patick, A.K.; Mo, H.; Markowitz, M.; Appelt, K.; Wu, B.; Musick, L.; Kalish, V.; Kaldor, S.; Reich, S.; Ho, D.; Webber, S. Antiviral and resistance studies of AG1343, an orally bioavailable inhibitor of human immunodeficiency virus protease. Antimicrob Agents Chemother. 1996, 40, 292–297. [Google Scholar] [PubMed]
- Kozisek, M.; Bray, J.; Rezacova, P.; Saskova, K.; Brynda, J.; Pokorna, J.; Mammano, F.; Rulisek, L.; Konvalinka, J. Molecular analysis of the HIV-1 resistance development: enzymatic activities, crystal structures, and thermodynamics of nelfinavir-resistant HIV protease mutants. J. Mol. Biol. 2007, 374, 1005–1016. [Google Scholar] [CrossRef] [PubMed]
- Bardsley-Elliot, A.; Plosker, G.L. Nelfinavir: an update on its use in HIV infection. Drugs 2000, 59, 581–620. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.E.; Baker, C.T.; Dwyer, M.D.; Murcko, M.A.; Rao, B.G.; Tung, R.D.; Navia, M.A. Crystal structure of HIV-1 protease in complex with VX-478, a potent and orally bioavailable inhibitor of the enzyme. J. Am. Chem. Soc. 1995, 117, 1181–1182. [Google Scholar] [CrossRef]
- Maguire, M.F.; Guinea, R.; Griffin, P.; Macmanus, S.; Elston, R.C.; Wolfram, J.; Richards, N.; Hanlon, M.H.; Porter, D.J.; Wrin, T.; Parkin, N.; Tisdale, M.; Furfine, E.; Petropoulos, C.; Snowden, B.W.; Kleim, J.P. Changes in human immunodeficiency virus type 1 Gag at positions L449 and P453 are linked to I50V protease mutants in vivo and cause reduction of sensitivity to amprenavir and improved viral fitness in vitro. J. Virol. 2002, 76, 7398–7406. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.P.; Qian, D.; Edmondson-Melancon, H.; Sattler, F.R.; Goodwin, D.; Martinez, C.; Williams, V.; Johnson, D.; Buchanan, T.A. Prospective, intensive study of metabolic changes associated with 48 weeks of amprenavir-based antiretroviral therapy. Clin. Infect. Dis. 2002, 35, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Adkins, J.C.; Faulds, D. Amprenavir. Drugs 1998, 55, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Floridia, M.; Bucciardini, R.; Fragola, V.; Galluzzo, C.M.; Giannini, G.; Pirillo, M.F.; Amici, R.; Andreotti, M.; Ricciardulli, D.; Tomino, C.; Vella, S. Risk factors and occurrence of rash in HIV-positive patients not receiving nonnucleoside reverse transcriptase inhibitor: data from a randomized study evaluating use of protease inhibitors in nucleoside-experienced patients with very low CD4 levels (<50 cells/microL). HIV Med. 2004, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vierling, P.; Greiner, J. Prodrugs of HIV protease inhibitors. Curr. Pharm. Des. 2003, 9, 1755–1770. [Google Scholar] [CrossRef] [PubMed]
- Torres, H.A.; Arduino, R.C. Fosamprenavir calcium plus ritonavir for HIV infection. Expert Rev. Anti Infect. Ther. 2007, 5, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Sham, H.L.; Kempf, D.J.; Molla, A.; Marsh, K.C.; Kumar, G.N.; Chen, C.M.; Kati, W.; Stewart, K.; Lal, R.; Hsu, A.; Betebenner, D.; Korneyeva, M.; Vasavanonda, S.; McDonald, E.; Saldivar, A.; Wideburg, N.; Chen, X.; Niu, P.; Park, C.; Jayanti, V.; Grabowski, B.; Granneman, G.R.; Sun, E.; Japour, A.J.; Leonard, J.M.; Plattner, J.J.; Norbeck, D.W. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob. Agents Chemother. 1998, 42, 3218–3224. [Google Scholar] [PubMed]
- Kempf, D.J.; Isaacson, J.D.; King, M.S.; Brun, S.C.; Xu, Y.; Real, K.; Bernstein, B.M.; Japour, A.J.; Sun, E.; Rode, R.A. Identification of genotypic changes in human immunodeficiency virus protease that correlate with reduced susceptibility to the protease inhibitor lopinavir among viral isolates from protease inhibitor-experienced patients. J. Virol. 2001, 75, 7462–7469. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, A.; Stewart, K.D.; Sham, H.L.; Norbeck, D.W.; Kohlbrenner, W.E.; Leonard, J.M.; Kempf, D.J.; Molla, A. In vitro selection and characterization of human immunodeficiency virus type 1 variants with increased resistance to ABT-378, a novel protease inhibitor. J. Virol. 1998, 72, 7532–7541. [Google Scholar] [PubMed]
- de Mendoza, C.; Valer, L.; Bacheler, L.; Pattery, T.; Corral, A.; Soriano, V. Prevalence of the HIV-1 protease mutation I47A in clinical practice and association with lopinavir resistance. AIDS 2006, 20, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Saskova, K.G.; Kozisek, M.; Lepsik, M.; Brynda, J.; Rezacova, P.; Vaclavikova, J.; Kagan, R.M.; Machala, L.; Konvalinka, J. Enzymatic and structural analysis of the I47A mutation contributing to the reduced susceptibility to HIV protease inhibitor lopinavir. Protein Sci. 2008, 17, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Masse, S.; Lu, X.; Dekhtyar, T.; Lu, L.; Koev, G.; Gao, F.; Mo, H.; Kempf, D.; Bernstein, B.; Hanna, G.J.; Molla, A. In vitro selection and characterization of human immunodeficiency virus type 2 with decreased susceptibility to lopinavir. Antimicrob. Agents Chemother. 2007, 51, 3075–3080. [Google Scholar] [CrossRef] [PubMed]
- Kagan, R.M.; Shenderovich, M.D.; Heseltine, P.N.; Ramnarayan, K. Structural analysis of an HIV-1 protease I47A mutant resistant to the protease inhibitor lopinavir. Protein Sci. 2005, 14, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, R.S.; Goa, K.L. Lopinavir/ritonavir: a review of its use in the management of HIV infection. Drugs 2003, 63, 769–802. [Google Scholar] [CrossRef] [PubMed]
- Bold, G.; Fassler, A.; Capraro, H.G.; Cozens, R.; Klimkait, T.; Lazdins, J.; Mestan, J.; Poncioni, B.; Rosel, J.; Stover, D.; Tintelnot-Blomley, M.; Acemoglu, F.; Beck, W.; Boss, E.; Eschbach, M.; Hurlimann, T.; Masso, E.; Roussel, S.; Ucci-Stoll, K.; Wyss, D.; Lang, M. New aza-dipeptide analogues as potent and orally absorbed HIV-1 protease inhibitors: candidates for clinical development. J. Med. Chem. 1998, 41, 3387–3401. [Google Scholar] [CrossRef] [PubMed]
- Le Tiec, C.; Barrail, A.; Goujard, C.; Taburet, A.M. Clinical pharmacokinetics and summary of efficacy and tolerability of atazanavir. Clin. Pharmacokinet. 2005, 44, 1035–1050. [Google Scholar] [CrossRef] [PubMed]
- Poppe, S.M.; Slade, D.E.; Chong, K.T.; Hinshaw, R.R.; Pagano, P.J.; Markowitz, M.; Ho, D.D.; Mo, H.; Gorman, R.R.; Dueweke, T.J.; Thaisrivongs, S.; Tarpley, W.G. Antiviral activity of the dihydropyrone PNU-140690, a new nonpeptidic human immunodeficiency virus protease inhibitor. Antimicrob. Agents Chemother. 1997, 41, 1058–1063. [Google Scholar] [PubMed]
- Muzammil, S.; Armstrong, A.A.; Kang, L.W.; Jakalian, A.; Bonneau, P.R.; Schmelmer, V.; Amzel, L.M.; Freire, E. Unique thermodynamic response of tipranavir to human immunodeficiency virus type 1 protease drug resistance mutations. J. Virol. 2007, 81, 5144–5154. [Google Scholar] [CrossRef] [PubMed]
- Larder, B.A.; Hertogs, K.; Bloor, S.; van den Eynde, C.H.; DeCian, W.; Wang, Y.; Freimuth, W.W.; Tarpley, G. Tipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samples. AIDS 2000, 14, 1943–1948. [Google Scholar] [CrossRef] [PubMed]
- Plosker, G.L.; Figgitt, D.P. Tipranavir. Drugs 2003, 63, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Macias, J.; Orihuela, F.; Rivero, A.; Viciana, P.; Marquez, M.; Portilla, J.; Rios, M.J.; Munoz, L.; Pasquau, J.; Castano, M.A.; Abdel-Kader, L.; Pineda, J.A. Hepatic safety of tipranavir plus ritonavir (TPV/r)-based antiretroviral combinations: effect of hepatitis virus co-infection and pre-existing fibrosis. J. Antimicrob. Chemother. 2009, 63, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Arbuthnot, C.; Wilde, J.T. Increased risk of bleeding with the use of tipranavir boosted with ritonavir in haemophilic patients. Haemophilia 2008, 14, 140–141. [Google Scholar] [PubMed]
- King, J.R.; Acosta, E.P. Tipranavir: a novel nonpeptidic protease inhibitor of HIV. Clin. Pharmacokinet. 2006, 45, 665–682. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.; Nakata, H.; Maeda, K.; Ogata, H.; Bilcer, G.; Devasamudram, T.; Kincaid, J.F.; Boross, P.; Wang, Y.F.; Tie, Y.; Volarath, P.; Gaddis, L.; Harrison, R.W.; Weber, I.T.; Ghosh, A.K.; Mitsuya, H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents. Chemother. 2003, 47, 3123–3129. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J. Mol. Biol. 2000, 301, 1207–1220. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, S.; Kiran Kumar Reddy, G.S.; Ali, A.; Nalam, M.N.; Anjum, S.G.; Cao, H.; Kairys, V.; Fernandes, M.X.; Altman, M.D.; Tidor, B.; Rana, T.M.; Schiffer, C.A.; Gilson, M.K. Design of mutation-resistant HIV protease inhibitors with the substrate envelope hypothesis. Chem. Biol. Drug Des. 2007, 69, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, E.; Schiffer, C.A. Resilience to resistance of HIV-1 protease inhibitors: profile of darunavir. AIDS Rev 2008, 10, 131–142. [Google Scholar] [PubMed]
- Kovalevsky, A.Y.; Liu, F.; Leshchenko, S.; Ghosh, A.K.; Louis, J.M.; Harrison, R.W.; Weber, I.T. Ultra-high resolution crystal structure of HIV-1 protease mutant reveals two binding sites for clinical inhibitor TMC114. J. Mol. Biol. 2006, 363, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Kovalevsky, A.Y.; Ghosh, A.K.; Weber, I.T. Solution kinetics measurements suggest HIV-1 protease has two binding sites for darunavir and amprenavir. J. Med. Chem. 2008, 51, 6599–6603. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.; Matsumi, S.; Das, D.; Amano, M.; Davis, D.A.; Li, J.; Leschenko, S.; Baldridge, A.; Shioda, T.; Yarchoan, R.; Ghosh, A.K.; Mitsuya, H. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 2007, 282, 28709–28720. [Google Scholar] [CrossRef] [PubMed]
- King, N.M.; Prabu-Jeyabalan, M.; Nalivaika, E.A.; Wigerinck, P.; de Bethune, M.P.; Schiffer, C.A. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J. Virol. 2004, 78, 12012–12021. [Google Scholar] [CrossRef] [PubMed]
- Dierynck, I.; Keuleers, I.; de Wit, M.; Tahri, A.; Surleraux, D.L.; Peeters, A.; Hertogs, K. Kinetic characterization of the potent activity of TMC114 on wild-type HIV-1 protease. Abstracts of 14th International HIV Drug Resistance Workshop 2005, 64. [Google Scholar]
- Clotet, B.; Bellos, N.; Molina, J.M.; Cooper, D.; Goffard, J.C.; Lazzarin, A.; Wohrmann, A.; Katlama, C.; Wilkin, T.; Haubrich, R.; Cohen, C.; Farthing, C.; Jayaweera, D.; Markowitz, M.; Ruane, P.; Spinosa-Guzman, S.; Lefebvre, E. Efficacy and safety of darunavir-ritonavir at week 48 in treatment-experienced patients with HIV-1 infection in POWER 1 and 2: a pooled subgroup analysis of data from two randomised trials. Lancet 2007, 369, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Madruga, J.V.; Berger, D.; McMurchie, M.; Suter, F.; Banhegyi, D.; Ruxrungtham, K.; Norris, D.; Lefebvre, E.; de Bethune, M.P.; Tomaka, F.; de Pauw, M.; Vangeneugden, T.; Spinosa-Guzman, S. Efficacy and safety of darunavir-ritonavir compared with that of lopinavir-ritonavir at 48 weeks in treatment-experienced, HIV-infected patients in TITAN: a randomised controlled phase III trial. Lancet 2007, 370, 49–58. [Google Scholar] [CrossRef] [PubMed]
- de Meyer, S.; Dierynck, I.; Lathouwers, E.; Van Baelen, B.; Vangeneugden T.; Spinosa-Guzman, S.; Peeters, M.; Picchio, G.; Bethune, M. P. Phenotypic and genotypic determinants of resistance to darunavir: analysis of data from treatment-experienced patients in POWER 1,2,3 and DUET-1 and 2 . Antivir. Ther. 2008, 13, A33. [Google Scholar]
- McKeage, K.; Perry, C.M.; Keam, S.J. Darunavir: a review of its use in the management of HIV infection in adults. Drugs 2009, 69, 477–503. [Google Scholar] [CrossRef] [PubMed]
- Dandache, S.; Sevigny, G.; Yelle, J.; Stranix, B.R.; Parkin, N.; Schapiro, J.M.; Wainberg, M.A.; Wu, J.J. In vitro antiviral activity and cross-resistance profile of PL-100, a novel protease inhibitor of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2007, 51, 4036–4043. [Google Scholar] [CrossRef] [PubMed]
- Nalam, M.N.; Peeters, A.; Jonckers, T.H.; Dierynck, I.; Schiffer, C.A. Crystal structure of lysine sulfonamide inhibitor reveals the displacement of the conserved flap water molecule in human immunodeficiency virus type 1 protease. J. Virol. 2007, 81, 9512–9518. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.; Harvey, R.; Ferris, R.; Craig, C.; Yates, P.; Griffin, P.; Miller, J.; Kaldor, I.; Ray, J.; Samano, V.; Furfine, E.; Spaltenstein, A.; Hale, M.; Tung, R.; St Clair, M.; Hanlon, M.; Boone, L. In vitro antiviral activity of the novel, tyrosyl-based human immunodeficiency virus (HIV) type 1 protease inhibitor brecanavir (GW640385) in combination with other antiretrovirals and against a panel of protease inhibitor-resistant HIV. Antimicrob. Agents Chemother. 2007, 51, 3147–3154. [Google Scholar] [CrossRef] [PubMed]
- Wynne, B.; Holland, A.; Ruff, D.; Guttendorf, R. A First-in-Human Study Evaluating the Safety, Tolerability, and Pharmacokinetics (PK) of SPI-256, a Novel HIV Protease Inhibitor (PI),Administered Alone and in Combination with Ritonavir (RTV) in HealthyAdult Subjects. 2008. [Google Scholar]
- Cihlar, T.; He, G.X.; Liu, X.; Chen, J.M.; Hatada, M.; Swaminathan, S.; McDermott, M.J.; Yang, Z.Y.; Mulato, A.S.; Chen, X.; Leavitt, S.A.; Stray, K.M.; Lee, W.A. Suppression of HIV-1 protease inhibitor resistance by phosphonate-mediated solvent anchoring. J. Mol. Biol. 2006, 363, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, C.; Stray, K.; Tsai, L.; Xu, L.; Lee, W.; Cihlar, T. In vitro HIV-1 resistance selection to GS-8374, novelphosphonate protease inhibitor: comparison with lopinavir, atazanavir and darunavir. 2007. [Google Scholar]
- Gulnik, S.V.; Eissenstat, M. Approaches to the design of HIV protease inhibitors with improved resistance profiles. Curr. Opin. HIV AIDS 2008, 3, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Sridhar, P.R.; Leshchenko, S.; Hussain, A.K.; Li, J.; Kovalevsky, A.Y.; Walters, D.E.; Wedekind, J.E.; Grum-Tokars, V.; Das, D.; Koh, Y.; Maeda, K.; Gatanaga, H.; Weber, I.T.; Mitsuya, H. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J. Med .Chem. 2006, 49, 5252–5261. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Leshchenko-Yashchuk, S.; Anderson, D.D.; Baldridge, A.; Noetzel, M.; Miller, H. J. Med. Chem. 2009, 52, 3902–3914. [CrossRef] [PubMed]
- Lam, P.Y.; Jadhav, P.K.; Eyermann, C.J.; Hodge, C.N.; Ru, Y.; Bacheler, L.T.; Meek, J.L.; Otto, M.J.; Rayner, M.M.; Wong, Y.N.; et al. Rational design of potent, bioavailable, nonpeptide cyclic ureas as HIV protease inhibitors. Science 1994, 263, 380–384. [Google Scholar] [PubMed]
- Erickson-Viitanen, S.; Klabe, R.M.; Cawood, P.G.; O'Neal, P.L.; Meek, J.L. Potency and selectivity of inhibition of human immunodeficiency virus protease by a small nonpeptide cyclic urea, DMP 323. Antimicrob. Agents Chemother. 1994, 38, 1628–1634. [Google Scholar] [PubMed]
- Lam, P.Y.; Ru, Y.; Jadhav, P.K.; Aldrich, P.E.; DeLucca, G.V.; Eyermann, C.J.; Chang, C.H.; Emmett, G.; Holler, E.R.; Daneker, W.F.; Li, L.; Confalone, P.N.; McHugh, R.J.; Han, Q.; Li, R.; Markwalder, J.A.; Seitz, S.P.; Sharpe, T.R.; Bacheler, L.T.; Rayner, M.M.; Klabe, R.M.; Shum,L.; Winslow, D.L.; Kornhauser, D.M.; Hodge, C.N.; et al. Cyclic HIV protease inhibitors: synthesis, conformational analysis, P2/P2' structure-activity relationship, and molecular recognition of cyclic ureas. J. Med. Chem. 1996, 39, 3514–3525. [Google Scholar] [CrossRef] [PubMed]
- Hulten, J.; Bonham, N.M.; Nillroth, U.; Hansson, T.; Zuccarello, G.; Bouzide, A.; Aqvist, J.; Classon, B.; Danielson, U.H.; Karlen, A.; Kvarnstrom, I.; Samuelsson, B.; Hallberg, A. Cyclic HIV-1 protease inhibitors derived from mannitol: synthesis, inhibitory potencies, and computational predictions of binding affinities. J. Med. Chem. 1997, 40, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Backbro, K.; Lowgren, S.; Osterlund, K.; Atepo, J.; Unge, T.; Hulten, J.; Bonham, N.M.; Schaal, W.; Karlen, A.; Hallberg, A. Unexpected binding mode of a cyclic sulfamide HIV-1 protease inhibitor. J. Med. Chem. 1997, 40, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Judd, D.A.; Nettles, J.H.; Nevins, N.; Snyder, J.P.; Liotta, D.C.; Tang, J.; Ermolieff, J.; Schinazi, R.F.; Hill, C.L. Polyoxometalate HIV-1 protease inhibitors. A new mode of protease inhibition. J. Am. Chem. Soc. 2001, 123, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Bosi, S.; Da Ros, T.; Spalluto, G.; Prato, M. Fullerene derivatives: an attractive tool for biological applications. Eur. J. Med. Chem. 2003, 38, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.H.; DeCamp, D.L.; Sijbesma, R.P.; Srdanov, G.; Wudl, F.; Kenyon, G.L. Inhibition of the HIV-1 protease by fullerene derivatives: model building studies and experimental verification. J. Am. Chem. Soc. 1993, 115, 6506–6509. [Google Scholar] [CrossRef]
- Sijbesma, R.; Srdanov, G.; Wudl, F.; Castoro, J.A.; Wilkins, C.; Friedman, S.H.; DeCamp, D.L.; Kenyon, G.L. Synthesis of a fullerene derivative for the inhibition of HIV enzymes. J. Am. Chem. Soc. 1993, 115, 6510–6512. [Google Scholar] [CrossRef]
- Cigler, P.; Kozisek, M.; Rezacova, P.; Brynda, J.; Otwinowski, Z.; Pokorna, J.; Plesek, J.; Gruner, B.; Doleckova-Maresova, L.; Masa, M.; Sedlacek, J.; Bodem, J.; Krausslich, H.G.; Kral, V.; Konvalinka, J. From nonpeptide toward noncarbon protease inhibitors: metallacarboranes as specific and potent inhibitors of HIV protease. Proc. Natl. Acad. Sci. U S A 2005, 102, 15394–15399. [Google Scholar] [CrossRef] [PubMed]
- Lesnikowski, Z.J. Boron Units as Pharmacophores ----- New Applications and Opportunities of Boron Cluster Chemistry. Coll. Czech CC. 2007, 72, 1646–1658. [Google Scholar] [CrossRef]
- Armstrong, A.F.; Valliant, J.F. The bioinorganic and medicinal chemistry of carboranes: from new drug discovery to molecular imaging and therapy. Dalton Trans. 2007, 4240–4251. [Google Scholar] [CrossRef]
- Kozisek, M.; Cigler, P.; Lepsik, M.; Fanfrlik, J.; Rezacova, P.; Brynda, J.; Pokorna, J.; Plesek, J.; Gruner, B.; Grantz Saskova, K.; Vaclavikova, J.; Kral, V.; Konvalinka, J. Inorganic polyhedral metallacarborane inhibitors of HIV protease: a new approach to overcoming antiviral resistance. J. Med. Chem. 2008, 51, 4839–4843. [Google Scholar] [CrossRef] [PubMed]
- Řezáčová, P.; Pokorná, J.; Brynda, J.; Kožíšek, M.; Cígler, P.; Lepšík, M.; Fanfrlík, J.; Řezáč, J.; Šašková, K. G.; Sieglová, I.; Plešek, J.; Šícha, V.; Grüner, B.; Oberwinkler, H.; Sedláček, J.; Kräusslich, H.G.; Hobza, P.; Král, V.; Konvalinka, J. Design of HIV protease inhibitors based on inorganic polyhedral metallacarboranes. J. Med. Chem. 2009, 52, 7132–7141. [Google Scholar] [CrossRef] [PubMed]
- Kairys, V.; Gilson, M.K.; Lather, V.; Schiffer, C.A.; Fernandes, M.X. Toward the design of mutation-resistant enzyme inhibitors: further evaluation of the substrate envelope hypothesis. Chem. Biol. Drug Des. 2009, 74, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Altman, M.D.; Ali, A.; Reddy, G.S.; Nalam, M.N.; Anjum, S.G.; Cao, H.; Chellappan, S.; Kairys, V.; Fernandes, M.X.; Gilson, M.K.; Schiffer, C.A.; Rana, T.M.; Tidor, B. HIV-1 protease inhibitors from inverse design in the substrate envelope exhibit subnanomolar binding to drug-resistant variants. J. Am. Chem. Soc. 2008, 130, 6099–6113. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, G.; Johnston, K.; Cohen, J.C.; Gallaher, W.R.; Robinson, J.; Luftig, R.B. PCR amplification of HIV-1 proteinase sequences directly from lab isolates allows determination of five conserved domains. Virology 1992, 190, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ingr, M.; Uhlikova, T.; Strisovsky, K.; Majerova, E.; Konvalinka, J. Kinetics of the dimerization of retroviral proteases: the "fireman's grip" and dimerization. Protein Sci. 2003, 12, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.S.; Yin, F.H.; Foundling, S.; Blomstrom, D.; Kettner, C.A. Stability and activity of human immunodeficiency virus protease: comparison of the natural dimer with a homologous, single-chain tethered dimer. Proc. Natl. Acad. Sci. U S A 1990, 87, 9660–9664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Poorman, R.A.; Maggiora, L.L.; Heinrikson, R.L.; Kezdy, F.J. Dissociative inhibition of dimeric enzymes. Kinetic characterization of the inhibition of HIV-1 protease by its COOH-terminal tetrapeptide. J. Biol. Chem. 1991, 266, 15591–15594. [Google Scholar] [PubMed]
- Jordan, S.P.; Zugay, J.; Darke, P.L.; Kuo, L.C. Activity and dimerization of human immunodeficiency virus protease as a function of solvent composition and enzyme concentration. J. Biol. Chem. 1992, 267, 20028–20032. [Google Scholar] [PubMed]
- Kuzmic, P. Kinetic assay for HIV proteinase subunit dissociation. Biochem. Biophys. Res. Commun. 1993, 191, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Darke, P.L.; Jordan, S.P.; Hall, D.L.; Zugay, J.A.; Shafer, J.A.; Kuo, L.C. Dissociation and association of the HIV-1 protease dimer subunits: equilibria and rates. Biochemistry 1994, 33, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Pargellis, C.A.; Morelock, M.M.; Graham, E.T.; Kinkade, P.; Pav, S.; Lubbe, K.; Lamarre, D.; Anderson, P.C. Determination of kinetic rate constants for the binding of inhibitors to HIV-1 protease and for the association and dissociation of active homodimer. Biochemistry 1994, 33, 12527–12534. [Google Scholar] [CrossRef] [PubMed]
- Uhlikova, T.; Konvalinka, J.; Pichova, I.; Soucek, M.; Krausslich, H.G.; Vondrasek, J. A modular approach to HIV-1 proteinase inhibitor design. Biochem. Biophys. Res. Commun. 1996, 222, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Weber, I.T. Comparison of the crystal structures and intersubunit interactions of human immunodeficiency and Rous sarcoma virus proteases. J. Biol. Chem. 1990, 265, 10492–10496. [Google Scholar] [PubMed]
- Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B.K.; Baldwin, E.; Weber, I.T.; Selk, L.M.; Clawson, L.; Schneider, J.; Kent, S.B. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. [Google Scholar] [PubMed]
- Todd, M.J.; Semo, N.; Freire, E. The structural stability of the HIV-1 protease. J. Mol. Biol. 1998, 283, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Babe, L.M.; Rose, J.; Craik, C.S. Synthetic "interface" peptides alter dimeric assembly of the HIV 1 and 2 proteases. Protein Sci. 1992, 1, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Schramm, H.J.; Boetzel, J.; Buttner, J.; Fritsche, E.; Gohring, W.; Jaeger, E.; Konig, S.; Thumfart, O.; Wenger, T.; Nagel, N.E.; Schramm, W. The inhibition of human immunodeficiency virus proteases by 'interface peptides'. Antiviral Res . 1996, 30, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Zutshi, R.; Franciskovich, J.; Shultz, M.; Schweitzer, B.; Bishop, P.; Wilson, M.; Chmielewski, J. Targetting the Dimerization Interface of HIV-1 Protease: Inhibition with Cross-Linked Interfacial Peptides. J. Am. Chem. Soc. 1997, 119, 4841–4845. [Google Scholar] [CrossRef]
- Shultz, M.D.; Chmielewski, J. Probing the role of interfacial residues in a dimerization inhibitor of HIV-1 protease. Bioorg. Med. Chem. Lett. 1999, 9, 2431–2436. [Google Scholar] [CrossRef]
- Bouras, A.; Boggetto, N.; Benatalah, Z.; de Rosny, E.; Sicsic, S.; Reboud-Ravaux, M. Design, synthesis, and evaluation of conformationally constrained tongs, new inhibitors of HIV-1 protease dimerization. J. Med. Chem. 1999, 42, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, L.; Kessler, A.; Pethe, S.; Collinet, B.; Merabet, N.; Boggetto, N.; Sicsic, S.; Reboud-Ravaux, M.; Ongeri, S. Molecular tongs containing amino acid mimetic fragments: new inhibitors of wild-type and mutated HIV-1 protease dimerization. J. Med. Chem. 2006, 49, 4657–4664. [Google Scholar] [CrossRef] [PubMed]
- Bowman, M.J.; Chmielewski, J. Sidechain-linked inhibitors of HIV-1 protease dimerization. Bioorg. Med. Chem. 2009, 17, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Schramm, H.J.; de Rosny, E.; Reboud-Ravaux, M.; Buttner, J.; Dick, A.; Schramm, W. Lipopeptides as dimerization inhibitors of HIV-1 protease. Biol. Chem. 1999, 380, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Dumond, J.; Boggetto, N.; Schramm, H.J.; Schramm, W.; Takahashi, M.; Reboud-Ravaux, M. Thyroxine-derivatives of lipopeptides: bifunctional dimerization inhibitors of human immunodeficiency virus-1 protease. Biochem. Pharmacol. 2003, 65, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, L.; Rose, T.; Dufau, L.; Vanderesse, R.; Dumond, J.; Jamart-Gregoire, B.; Pannecouque, C.; de Clercq, E.; Reboud-Ravaux, M. Dimer disruption and monomer sequestration by alkyl tripeptides are successful strategies for inhibiting wild-type and multidrug-resistant mutated HIV-1 proteases. Biochemistry 2009, 48, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Zutshi, R.; Chmielewski, J. Targeting the dimerization interface for irreversible inhibition of HIV-1 protease. Bioorg. Med. Chem. Lett. 2000, 10, 1901–1903. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.; Brown, C.A.; Singer, K.E.; Wang, V.; Kaufman, J.; Stahl, S.J.; Wingfield, P.; Maeda, K.; Harada, S.; Yoshimura, K.; Kosalaraksa, P.; Mitsuya, H.; Yarchoan, R. Inhibition of HIV-1 replication by a peptide dimerization inhibitor of HIV-1 protease. Antiviral Res. 2006, 72, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.A.; Tebbs, I.R.; Daniels, S.I.; Stahl, S.J.; Kaufman, J.D.; Wingfield, P.; Bowman, M.J.; Chmielewski, J.; Yarchoan, R. Analysis and characterization of dimerization inhibition of a multi-drug-resistant human immunodeficiency virus type 1 protease using a novel size-exclusion chromatographic approach. Biochem. J. 2009, 419, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Lescar, J.; Brynda, J.; Rezacova, P.; Stouracova, R.; Riottot, M.M.; Chitarra, V.; Fabry, M.; Horejsi, M.; Sedlacek, J.; Bentley, G.A. Inhibition of the HIV-1 and HIV-2 proteases by a monoclonal antibody. Protein Sci. 1999, 8, 2686–2696. [Google Scholar] [CrossRef]
- Bartonova, V.; Kral, V.; Sieglova, I.; Brynda, J.; Fabry, M.; Horejsi, M.; Kozisek, M.; Saskova, K.G.; Konvalinka, J.; Sedlacek, J.; Rezacova, P. Potent inhibition of drug-resistant HIV protease variants by monoclonal antibodies. Antiviral Res. 2008, 78, 275–277. [Google Scholar] [PubMed]
- Rezacova, P.; Lescar, J.; Brynda, J.; Fabry, M.; Horejsi, M.; Sedlacek, J.; Bentley, G.A. Structural basis of HIV-1 and HIV-2 protease inhibition by a monoclonal antibody. Structure 2001, 9, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Rezacova, P.; Brynda, J.; Lescar, J.; Fabry, M.; Horejsi, M.; Sieglova, I.; Sedlacek, J.; Bentley, G.A. Crystal structure of a cross-reaction complex between an anti-HIV-1 protease antibody and an HIV-2 protease peptide. J. Struct. Biol. 2005, 149, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Frutos, S.; Rodriguez-Mias, R.A.; Madurga, S.; Collinet, B.; Reboud-Ravaux, M.; Ludevid, D.; Giralt, E. Disruption of the HIV-1 protease dimer with interface peptides: structural studies using NMR spectroscopy combined with [2-(13)C]-Trp selective labeling. Biopolymers 2007, 88, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Busschots, K.; De Rijck, J.; Christ, F.; Debyser, Z. In search of small molecules blocking interactions between HIV proteins and intracellular cofactors. Mol. Biosyst. 2009, 5, 21–31. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Bio. 2008, 18, 203–217. [Google Scholar] [CrossRef]
- Shimba, N.; Nomura, A.M.; Marnett, A.B.; Craik, C.S. Herpesvirus protease inhibition by dimer disruption. J. Virol. 2004, 78, 6657–6665. [Google Scholar] [CrossRef] [PubMed]
- Camarasa, M.J.; Velazquez, S.; San-Felix, A.; Perez-Perez, M.J.; Gago, F. Dimerization inhibitors of HIV-1 reverse transcriptase, protease and integrase: a single mode of inhibition for the three HIV enzymes? Antiviral Res. 2006, 71, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Simmerling, C. Targeting structural flexibility in HIV-1 protease inhibitor binding. Drug Discov. Today 2007, 12, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ishima, R.; Freedberg, D.I.; Wang, Y.X.; Louis, J.M.; Torchia, D.A. Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease, and their implications for function. Structure 1999, 7, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Liu, Q.Z.; Alzari, P.M.; Hirel, P.H.; Poljak, R.J. The three-dimensional structure of the aspartyl protease from the HIV-1 isolate BRU. Biochimie 1991, 73, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, J.; Blum, A.; Dorr, S.; Heine, A.; Diederich, W.E.; Klebe, G. Targeting the open-flap conformation of HIV-1 protease with pyrrolidine-based inhibitors. Chem. Med. Chem. 2008, 3, 1337–1344. [Google Scholar]
- Lescar, J.; Stouracova, R.; Riottot, M.M.; Chitarra, V.; Brynda, J.; Fabry, M.; Horejsi, M.; Sedlacek, J.; Bentley, G.A. Preliminary crystallographic studies of an anti-HIV-1 protease antibody that inhibits enzyme activity. Protein Sci. 1996, 5, 966–968. [Google Scholar] [PubMed]
- Lescar, J.; Stouracova, R.; Riottot, M.M.; Chitarra, V.; Brynda, J.; Fabry, M.; Horejsi, M.; Sedlacek, J.; Bentley, G.A. Three-dimensional structure of an Fab-peptide complex: structural basis of HIV-1 protease inhibition by a monoclonal antibody. J. Mol. Biol. 1997, 267, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Montero, J.V.; Barreiro, P.; Soriano, V. HIV protease inhibitors: recent clinical trials and recommendations on use. Expert Opin. Pharmacother. 2009, 10, 1615–1629. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; Pujari, S. Protease inhibitor therapy in resource-limited settings. Curr. Opin. HIV 2008, 3, 612–619. [Google Scholar] [CrossRef]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Pokorná, J.; Machala, L.; Řezáčová, P.; Konvalinka, J. Current and Novel Inhibitors of HIV Protease. Viruses 2009, 1, 1209-1239. https://doi.org/10.3390/v1031209
Pokorná J, Machala L, Řezáčová P, Konvalinka J. Current and Novel Inhibitors of HIV Protease. Viruses. 2009; 1(3):1209-1239. https://doi.org/10.3390/v1031209
Chicago/Turabian StylePokorná, Jana, Ladislav Machala, Pavlína Řezáčová, and Jan Konvalinka. 2009. "Current and Novel Inhibitors of HIV Protease" Viruses 1, no. 3: 1209-1239. https://doi.org/10.3390/v1031209
APA StylePokorná, J., Machala, L., Řezáčová, P., & Konvalinka, J. (2009). Current and Novel Inhibitors of HIV Protease. Viruses, 1(3), 1209-1239. https://doi.org/10.3390/v1031209