Interplay between Herpesvirus Infection and Host Defense by PML Nuclear Bodies


Abstract
1. Introduction
2. Structural Aspects of ND10
3. The Major ND10 Constituents and Their Functions
3.1. PML
3.2. hDaxx
3.3. Sp100
4. Functions of ND10
5. ND10 during Herpesvirus Infections
6. Alpha- Herpesviruses and ND10
6.1. Herpes Simplex Virus Type 1 (HSV-1)
6.2. Varicella Zoster Virus (VZV)
7. Beta-Herpesviruses and ND10
8. Gamma-Herpesviruses and ND10
8.1. Epstein-Barr Virus (EBV)
8.2. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV)
9. Concluding Remarks
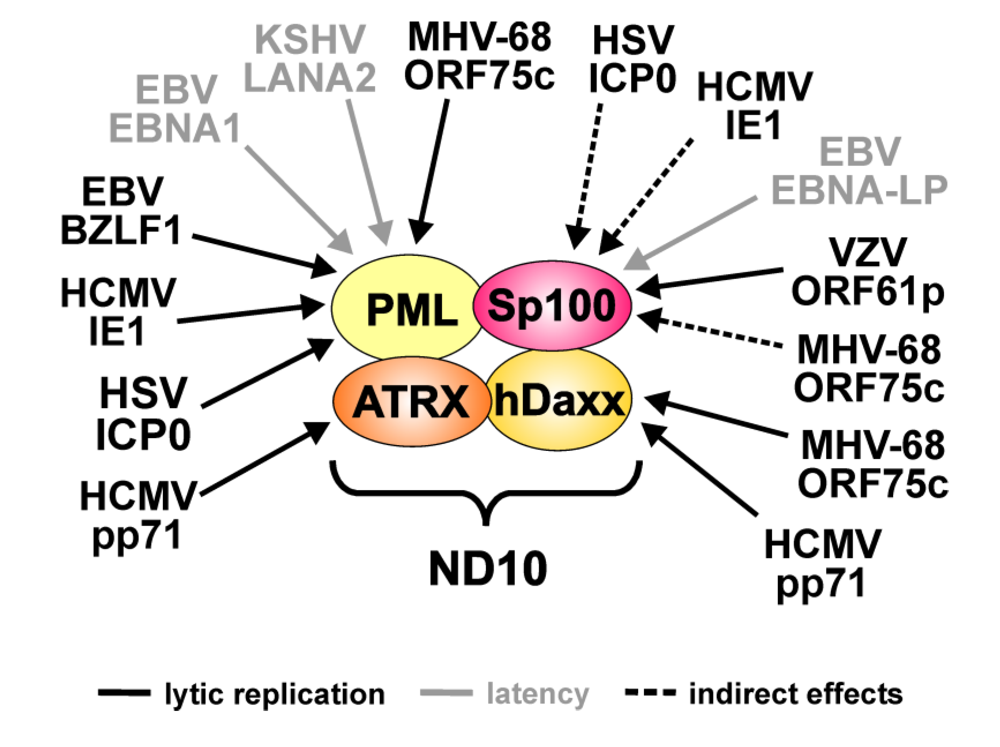

{kind=link}
| Virus | Viral protein | ND10 target | Effects |
|---|---|---|---|
| HSV-1 | ICP0 | PML | Proteasomal degradation of all isoforms |
| Sp100 | Loss of low mobility isoforms (indirect effect: linkage to PML metabolism) | ||
| VZV | ORF61p | Sp100 | Downregulation of all isoforms |
| HCMV | IE1 | PML | Loss of SUMOylated variants |
| Sp100 | Loss of low mobility isoforms (indirect effect: linkage to PML metabolism) | ||
| pp71 | hDaxx | Proteasomal degradation | |
| ATRX | Release from ND10 | ||
| EBV | BZLF-1 | PML | Loss of SUMOylated variants during lytic replication |
| EBNA-LP | Sp100 | Release from ND10 | |
| EBNA1 | PML | Proteasomal degradation during latent infection | |
| KSHV | LANA2 | PML | Proteasomal degradation during latent infection |
| MHV-68 | ORF75c | PML | Proteasomal degradation of all isoforms |
| Sp100 | Loss of low mobility isoforms (indirect effect: linkage to PML metabolism) | ||
| hDaxx | Downregulation |
Acknowledgments
References
- Bieniasz, P.D. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Negorev, D.; Maul, G.G. Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene 2001, 20, 7234–7242. [Google Scholar] [CrossRef] [PubMed]
- Stuurman, N.; de Graaf, A.; Floore, A.; Josso, A.; Humbel, B.; de, J.L.; van Driel, R. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J. Cell Sci. 1992, 101, 773–784. [Google Scholar] [PubMed]
- Ascoli, C.A.; Maul, G.G. Identification of a novel nuclear domain. J. Cell Biol. 1991, 112, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. Interactions between DNA viruses, ND10 and the DNA damage response. Cell Microbiol. 2006, 8, 365–374. [Google Scholar] [CrossRef]
- Dellaire, G.; Bazett-Jones, D.P. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays 2004, 26, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Dellaire, G.; Bazett-Jones, D.P. Beyond repair foci: subnuclear domains and the cellular response to DNA damage. Cell Cycle 2007, 6, 1864–1872. [Google Scholar] [PubMed]
- Maul, G.G.; Jensen, D.E.; Ishov, A.M.; Herlyn, M.; Rauscher III, F.J. Nuclear redistribution of BRCA1 during viral infection. Cell Growth Differ. 1998, 9, 743-755. [Google Scholar] [PubMed]
- Wileman, T. Aggresomes and pericentriolar sites of virus assembly: cellular defense or viral design? Annu. Rev. Microbiol. 2007, 61, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Gao, Y.S.; Tousson, A.; Shah, A.; Chen, T.L.; Vertel, B.M.; Sztul, E. Nuclear aggresomes form by fusion of PML-associated aggregates. Mol. Biol. Cell 2005, 16, 4905–4917. [Google Scholar] [CrossRef] [PubMed]
- Szostecki, C.; Guldner, H.H.; Netter, H.J.; Will, H. Isolation and characterization of cDNA encoding a human nuclear antigen predominantly recognized by autoantibodies from patients with primary biliary cirrhosis. J. Immunol. 1990, 145, 4338–4347. [Google Scholar] [PubMed]
- Zhong, S.; Muller, S.; Ronchetti, S.; Freemont, P.S.; Dejean, A.; Pandolfi, P.P. Role of SUMO-1-modified PML in nuclear body formation. Blood 2000, 95, 2748–2753. [Google Scholar] [PubMed]
- Ishov, A.M.; Sotnikov, A.G.; Negorev, D.; Vladimirova, O.V.; Neff, N.; Kamitani, T.; Yeh, E.T.H.; Strauss III, J.F.; Maul, G.G. PML is critical for ND10 formation and recruits the PML-interacting protein Daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 1999, 147, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Rechter, S.; Papior, P.; Tavalai, N.; Stamminger, T.; Orr, A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 2006, 80, 7995–8005. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Leis, M.; Stamminger, T. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 2006, 80, 8006–8018. [Google Scholar] [CrossRef] [PubMed]
- Kakizuka, A.; Miller Jr., W.H.; Umesono, K.; Warrell Jr., R.P.; Frankel, S.R.; Murty, V.V.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991, 66, 663-674. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Shiels, C.; Freemont, P.S. PML protein isoforms and the RBCC/TRIM motif. Oncogene 2001, 20, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Le, X.F.; Yang, P.; Chang, K.S. Analysis of the growth and transformation suppressor domains of promyelocytic leukemia gene, PML. J. Biol. Chem. 1996, 271, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Nojima, T.; Oshiro-Ideue, T.; Nakanoya, H.; Kawamura, H.; Morimoto, T.; Kawaguchi, Y.; Kataoka, N.; Hagiwara, M. Herpesvirus protein ICP27 switches PML isoform by altering mRNA splicing. Nucleic Acids Res. 2009, 37, 6515–6527. [Google Scholar] [CrossRef] [PubMed]
- McNally, B.A.; Trgovcich, J.; Maul, G.G.; Liu, Y.; Zheng, P. A role for cytoplasmic PML in cellular resistance to viral infection. PLoS ONE 2008, 3, e2277. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Lomonte, P.; Sternsdorf, T.; van, D.R.; Orr, A. Cell cycle regulation of PML modification and ND10 composition. J. Cell Sci. 1999, 112, 4581–4588. [Google Scholar] [PubMed]
- Kamitani, T.; Nguyen, H.P.; Kito, K.; Fukuda-Kamitani, T.; Yeh, E.T. Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 1998, 273, 3117–3120. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Matunis, M.J.; Dejean, A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 1998, 17, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, T.; Kito, K.; Nguyen, H.P.; Wada, H.; Fukuda-Kamitani, T.; Yeh, E.T. Identification of three major sentrinization sites in PML. J. Biol. Chem. 1998, 273, 26675–26682. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.H.; Lin, H.K.; Scaglioni, P.P.; Yung, T.M.; Pandolfi, P.P. The mechanisms of PML-nuclear body formation. Mol. Cell 2006, 24, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.Y.; Huang, Y.S.; Jeng, J.C.; Kuo, H.Y.; Chang, C.C.; Chao, T.T.; Ho, C.C.; Chen, Y.C.; Lin, T.P.; Fang, H.I. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 2006, 24, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, A.D.; Sublett, J.E.; McPherson, C.J.; Grosveld, G. The Pax3-FKHR oncoprotein is unresponsive to the Pax3-associated repressor hDaxx. EMBO J. 1999, 18, 3702–3711. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidou, M.; Driscoll, D.A.; Lopez-Guisa, J.M.; Strauss III, J.F. Cloning and expression of primate Daxx cDNAs and mapping of the human gene to chromosome 6p21.3 in the MHC region. DNA Cell Biol. 1997, 16, 1289-1298. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leo, C.; Zhu, J.; Wu, X.; O'Neil, J.; Park, E.J.; Chen, J.D. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell Biol. 2000, 20, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.F.; Earnshaw, W.C.; Goldberg, I.G. Interphase-specific association of intrinsic centromere protein CENP-C with HDaxx, a death domain-binding protein implicated in Fas-mediated cell death. J. Cell Sci. 1998, 111, 2029–2041. [Google Scholar] [PubMed]
- Salomoni, P.; Khelifi, A.F. Daxx: death or survival protein? Trends Cell Biol. 2006, 16, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Pei, H.; Watson, D.K.; Papas, T.S. EAP1/Daxx interacts with ETS1 and represses transcriptional activation of ETS1 target genes. Oncogene 2000, 19, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, J.H.; La, M.; Jang, M.J.; Chae, G.W.; Kim, S.B.; Tak, H.; Jung, Y.; Byun, B.; Ahn, J.K. Inhibition of NF-kappaB acetylation and its transcriptional activity by Daxx. J. Mol. Biol. 2007, 368, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, J.S.; Leder, P. RNAi reveals anti-apoptotic and transcriptionally repressive activities of DAXX. J. Cell Sci. 2003, 116, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Lin, D.Y.; Fang, H.I.; Chen, R.H.; Shih, H.M. Daxx mediates the small ubiquitin-like modifier-dependent transcriptional repression of Smad4. J. Biol. Chem. 2005, 280, 10164–10173. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Park, J.S.; Um, S.J. Identification of Daxx interacting with p73, one of the p53 family, and its regulation of p53 activity by competitive interaction with PML. Nucleic Acids Res. 2003, 31, 5356–5367. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.Y.; Fang, H.I.; Ma, A.H.; Huang, Y.S.; Pu, Y.S.; Jenster, G.; Kung, H.J.; Shih, H.M. Negative modulation of androgen receptor transcriptional activity by Daxx. Mol. Cell Biol. 2004, 24, 10529–10541. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, A.D.; McPherson, C.J.; Mientjes, E.J.; Iyengar, R.; Grosveld, G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 2002, 115, 3319–3330. [Google Scholar] [PubMed]
- Muromoto, R.; Sugiyama, K.; Takachi, A.; Imoto, S.; Sato, N.; Yamamoto, T.; Oritani, K.; Shimoda, K.; Matsuda, T. Physical and functional interactions between Daxx and DNA methyltransferase 1-associated protein, DMAP1. J. Immunol. 2004, 172, 2985–2993. [Google Scholar] [PubMed]
- Ishov, A.M.; Vladimirova, O.V.; Maul, G.G. Heterochromatin and ND10 are cell-cycle regulated and phosphorylation-dependent alternate nuclear sites of the transcription repressor Daxx and SWI/SNF protein ATRX. J. Cell Sci. 2004, 117, 3807–3820. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Gibbons, R.; Yan, Z.; Yang, D.; McDowell, T.L.; Sechi, S.; Qin, J.; Zhou, S.; Higgs, D.; Wang, W. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 10635–10640. [Google Scholar] [CrossRef]
- Lin, D.Y.; Shih, H.M. Essential role of the 58-kDa microspherule protein in the modulation of Daxx-dependent transcriptional repression as revealed by nucleolar sequestration. J. Biol. Chem. 2002, 277, 25446–25456. [Google Scholar] [CrossRef] [PubMed]
- Dent, A.L.; Yewdell, J.; Puvion-Dutilleul, F.; Koken, M.H.; de Thé, H.; Staudt, L.M. LYSP100-associated nuclear domains (LANDs): description of a new class of subnuclear structures and their relationship to PML nuclear bodies. Blood 1996, 88, 1423–1426. [Google Scholar] [PubMed]
- Seeler, J.S.; Marchio, A.; Losson, R.; Desterro, J.M.; Hay, R.T.; Chambon, P.; Dejean, A. Common properties of nuclear body protein SP100 and TIF1alpha chromatin factor: role of SUMO modification. Mol. Cell Biol. 2001, 21, 3314–3324. [Google Scholar] [CrossRef] [PubMed]
- Seeler, J.S.; Marchio, A.; Sitterlin, D.; Transy, C.; Dejean, A. Interaction of SP100 with HP1 proteins: a link between the promyelocytic leukemia-associated nuclear bodies and the chromatin compartment. Proc. Natl. Acad. Sci. USA 1998, 95, 7316–7321. [Google Scholar] [CrossRef]
- Guldner, H.H.; Szostecki, C.; Schroder, P.; Matschl, U.; Jensen, K.; Luders, C.; Will, H.; Sternsdorf, T. Splice variants of the nuclear dot-associated Sp100 protein contain homologies to HMG-1 and a human nuclear phosphoprotein-box motif. J. Cell Sci. 1999, 112, 733–747. [Google Scholar] [PubMed]
- Bottomley, M.J.; Collard, M.W.; Huggenvik, J.I.; Liu, Z.; Gibson, T.J.; Sattler, M. The SAND domain structure defines a novel DNA-binding fold in transcriptional regulation. Nat. Struct. Biol. 2001, 8, 626–633. [Google Scholar] [CrossRef]
- Wilcox, K.W.; Sheriff, S.; Isaac, A.; Taylor, J.L. SP100B is a repressor of gene expression. J. Cell Biochem. 2005, 95, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Isaac, A.; Wilcox, K.W.; Taylor, J.L. SP100B, a repressor of gene expression preferentially binds to DNA with unmethylated CpGs. J. Cell Biochem. 2006, 98, 1106–1122. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.S.; Ryu, S.W.; Kim, E. Modification of Daxx by small ubiquitin-related modifier-1. Biochem. Biophys. Res. Commun. 2002, 295, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Sternsdorf, T.; Jensen, K.; Will, H. Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J. Cell Biol. 1997, 139, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Sternsdorf, T.; Jensen, K.; Reich, B.; Will, H. The nuclear dot protein sp100, characterization of domains necessary for dimerization, subcellular localization, and modification by small ubiquitin-like modifiers. J. Biol. Chem. 1999, 274, 12555–12566. [Google Scholar] [CrossRef] [PubMed]
- Salomoni, P.; Pandolfi, P.P. The role of PML in tumor suppression. Cell 2002, 108, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Lallemand-Breitenbach, V.; Zhu, J.; de Thé, H. PML nuclear bodies and apoptosis. Oncogene 2004, 23, 2819–2824. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Pandolfi, P.P. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene 2003, 22, 9048–9057. [Google Scholar] [CrossRef] [PubMed]
- Maul, G.G.; Yu, E.; Ishov, A.M.; Epstein, A.L. Nuclear domain 10 (ND10) associated proteins are also present in nuclear bodies and redistribute to hundreds of nuclear sites after stress. J. Cell Biochem. 1995, 59, 498–513. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O.; Kirsh, O.; Pearson, M.; Itahana, K.; Pelicci, P.G.; Dejean, A. Deconstructing PML-induced premature senescence. EMBO J. 2002, 21, 3358–3369. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; Zhu, J.; Puvion, F.; Koken, M.; Honore, N.; Doubeikovsky, A.; Duprez, E.; Pandolfi, P.P.; Puvion, E.; Freemont, P.; et al. et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 2001, 193, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Salomoni, P.; Pandolfi, P.P. The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2000, 2, E85-E90. [Google Scholar] [PubMed]
- Borden, K.L. Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol. Cell Biol. 2002, 22, 5259–5269. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef]
- Eskiw, C.H.; Bazett-Jones, D.P. The promyelocytic leukemia nuclear body: sites of activity? Biochem. Cell Biol. 2002, 80, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; Hendzel, M.J.; Bazett-Jones, D.P. Promyelocytic leukemia (PML) nuclear bodies are protein structures that do not accumulate RNA. J. Cell Biol. 2000, 148, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.M.; Chang, C.C.; Kuo, H.Y.; Lin, D.Y. Daxx mediates SUMO-dependent transcriptional control and subnuclear compartmentalization. Biochem. Soc. Trans. 2007, 35, 1397–1400. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.S.; Vallian, S.; Seto, E.; Yang, W.M.; Edmondson, D.; Roth, S.; Chang, K.S. The growth suppressor PML represses transcription by functionally and physically interacting with histone deacetylases. Mol. Cell Biol. 2001, 21, 2259–2268. [Google Scholar] [CrossRef] [PubMed]
- Puto, L.A.; Reed, J.C. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008, 22, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Di Croce, L.; Raker, V.A.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo, C.F.; Kouzarides, T.; Nervi, C. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Nomura, T.; Kim, H.; Kaul, S.C.; Wadhwa, R.; Shinagawa, T.; Ichikawa-Iwata, E.; Zhong, S.; Pandolfi, P.P.; Ishii, S. Role of PML and PML-RARalpha in Mad-mediated transcriptional repression. Mol. Cell 2001, 7, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, S.; Wiemann, S.; Will, H.; Hofmann, T.G. PML-associated repressor of transcription (PAROT), a novel KRAB-zinc finger repressor, is regulated through association with PML nuclear bodies. Exp. Cell Res. 2006, 312, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Carbone, R.; Botrugno, O.A.; Ronzoni, S.; Insinga, A.; Di Croce, L.; Pelicci, P.G.; Minucci, S. Recruitment of the histone methyltransferase SUV39H1 and its role in the oncogenic properties of the leukemia-associated PML-retinoic acid receptor fusion protein. Mol. Cell Biol. 2006, 26, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Villa, R.; Pasini, D.; Gutierrez, A.; Morey, L.; Occhionorelli, M.; Vire, E.; Nomdedeu, J.F.; Jenuwein, T.; Pelicci, P.G.; Minucci, S.; et al. et al. Role of the polycomb repressive complex 2 in acute promyelocytic leukemia. Cancer Cell 2007, 11, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; Kruhlak, M.J.; Box, A.K.; Hendzel, M.J.; Bazett-Jones, D.P. The transcription coactivator CBP is a dynamic component of the promyelocytic leukemia nuclear body. J. Cell Biol. 2001, 152, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- LaMorte, V.J.; Dyck, J.A.; Ochs, R.L.; Evans, R.M. Localization of nascent RNA and CREB binding protein with the PML-containing nuclear body. Proc. Natl. Acad. Sci. USA 1998, 95, 4991–4996. [Google Scholar] [CrossRef]
- Everett, R.D. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 2001, 20, 7266–7273. [Google Scholar] [CrossRef] [PubMed]
- Moller, A.; Schmitz, M.L. Viruses as hijackers of PML nuclear bodies. Arch. Immunol. Ther. Exp. (Warsz.) 2003, 51, 295–300. [Google Scholar] [PubMed]
- Maul, G.G. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 1998, 20, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Regad, T.; Chelbi-Alix, M.K. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 2001, 20, 7274–7286. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Chelbi-Alix, M.K.; Pelicano, L.; Quignon, F.; Koken, M.H.; Venturini, L.; Stadler, M.; Pavlovic, J.; Degos, L.; de Thé, H. Induction of the PML protein by interferons in normal and APL cells. Leukemia 1995, 9, 2027–2033. [Google Scholar] [PubMed]
- Guldner, H.H.; Szostecki, C.; Grotzinger, T.; Will, H. IFN enhance expression of Sp100, an autoantigen in primary biliary cirrhosis. J. Immunol. 1992, 149, 4067–4073. [Google Scholar] [PubMed]
- Stadler, M.; Chelbi-Alix, M.K.; Koken, M.H.; Venturini, L.; Lee, C.; Saib, A.; Quignon, F.; Pelicano, L.; Guillemin, M.C.; Schindler, C. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 1995, 11, 2565–2573. [Google Scholar] [PubMed]
- Grotzinger, T.; Jensen, K.; Will, H. The interferon (IFN)-stimulated gene Sp100 promoter contains an IFN-gamma activation site and an imperfect IFN-stimulated response element which mediate type I IFN inducibility. J. Biol. Chem. 1996, 271, 25253–25260. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Lee, J.S.; Oh, S.Y.; Jin, X.; Choi, Y.J.; Lee, T.H.; Lee, E.; Choi, Y.K.; You, S.; Chung, Y.G. Direct transcriptional activation of promyelocytic leukemia protein by IFN regulatory factor 3 induces the p53-dependent growth inhibition of cancer cells. Cancer Res. 2007, 67, 11133–11140. [Google Scholar] [CrossRef] [PubMed]
- Grotzinger, T.; Sternsdorf, T.; Jensen, K.; Will, H. Interferon-modulated expression of genes encoding the nuclear-dot-associated proteins Sp100 and promyelocytic leukemia protein (PML). Eur. J. Biochem. 1996, 238, 554–560. [Google Scholar] [PubMed]
- Maul, G.G.; Guldner, H.H.; Spivack, J.G. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 1993, 74, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Maul, G.G. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 1996, 134, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Maul, G.G.; Ishov, A.M.; Everett, R.D. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 1996, 217, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Maul, G.G. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994, 13, 5062–5069. [Google Scholar] [PubMed]
- Maul, G.G.; Everett, R.D. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 1994, 75, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 2000, 22, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Chelbi-Alix, M.K.; de Thé, H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 1999, 18, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Freemont, P.; Saitoh, H.; Dasso, M.; Orr, A.; Kathoria, M.; Parkinson, J. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 1998, 72, 6581–6591. [Google Scholar] [PubMed]
- Muller, S.; Dejean, A. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 1999, 73, 5137–5143. [Google Scholar] [PubMed]
- Parkinson, J.; Everett, R.D. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J. Virol. 2000, 74, 10006–10017. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Roizman, B. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 2003, 100, 8963–8968. [Google Scholar] [CrossRef]
- Harle, P.; Sainz Jr., B.; Carr, D.J.; Halford, W.P. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology 2002, 293, 295-304. [Google Scholar] [CrossRef] [PubMed]
- Mossman, K.L.; Saffran, H.A.; Smiley, J.R. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 2000, 74, 2052–2056. [Google Scholar] [CrossRef] [PubMed]
- Halford, W.P.; Weisend, C.; Grace, J.; Soboleski, M.; Carr, D.J.; Balliet, J.W.; Imai, Y.; Margolis, T.P.; Gebhardt, B.M. ICP0 antagonizes Stat 1-dependent repression of herpes simplex virus: implications for the regulation of viral latency. Virol. J. 2006, 3, 44. [Google Scholar] [CrossRef]
- Chee, A.V.; Lopez, P.; Pandolfi, P.P.; Roizman, B. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J. Virol. 2003, 77, 7101–7105. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Zafiropoulos, A. Visualization by live-cell microscopy of disruption of ND10 during herpes simplex virus type 1 infection. J. Virol. 2004, 78, 11411–11415. [Google Scholar] [CrossRef] [PubMed]
- Lopez, P.; Jacob, R.J.; Roizman, B. Overexpression of promyelocytic leukemia protein precludes the dispersal of ND10 structures and has no effect on accumulation of infectious herpes simplex virus 1 or its proteins. J. Virol. 2002, 76, 9355–9367. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Parada, C.; Gripon, P.; Sirma, H.; Orr, A. Replication of ICP0 null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 2007. [Google Scholar] [CrossRef]
- Negorev, D.G.; Vladimirova, O.V.; Maul, G.G. Differential functions of interferon-upregulated Sp100 isoforms: herpes simplex virus type 1 promoter-based immediate-early gene suppression and PML protection from ICP0-mediated degradation. J. Virol. 2009, 83, 5168–5180. [Google Scholar] [CrossRef] [PubMed]
- Negorev, D.G.; Vladimirova, O.V.; Ivanov, A.; Rauscher III, F.; Maul, G.G. Differential role of Sp100 isoforms in interferon-mediated repression of herpes simplex virus type 1 immediate-early protein expression. J. Virol. 2006, 80, 8019-8029. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M. Varicella-zoster virus. Clin. Microbiol. Rev. 1996, 9, 361–381. [Google Scholar] [PubMed]
- Kyratsous, C.A.; Silverstein, S.J. Components of nuclear domain 10 bodies regulate varicella-zoster virus replication. J. Virol. 2009, 83, 4262–4274. [Google Scholar] [CrossRef] [PubMed]
- Moriuchi, H.; Moriuchi, M.; Smith, H.A.; Straus, S.E.; Cohen, J.I. Varicella-zoster virus open reading frame 61 protein is functionally homologous to herpes simplex virus type 1 ICP0. J. Virol. 1992, 66, 7303–7308. [Google Scholar] [PubMed]
- Moriuchi, H.; Moriuchi, M.; Straus, S.E.; Cohen, J.I. Varicella-zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. J. Virol. 1993, 67, 4290–4295. [Google Scholar] [PubMed]
- Kyratsous, C.A.; Walters, M.S.; Panagiotidis, C.A.; Silverstein, S.J. Complementation of a Herpes Simplex Virus ICP0 null mutant by Varicella Zoster Virus ORF61p. J. Virol. 2009, 83, 10637–10643. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Howley, P.; Griffin, D.E.; Lamb, R.A.; Martin, M.A.; Roizman, B.; Straus, S.E. Fields Virology, 5th ed2007; Lippincott-Williams and Wilkins: Philadelphia, PA, USA. [Google Scholar]
- Ahn, J.H.; Hayward, G.S. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol. 1997, 71, 4599–4613. [Google Scholar] [PubMed]
- Ishov, A.M.; Stenberg, R.M.; Maul, G.G. Human cytomegalovirus immediate early interaction with host nuclear structures: definition of an immediate transcript environment. J. Cell Biol. 1997, 138, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.W.; Kelly, C.; Sinclair, J.H.; Rickards, C. Disruption of PML-associated nuclear bodies mediated by the human cytomegalovirus major immediate early gene product. J. Gen. Virol. 1998, 79, 1233–1245. [Google Scholar] [PubMed]
- Korioth, F.; Maul, G.G.; Plachter, B.; Stamminger, T.; Frey, J. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp. Cell Res. 1996, 229, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Brignole III, E.J.; Hayward, G.S. Disruption of PML subnuclear domains by the acidic IE1 protein of human cytomegalovirus is mediated through interaction with PML and may modulate a RING finger-dependent cryptic transactivator function of PML. Mol. Cell Biol. 1998, 18, 4899-4913. [Google Scholar] [PubMed]
- Lee, H.R.; Kim, D.J.; Lee, J.M.; Choi, C.Y.; Ahn, B.Y.; Hayward, G.S.; Ahn, J.H. Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. J. Virol. 2004, 78, 6527–6542. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ahn, J.H.; Cheng, M.; apRhys, C.M.; Chiou, C.J.; Zong, J.; Matunis, M.J.; Hayward, G.S. Proteasome-independent disruption of PML oncogenic domains (PODs), but not covalent modification by SUMO-1, is required for human cytomegalovirus immediate-early protein IE1 to inhibit PML-mediated transcriptional repression. J. Virol. 2001, 75, 10683–10695. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, E.T.; Lee, H.R.; Park, J.J.; Go, Y.Y.; Choi, C.Y.; Ahn, J.H. Inhibition of SUMO-independent PML oligomerization by the human cytomegalovirus IE1 protein. J. Gen. Virol. 2006, 87, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Hayward, G.S. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology 2000, 274, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Sindre, H.; Stamminger, T. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 2002, 76, 5769–5783. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Vladimirova, O.V.; Maul, G.G. Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J. Virol. 2002, 76, 7705–7712. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.R.; Rowley, K.V.; Rinaldi, A.; Nicholson, I.P.; Ishov, A.M.; Maul, G.G.; Preston, C.M. Activity and intracellular localization of the human cytomegalovirus protein pp71. J. Gen. Virol. 2002, 83, 1601–1612. [Google Scholar] [PubMed]
- Cantrell, S.R.; Bresnahan, W.A. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J. Virol. 2005, 79, 7792–7802. [Google Scholar] [CrossRef] [PubMed]
- Woodhall, D.L.; Groves, I.J.; Reeves, M.B.; Wilkinson, G.; Sinclair, J.H. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 2006, 281, 37652–37660. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Kalejta, R.F. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 2006, 80, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.M.; Nicholl, M.J. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 2006, 87, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, S.R.; Bresnahan, W.A. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 2006, 80, 6188–6191. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Stamminger, T. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 2008, 82, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 2007, 367, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Kaspari, M.; Tavalai, N.; Stamminger, T.; Zimmermann, A.; Schilf, R.; Bogner, E. Proteasome inhibitor MG132 blocks viral DNA replication and assembly of human cytomegalovirus. FEBS Lett. 2008, 582, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J. Virol. 2009, 83, 6591–6598. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, V.; McFarlane, S.; Everett, R.D.; Preston, C.M. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008, 82, 12543–12554. [Google Scholar] [CrossRef] [PubMed]
- Bresnahan, W.A.; Shenk, T.E. UL82 virion protein activates expression of immediate early viral genes in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14506–14511. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 2007, 81, 9109–9120. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Sinclair, J.H. Knockdown of hDaxx in normally non-permissive undifferentiated cells does not permit human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 2007, 88, 2935–2940. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, J. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys Acta 2009. [Google Scholar]
- Torok, D.; Ching, R.W.; Bazett-Jones, D.P. PML nuclear bodies as sites of epigenetic regulation. Front Biosci. 2009, 14, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Reeves, M.B.; Sinclair, J.H. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic 'pre-immediate-early' repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 2009, 90, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Bennett, C.; Shenk, T. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J. Virol. 2008, 82, 9525–9536. [Google Scholar] [CrossRef] [PubMed]
- Raab-Traub, N. Epstein-Barr virus in the pathogenesis of NPC. Semin. Cancer Biol 2002, 12, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Bell, P.; Lieberman, P.M.; Maul, G.G. Lytic but not latent replication of epstein-barr virus is associated with PML and induces sequential release of nuclear domain 10 proteins. J. Virol. 2000, 74, 11800–11810. [Google Scholar] [CrossRef] [PubMed]
- Amon, W.; White, R.E.; Farrell, P.J. Epstein-Barr virus origin of lytic replication mediates association of replicating episomes with promyelocytic leukaemia protein nuclear bodies and replication compartments. J. Gen. Virol. 2006, 87, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Adamson, A.L.; Kenney, S. Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 2001, 75, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Chen, C.J.; Zerby, D.; Delecluse, H.J.; Lieberman, P.M. Identification of acidic and aromatic residues in the Zta activation domain essential for Epstein-Barr virus reactivation. J. Virol. 2001, 75, 10334–10347. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Moses, S.C.; Tan, J.; Kremmer, E.; Ling, P.D. The Epstein-Barr virus EBNA-LP protein preferentially coactivates EBNA2-mediated stimulation of latent membrane proteins expressed from the viral divergent promoter. J. Virol. 2005, 79, 4492–4505. [Google Scholar] [CrossRef] [PubMed]
- Szekely, L.; Pokrovskaja, K.; Jiang, W.Q.; de Thé, H.; Ringertz, N.; Klein, G. The Epstein-Barr virus-encoded nuclear antigen EBNA-5 accumulates in PML-containing bodies. J. Virol. 1996, 70, 2562–2568. [Google Scholar] [PubMed]
- Ling, P.D.; Peng, R.S.; Nakajima, A.; Yu, J.H.; Tan, J.; Moses, S.M.; Yang, W.H.; Zhao, B.; Kieff, E.; Bloch, K.D. Mediation of Epstein-Barr virus EBNA-LP transcriptional coactivation by Sp100. EMBO J. 2005, 24, 3565–3575. [Google Scholar] [CrossRef] [PubMed]
- Lehming, N.; Le, S.A.; Schuller, J.; Ptashne, M. Chromatin components as part of a putative transcriptional repressing complex. Proc. Natl. Acad. Sci. USA 1998, 95, 7322–7326. [Google Scholar] [CrossRef]
- Echendu, C.W.; Ling, P.D. Regulation of Sp100A subnuclear localization and transcriptional function by EBNA-LP and interferon. J. Interferon Cytokine Res. 2008, 28, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Sivachandran, N.; Sarkari, F.; Frappier, L. Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog. 2008, 4, e1000170. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Y.; Ahn, J.H.; Alcendor, D.J.; Jang, W.J.; Xiao, J.; Hayward, S.D.; Hayward, G.S. Origin-independent assembly of Kaposi's sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J. Virol. 2001, 75, 1487–1506. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Ogawa-Goto, K.; Hasegawa, H.; Kurata, T.; Sata, T. Human-herpesvirus-8-encoded K8 protein colocalizes with the promyelocytic leukemia protein (PML) bodies and recruits p53 to the PML bodies. Virology 2001, 286, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Villar, L.; Lopitz-Otsoa, F.; Gallego, P.; Munoz-Fontela, C.; Gonzalez-Santamaria, J.; Campagna, M.; Shou-Jiang, G.; Rodriguez, M.S.; Rivas, C. Kaposi's sarcoma-associated herpesvirus protein LANA2 disrupts PML oncogenic domains and inhibits PML-mediated transcriptional repression of the survivin gene. J. Virol. 2009, 83, 8849–8858. [Google Scholar] [CrossRef] [PubMed]
- Ling, P.D.; Tan, J.; Sewatanon, J.; Peng, R. The murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J. Virol. 2008. [Google Scholar]
- Gaspar, M.; Gill, M.B.; Losing, J.B.; May, J.S.; Stevenson, P.G. Multiple functions for ORF75c in murid herpesvirus-4 infection. PLoS ONE 2008, 3, e2781. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Tavalai, N.; Stamminger, T. Interplay between Herpesvirus Infection and Host Defense by PML Nuclear Bodies. Viruses 2009, 1, 1240-1264. https://doi.org/10.3390/v1031240
Tavalai N, Stamminger T. Interplay between Herpesvirus Infection and Host Defense by PML Nuclear Bodies. Viruses. 2009; 1(3):1240-1264. https://doi.org/10.3390/v1031240
Chicago/Turabian StyleTavalai, Nina, and Thomas Stamminger. 2009. "Interplay between Herpesvirus Infection and Host Defense by PML Nuclear Bodies" Viruses 1, no. 3: 1240-1264. https://doi.org/10.3390/v1031240
APA StyleTavalai, N., & Stamminger, T. (2009). Interplay between Herpesvirus Infection and Host Defense by PML Nuclear Bodies. Viruses, 1(3), 1240-1264. https://doi.org/10.3390/v1031240